
Introduction
The mammalian epidermis undergoes continuous self-renewal to repair damaged tissue and replace aged cells. A single layer of proliferative keratinocytes at the inner base of the epidermis gives rise to non-proliferative keratinocytes which renew the suprabasal (outer) layers, until they reach the outermost layer where they terminally differentiate and are shed [1].
In the human epidermis 14-3-3σ expression gradually increases along the axis of keratinocyte differentiation. Suppression of 14-3-3σ expression abrogates terminal keratinocyte differentiation and immortalizes human primary keratinocytes in cell culture [2, 3]. This suggests that elevated 14-3-3σ protein levels inhibit clonal expansion of cells within the epidermal basal layer and support keratinocyte differentiation.
The p63 protein is a master regulator of epidermal morphogenesis and is exclusively expressed in the basal cell layer. p63-null mice lack suprabasal layers and do not express differentiation markers [4]. ∆Np63α, an alternative p63 splice variant, transcriptionally represses 14-3-3σ expression [5]. Thus, p63 and 14-3-3σ exercise opposite functions in the epidermis. In line with this scenario, it has been reported that 14-3-3σ binds to ∆Np63α upon DNA damage and sequesters ∆Np63α to the cytoplasm to facilitate its proteasomal degradation [6]. Thereby, an elevated 14-3-3σ expression could prevent expansion of the basal keratinocyte layer.
In addition, many studies have demonstrated that 14-3-3σ expression is deregulated or lost in several types of human epithelial cancers [7, 8]. Silencing of the 14-3-3σ gene by CpG-methylation also frequently occurs in breast cancer [9]. Hence, loss of 14-3-3σ could be a necessary step for the initiation and/or progression of epithelial tumorigenesis.
14-3-3σ has been characterized as a p53-target genes that is sufficient and necessary to mediate a G2/M-arrest after DNA damage [10, 11]. 14-3-3σ proteins only form homodimers and have a distinct repertoire of ligand interactions when compared to the six other 14-3-3 isoforms expressed in human and murine cells [12, 13]. 14-3-3σ dimers bind to a large number of ligands via phosphorylated serine/threonine residues and thereby presumably affect a plethora of cellular processes [14]. Numerous studies based on cellular analyses have implicated 14-3-3σ in the regulation of diverse processes, such as cell cycle progression, signaling, differentiation, apoptosis and metabolism [7, 15-17]. However, relatively few studies of the organismal function and relevance of 14-3-3σ employing genetically engineered mouse models have been reported. By studying a mammary epithelial-specific 14-3-3σ knock-out mouse it was shown that 14-3-3σ-deficient mammary epithelial cells lose their polarity and show increased proliferation [18]. Amplification and elevated expression of the erbB2 proto-oncogene is associated with poor outcome in patients with breast cancer [19]. It has been shown that the Cre-mediated deletion of 14-3-3σ in an erbB2-driven breast cancer mouse model enhances tumor initiation and metastases [20]. Additionally, ectopic 14-3-3σ expression reduces the metastatic capacity of a human breast cancer cell line in a xenograft mouse model [21]. Therefore, 14-3-3σ shows tumor suppressive capacities in vivo.
The repeated epilation (ER) mouse model has been used multiple times for studying the role of 14-3-3σ in the epidermis. ER mutant mice harbor a single nucleotide insertion in the 14-3-3σ gene causing a frame shift which leads to the expression of a C-terminally truncated 14-3-3σ protein that lacks the nuclear export signal and several phosphopeptide-binding residues [22, 23]. Homozygous ER mutant mice (ER/ER) die at birth as a result of respiratory stress [22-24]: The oral cavity is fused, limbs and tail are shortened, defined digits and nails are lacking. Their epidermis is strongly hyper-proliferative and thickened accompanied with failures in terminal differentiation. The hair follicles are underdeveloped and sparse. Heterozygous ER mutant mice (+/ER) are viable. However, at postnatal day 7 they display a hyper-proliferative epidermis, however not as pronounced as observed in ER/ER mutant mice at embryonic day E18.5. Furthermore, +/ER mutant mice show defects in hair shaft differentiation, resulting in destruction of the hair shaft and a cyclic hair loss [25]. Studies analyzing this hair defect showed that the heterozygous ER-mutation in 14-3-3σ specifically affects the club hair retention [26] and causes severe defects in hair shaft differentiation during morphogenesis and abnormal cycling of the hair follicle stem cells in the bulge [25]. In addition, heterozygous ER-mice show an extensive hyper-proliferation of the epidermis followed by the development of multiple papillomas and squamous cell carcinoma at the age of 6 month [27]. ER-mice display increased levels of p63 in the epidermal suprabasal layers, suggesting that truncated 14-3-3σ fails to block the transcription of TP63 which may contribute to hyper-proliferation of the epidermis [28]. In addition, Yap1, an essential factor of the Hippo pathway controlling organ size and tissue homeostasis [29], is expressed in the epidermal suprabasal layers of ER-mice [30]. In keratinocytes derived from ER mice truncated 14-3-3σ fails to bind and sequester Yap1 in the cytoplasm, suggesting that 14-3-3σ inhibits Yap1-dependent gene expression to control epidermal proliferation and differentiation.
To date the in vivo role of 14-3-3σ expression in epidermal homeostasis and tumorigenesis has been mainly inferred from results obtained with ER-mice. However, truncation of the 14-3-3σ protein in ER-mice may not be equivalent to deletion and therefore complete inactivation of 14-3-3σ function.
In this study we provide evidence that the ER phenotype may rather result from a gain of function mutation since we determined that 14-3-3σ deficiency does not affect homeostasis of normal epidermal tissues and does not result in perinatal lethality seen in ER/ER mice. Nonetheless, loss of 14-3-3σ sensitizes mice to chemically-induced skin carcinogenesis. Therefore, 14-3-3σ may indeed represent a mediator of tumor suppression in the skin.
Results and Discussion
14-3-3σ-deficient mice display disorganized hair but lack obvious epidermal defects
In order to analyze whether the reported phenotypes of ER-mice are caused by the expression of the truncated 14-3-3σ protein or are due to 14-3-3σ loss of function, we generated mice with a floxed 14-3-3σ allele and inactivated the 14-3-3σ gene in the germ-line by crossing these with deleter-Cre mice (see also Supplemental Figure S1). By further back-crossing we obtained 14-3-3σ-deficient mice with an FVB background. FVB mice are prone to develop tumors of the skin when subjected to chemical carcinogenesis [31]. Mice with heterozygous (14-3-3σ+/-) and homozygous (14-3-3σ-/-) deletion of 14-3-3σ were viable, fertile and born at the normal Mendelian ratio, had a normal life-span and did not display an increase in tumor formation (data not shown). 14-3-3σ+/- mice were indistinguishable from their wild-type littermates. However, 14-3-3σ-/- mice showed a disorganized fur beginning around postnatal day 17 (Figure 1). Nonetheless, the histological analysis of skin from postnatal day 17 animals did not reveal obvious alterations in hair follicle density or hair shaft shape (Figure 2). Immunohistochemical detection of Loricrin indicated no difference in epidermal thickness either. Ki-67 staining did not reveal differences in proliferation of keratinocytes between the genotypes. Therefore, 14-3-3σ-/- mice and 14-3-3σ+/ER mutant mice have at least one phenotype in common: a disorganized fur onwards from approximately 2 weeks after birth. However, other phenotypes characteristic for 14-3-3σ+/ER mice were not observed in 14-3-3σ-deficient mice: the frequency of Ki-67-positive hair follicle matrix cells (Figure 2A) or basal epidermal cells (Figure 2B) was unchanged. Additionally, the number of hair follicles and the epidermal thickness showed no differences in 14-3-3σ-/- tissues (Figure 2). Similar results were obtained after tissue specific 14-3-3σ knockout in the epidermis of mice with a the C57BL/6J background (Supplementary Figure S2A). This suggested that the epidermal hyper-proliferation observed in 14-3-3σ+/ER mice is caused by the expression of the truncated 14-3-3σ protein, which acquired additional functions and presumably an altered ligand specificity, and not by the mere loss of 14-3-3σ function. Notably, p63 and YAP1 have been shown to functionally interact with the ER mutant of 14-3-3σ in murine epidermis [28, 30]. However, we could not detect an effect of 14-3-3σ loss on the localization and expression level of p63 (Supplemental Figure 2B). In addition, we were unable to detect a functional or direct interaction or colocalization of 14-3-3σ with YAP1 in cells ex vivo (data not shown). Also YAP1 expression and localization in chemically induced skin papillomas was not affected by deletion of 14-3-3σ in mice (data not shown). The expression of a truncated ER 14-3-3σ mutant may therefore indirectly promote the deregulation of p63 and YAP1, whereas complete loss of 14-3-3σ function does not affect the function and expression of p63 or YAP1. This divergence could, at least in part, explain the phenotypic differences between the ER mice and 14-3-3σ knock-out mice.
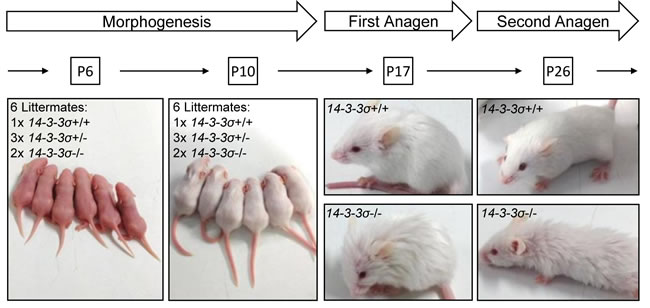
Figure 1: 14-3-3σ knock-out mice show a disorganized fur from postnatal day 17 onwards. Heterozygous 14-3-3σ knock-out mice were intercrossed and pictures of the offspring with the indicated genotypes were taken at the indicated time-points.
Unchanged frequency of spontaneous epidermal tumors in 14-3-3σ deficient mice
To exclude the possibility that mice lacking 14-3-3σ exhibit an attenuated epidermal hyper-proliferation phenotype similar to that of 14-3-3σ+/ER mice, we analyzed the long-term consequences of 14-3-3σ deficiency in mice. 14-3-3σ+/+, 14-3-3σ+/- and 14-3-3σ-/- (40 mice / genotype) mice were maintained for a prolonged time period (up to 32 months) and were inspected twice a week for the presence of neoplastic lesions or other pathologies. Mice with tumors larger than 2 cm in diameter or with other signs of illness were euthanized. Dead mice were analyzed by necropsy. Tumor burden of dead mice was recorded (Figure 3A). Time points of death were noted and used for calculation of the survival probability of each genotype (Figure 3A, 3B). No significant difference in survival between wild-type, heterozygous and knockout 14-3-3σ mice was found. In addition, 14-3-3σ knockout mice did not show a significantly increased risk of spontaneous tumor development (Figure 3A, 3B). Furthermore, the tumor burden was independent from the genotype (Figure 3A). These results show that 14-3-3σ deficiency is not sufficient to initiate spontaneous skin carcinogenesis. Therefore, the development of multiple papillomas and squamous cell carcinomas observed in 14-3-3σ+/ER mice [27] is presumably not caused by the loss of 14-3-3σ function but rather by the expression of a truncated 14-3-3σ protein, which presumably acquired additional, non-physiologic properties.
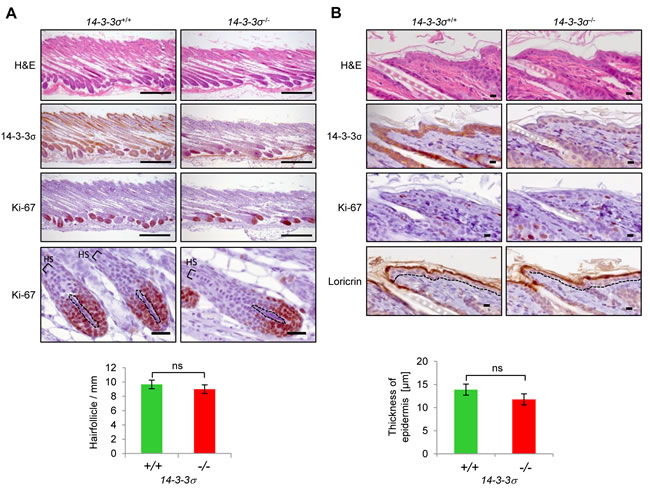
Figure 2: Comparison of back skin morphology in 14-3-3σ+/+ and 14-3-3σ-/- mice. Back skins from wild-type and 14-3-3σ knock-out mice were resected at postnatal day 17, paraffin embedded, vertically sectioned and stained with haematoxilin/eosin (H&E, upper panels) or subjected to immunohistochemistry with 14-3-3σ, Ki-67, and Loricrin specific antibodies. A. Hair follicle density (scale bar = 300 µm) and hair shaft (HS) morphology (scale bar = 30 µm) are visible. The dashed line indicates the dermal papilla. Bar-chart: The average number of hair follicles per millimeter skin section was evaluated (n = 3). B. A higher magnification of the interfollicular epidermis and the underlying dermis is shown (scale bar = 10 µm). The dashed line indicates the border between epidermis and dermis. The Loricrin staining shows the outermost layer of the epidermis. Bar-chart: The average epidermal thickness was evaluated (n = 3).
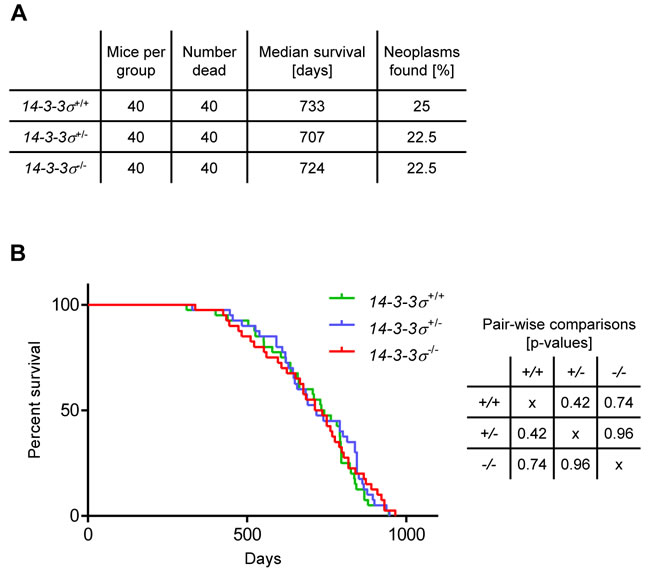
Figure 3 : Survival analysis of mice with varying 14-3-3σ genotype. A. Experimental groups (20 female and 20 male mice per group) and summary of results. B. Kaplan-Meier curve (n = 40) and p-values of the curve comparisons calculated using the GraphPad Prism5 software.
14-3-3σ deficiency increases number and size of DMBA/TPA induced papillomata
The 14-3-3σ gene is frequently silenced in basal cell and squamous cell carcinoma [8, 32, 33]. Nevertheless, the pathophysiological relevance of 14-3-3σ inactivation for epithelial skin cancer formation is largely unknown. In order to study the effect of 14-3-3σ inactivation on the onset, frequency and progression of chemically-induced papillomas of the skin, which ultimately progress to squamous cell carcinomas (SCC), we employed a well-established two-stage model of chemical skin-carcinogenesis which is based on DMBA/TPA treatments [34]. The back skin of male wild-type and 14-3-3σ knockout mice (7 to 9 weeks old) was treated in the pause phase of the hair cycle with a single dose of 7.12-dimethylbenz(a)anthracene (DMBA) for tumor initiation (Figure 4A). Two weeks later, both groups were treated twice a week with 12-O-tetradecanoylphorbol-13 acetate (TPA) for a total period of 20 weeks to achieve tumor promotion. The time points of tumor occurrence, number and size were quantified throughout the complete treatment period (Figure 4C-4F). After 20 weeks the back skin was resected (Figure 4B) and the number (4C) and size (4D) of tumors present in these specimen was determined. Subsequently, tumors were analyzed histologically (Figure 5). ~7 weeks after the first TPA administration the first skin tumors (≥1 mm diameter) occurred in both groups (Figure 4E). Over the subsequent 13 weeks of tumor promotion, the number of mice with tumors (Figure 4E) and the number of tumors per mouse (Figure 4F) increased significantly faster in 14-3-3σ -deficient mice. By the end of the TPA treatments, 100% of the 14-3-3σ-deficient and 89% of the wild-type mice had developed numerous tumors. 60% of the 14-3-3σ deficient and only 11% of the wild-type mice had 16 or more tumors (Figure 4C). Counting of tumors with an area of ≥15mm2 revealed that 14-3-3σ wild-type mice developed on average 2.8, whereas 14-3-3σ knock-out mice displayed 5.5 papilloma of this size (Figure 4D). Taken together, 14-3-3σ-deficient mice developed 1.8-fold more papillomas per mouse in total and 1.98-fold more larger tumors (≥15mm2) per mouse than wild-type mice (Figure 4F, 4D). Further examination revealed that all induced tumors from wild-type and 14-3-3σ knock-out mice projected outwardly, with a cauliflower-like shape, which is a characteristic of benign papillomas (see example in Figure 5). Next we obtained tissues sections containing tumors of different sizes from wild-type and 14-3-3σ-deficient mice and analyzed them microscopically after haematoxilin/eosin staining and by immunohistochemistry (Figure 5). Like we already had deduced from the external examination of the tumors (Figure 4B), inspection of the histological sections confirmed a tumor growth directed towards the outer side of the epidermis (Figure 5). The tumors resemble the architecture of normal skin with a basal layer, stratified squamous epithelium and an outer keratinized layer (Figure 5A). No invasion was observed (Figure 5B). Taken together our results show that 14-3-3σ deficiency significantly increases the number and size of DMBA/TPA-induced papillomas. Thus, 14-3-3σ fulfills a tumor suppressive function in vivo, which presumably prevents the initiation and growth of epidermal tumors. This is in line with the timing of epigenetic inactivation of 14-3-3σ in tumors, which has been described to occur in the early phases of tumor progression [35].
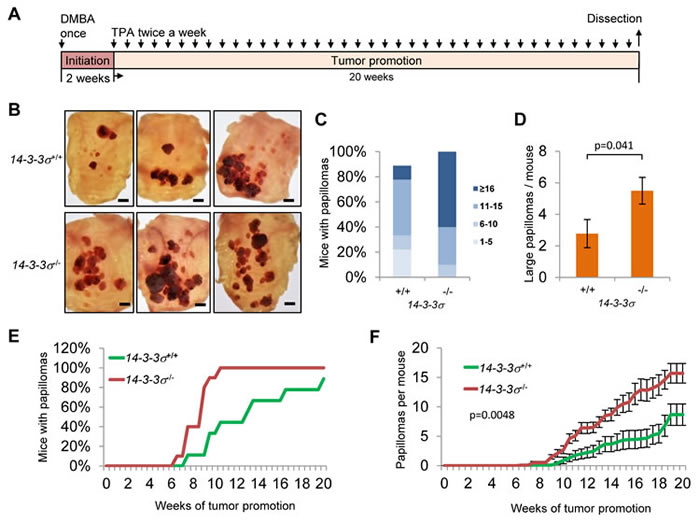
Figure 4: Effect of 14-3-3σ-deficiency on the number and size of DMBA/TPA-induced skin tumors. A. 14-3-3σ+/+; n = 9 and 14-3-3σ-/-; n = 10 male mice were treated once with 100 nmol DMBA. After 2 weeks all mice were treated twice a week with 13.6 nmol TPA for 20 weeks. B. Overview of representative back skin resections (scale bar = 5 mm). C. Cumulative percentage of mice carrying the indicated numbers of tumors. D. Average numbers of large tumors (surface ≥ 15 mm2). Results are the mean ± s.e.m.; p = 0.041 by two-sided t test. E. Percentage of mice with detectable papillomas during the period of tumor promotion. F. Average number of tumors during the period of tumor promotion. Results are the mean ± s.e.m. with p = 0.0048 by Mann-Whitney U test.
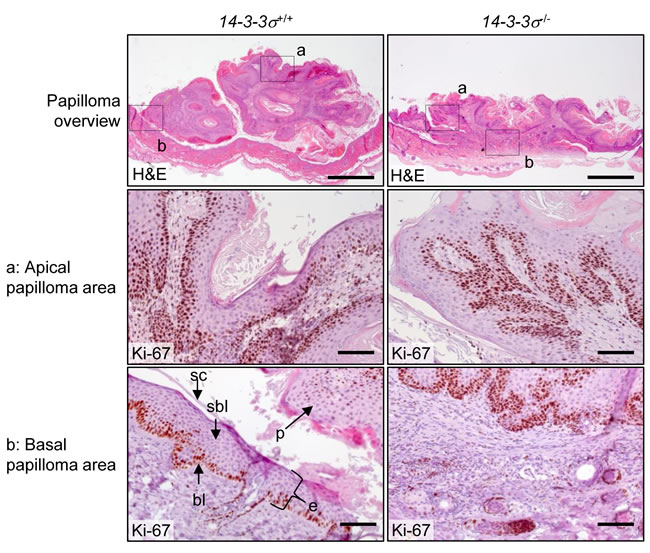
Figure 5: Morphology of DMBA/TPA-induced epidermal tumors in mice with varying 14-3-3σ genotype. Skin sections containing papillomas from 14-3-3σ+/+ and 14-3-3σ-/- were stained with haematoxilin/eosin and probed for Ki-67. The upper panel shows an overview of the whole papilloma/skin area (scale bar = 1 mm). The dashed boxes in the overview indicate the magnified areas of a. an apical tumor region, b. a tumor attachment site shown in the lower panels (scale bar = 100 µm).
Materials and Methods
Generation and handling of mice
The detailed description of the generation of 14-3-3σ deficient mice can be found in supplementary material and methods (Figure S1). 14-3-3σ-/- mice were transferred into an FVB background by backcrossing for at least 6 generations with FVB wild-type mice. For the experiments, FVB littermates of the genotypes 14-3-3σ+/+, 14-3-3σ+/- and 14-3-3σ-/- were generated by intercrossing of FVB 14-3-3σ+/- mice. All mice were maintained in the animal facility at the Pathology Institute of the Ludwig-Maximilians-University Munich in individually ventilated cages (IVC). All animal experiments were approved by the Regierung von Oberbayern (AZ: 55.2-1-54-2532-11-13).
Genotyping PCR
Genomic DNA was obtained by overnight digest of ear punches in lysis buffer (50 mM KCl, 1.5 mM MgCl2, 10 mM Tris pH8.5, 0.01% gelatin, 0.45% NP40, 0.45% Tween 20, 100 µg/ml proteinase K). PCR was performed for 35 cycles with an annealing temperature of 65°C. Primers for the detection of the 14-3-3σ wild-type and floxed alleles (forward: 5’-CAC TAC CGT GGT CTT CCC TAA CTT GAT G-3’; reverse: 5’-TCC CAG GAA GCA GAT GGG ATT TCT GTC C-3’) and the 14-3-3σ knock-out allele (reverse: 5’-AGG CAC TAT GCC CCT GCC TCA GAT-3’) were added in the same ratio to the reaction mix and PCR products were resolved on 2% agarose or 8% polyacrylamide gels. The PCR-products were 106 bp (wild-type allele), 159 bp (floxed allele), and 183 bp (14-3-3σ knock-out allele).
Determination of hair follicle numbers and epidermal thickness
Dorsal skin from male 14-3-3σ+/+ and 14-3-3σ-/-
FVB littermates was dissected, fixed in 4% formalin, paraffin embedded, vertically sectioned at 5 µm distances, stained with haematoxilin and eosin (H&E), mounted and analyzed by light microscopy. Hair follicles were counted over an epidermis length of 1 mm from 3 individuals per genotype. In addition, the epidermal thickness was measured at 3 randomly chosen positions of the epidermis of 350 µm length using the AxioVison Rel4.8 software. Average numbers, standard deviation and significance were calculated using a Student’s test (two-sided; n = 3).
Survival probability
40 mice (20 male, 20 female) per genotype (14-3-3σ+/+, 14-3-3σ+/-, 14-3-3σ-/-) with a FVB background were maintained and monitored over a period of more than 2 years. Moribund mice were euthanized and skin sections were analyzed immunohistochemically. Tumor burden [No = 0, Yes = 1] and age at death [in days] was documented. Survival probability was calculated according to Kaplan-Meier using GraphPad Prism5 software. Additionally, the percentage of mice with tumors was calculated.
Chemically-induced skin carcinogenesis
The back skin of male 14-3-3σ+/+ (n = 9) and 14-3-3σ-/- FVB (n = 10) littermates (7-9 weeks of age) was shaved 2 days prior to tumor initiation. Tumors were initiated with a single topical application of DMBA (Sigma-Aldrich, Munich, Germany, order no. 40567, 100 nmol in 50 µl methanol) to the back skins of the mice. Two weeks later, tumor promotion was achieved by topical application of TPA (Sigma-Aldrich, Munich, Germany, order no. P1585, 13.6 nmol in 200 µl acetone) twice weekly for 20 weeks. Tumor incidence (percentage of tumor-bearing mice) and multiplicity (papillomas per mouse) were recorded twice weekly throughout the experiment. Pictures of dissected back skin were taken and used to determine the sizes of papillomas [mm2] with the ImageJ software.
Immunohistochemistry
Tissues were fixed in 4% formalin and embedded in paraffin. 5 µm sections were prepared, deparaffinized in xylene and rehydrated in serial ethanol dilutions. Antigen retrieval was carried out by boiling in DAKO citrate buffer (pH 6.0) twice for 15 minutes in a microwave oven at 750 Watts. After quenching endogenous peroxidase activity by 10 min. exposure to 7.5% H2O2 solution, tissue sections were blocked in either 10% goat or rabbit serum, or 10% BSA for 30 min. at room temperature depending on the origin of the primary antibody. Primary antibodies were used at 1:20 dilution for anti-14-3-3σ (C-18, goat polyclonal anti-mouse, Santa Cruz), 1:500 for anti-Ki67 (D3B5, rabbit monoclonal anti-mouse, Cell Signaling), 1:500 for anti-Loricrin (AF62, rabbit polyclonal anti-mouse, Covance) and 1:100 for anti-p63 (4A4, Dako, supplementary data). Biotinylated secondary antibodies and streptavidin/HRP complexes were used according to the provided manual (Vectastain kit, Vector Labs). Sections were treated with DAB chromogenic substrate (DAKO), counterstained with haematoxilin and mounted in xylene-based mounting medium.
Abbreviations
ER, repeated epilation; DMBA, 7.12-dimethylbenz(a)anthracene; TPA, 12-O-tetradecanoylphorbol-13 acetate
Acknowledgments
We thank Ursula Götz for technical assistance and Dr. Thomas Herzinger for reading the manuscript. The project was funded by Deutsche Forschungsgemeinschaft (Grant No. He 2701/6-1) and the Deutsche Krebshilfe (Grant No. 110221).
Conflicts of Interest
The authors declare no conflict of interest.
References
1. Hsu YC, Li LS and Fuchs E. Emerging interactions between skin stem cells and their niches. Nature Medicine. 2014; 20:847-856.
2. Dellambra E, Golisano O, Bondanza S, Siviero E, Lacal P, Molinari M, D’Atri S and De Luca M. Downregulation of 14-3-3sigma prevents clonal evolution and leads to immortalization of primary human keratinocytes. J Cell Biol. 2000; 149:1117-1130.
3. Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, Ponzin D, McKeon F and De Luca M. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001; 98:3156-3161.
4. Mills AA, Zheng BH, Wang XJ, Vogel H, Roop DR and Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999; 398:708-713.
5. Westfall MD, Mays DJ, Sniezek JC and Pietenpol JA. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol. 2003; 23:2264-2276.
6. Fomenkov A, Zangen R, Huang YP, Osada M, Guo Z, Fomenkov T, Trink B, Sidransky D and Ratovitski EA. RACK1 and stratifin target DeltaNp63alpha for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2004; 3:1285-1295.
7. Hermeking H. The 14-3-3 cancer connection. Nat Rev Cancer. 2003; 3:931-943.
8. Lodygin D and Hermeking H. Epigenetic silencing of 14-3-3sigma in cancer. Semin Cancer Biol. 2006; 16:214-224.
9. Ferguson AT, Evron E, Umbricht CB, Pandita TK, Chan TA, Hermeking H, Marks JR, Lambers AR, Futreal PA, Stampfer MR and Sukumar S. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A. 2000; 97:6049-6054.
10. Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW and Vogelstein B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997; 1:3-11.
11. Chan TA, Hermeking H, Lengauer C, Kinzler KW and Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999; 401:616-620.
12. Benzinger A, Popowicz GM, Joy JK, Majumdar S, Holak TA and Hermeking H. The crystal structure of the non-liganded 14-3-3sigma protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res. 2005; 15:219-227.
13. Verdoodt B, Benzinger A, Popowicz GM, Holak TA and Hermeking H. Characterization of 14-3-3sigma dimerization determinants: requirement of homodimerization for inhibition of cell proliferation. Cell Cycle. 2006; 5:2920-2926.
14. Benzinger A, Muster N, Koch HB, Yates JR, 3rd and Hermeking H. Targeted proteomic analysis of 14-3-3 sigma, a p53 effector commonly silenced in cancer. Mol Cell Proteomics. 2005; 4:785-795.
15. Hermeking H. Extracellular 14-3-3sigma protein: a potential mediator of epithelial-mesenchymal interactions. J Invest Dermatol. 2005; 124:ix-x.
16. Hermeking H and Benzinger A. 14-3-3 proteins in cell cycle regulation. Semin Cancer Biol. 2006; 16:183-192.
17. Phan L, Chou PC, Velazquez-Torres G, Samudio I, Parreno K, Huang YL, Tseng C, Vu T, Gully C, Su CH, Wang E, Chen J, Choi HH, Fuentes-Mattei E, Shin JH, Shiang C, et al. The cell cycle regulator 14-3-3 sigma opposes and reverses cancer metabolic reprogramming. Nature Communications. 2015; 6.
18. Ling C, Zuo D, Xue B, Muthuswamy S and Muller WJ. A novel role for 14-3-3sigma in regulating epithelial cell polarity. Genes Dev. 2010; 24:947-956.
19. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A and McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987; 235:177-182.
20. Ling C, Su VM, Zuo D and Muller WJ. Loss of the 14-3-3sigma tumor suppressor is a critical event in ErbB2-mediated tumor progression. Cancer Discov. 2012; 2:68-81.
21. Ingles-Esteve J, Morales M, Dalmases A, Garcia-Carbonell R, Jene-Sanz A, Lopez-Bigas N, Iglesias M, Ruiz-Herguido C, Rovira A, Rojo F, Albanell J, Gomis RR, Bigas A and Espinosa L. Inhibition of Specific NF-kappa B Activity Contributes to the Tumor Suppressor Function of 14-3-3 sigma in Breast Cancer. Plos One. 2012; 7.
22. Herron BJ, Liddell RA, Parker A, Grant S, Kinne J, Fisher JK and Siracusa LD. A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat Genet. 2005; 37:1210-1212.
23. Li Q, Lu Q, Estepa G and Verma IM. Identification of 14-3-3sigma mutation causing cutaneous abnormality in repeated-epilation mutant mouse. Proc Natl Acad Sci U S A. 2005; 102:15977-15982.
24. Guenet JL, Salzgeber B and Tassin MT. Repeated epilation: a genetic epidermal syndrome in mice. J Hered. 1979; 70:90-94.
25. Hammond NL, Headon DJ and Dixon MJ. The cell cycle regulator protein 14-3-3sigma is essential for hair follicle integrity and epidermal homeostasis. J Invest Dermatol. 2012; 132:1543-1553.
26. Xin Y, Lu Q and Li Q. 14-3-3sigma is required for club hair retention. J Invest Dermatol. 2010; 130:1934-1936.
27. Lutzner MA, Guenet JL and Breitburd F. Multiple cutaneous papillomas and carcinomas that develop spontaneously in a mouse mutant, the repeated epilation heterozygote Er/+. J Natl Cancer Inst. 1985; 75:161-166.
28. Li Q, Sambandam SA, Lu HJ, Thomson A, Kim SH, Lu H, Xin Y and Lu Q. 14-3-3sigma and p63 play opposing roles in epidermal tumorigenesis. Carcinogenesis. 2011; 32:1782-1788.
29. Yu FX, Zhao B and Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell. 2015; 163:811-828.
30. Sambandam SA, Kasetti RB, Xue L, Dean DC, Lu Q and Li Q. 14-3-3sigma regulates keratinocyte proliferation and differentiation by modulating Yap1 cellular localization. J Invest Dermatol. 2015; 135:1621-1628.
31. Abel EL, Angel JM, Kiguchi K and DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009; 4:1350-1362.
32. Lodygin D, Yazdi AS, Sander CA, Herzinger T and Hermeking H. Analysis of 14-3-3sigma expression in hyperproliferative skin diseases reveals selective loss associated with CpG-methylation in basal cell carcinoma. Oncogene. 2003; 22:5519-5524.
33. Lodygin D and Hermeking H. The role of epigenetic inactivation of 14-3-3sigma in human cancer. Cell Res. 2005; 15:237-246.
34. DiGiovanni J. Multistage carcinogenesis in mouse skin. Pharmacol Ther. 1992; 54:63-128.
35. Umbricht CB, Evron E, Gabrielson E, Ferguson A, Marks J and Sukumar S. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene. 2001; 20:3348-3353.