
INTRODUCTION
Gastrointestinal stromal tumors (GISTs) potentially originate from interstitial Cajal cells or their precursors. They are usually genetically characterized through mutually exclusive gain-of-function mutations of KIT protooncogene receptor tyrosine kinase (KIT) or platelet-derived growth factor receptor alpha (PDGFRA) genes [1]. KIT and PDGFRA mutations elicit constitutive activation of the corresponding receptor tyrosine kinases, which drive tumor inception and dictate treatment response to imatinib [1-4]. The KIT/PDGFRA genotypes are variably associated with the aggressiveness of resected imatinib-naïve GISTs [3-5]; however, their prognostic value has not been uniformly validated in previous studies [6-8]. Although both the National Institutes of Health (NIH) and National Comprehensive Cancer Network (NCCN) risk schemes are prognostically useful [9, 10], more accurate prognostication is becoming a critical issue in the postimatinib era for counseling regarding outcomes and for the identification of targetable aberrant molecules other than receptor tyrosine kinases. Therefore, identifying candidate deregulated molecules of other signaling pathways is essential for resolving the current limitations in prognostication and therapy.
Compared with normal cells, cancer cells voraciously consume nutrients, including glucose, lipids, and amino acids, through unique signaling pathways; molecular aberrations in metabolism-associated enzymes may alter these pathways [11]. Despite being a cancer hallmark of renewed interest, extremely little has been clarified regarding the pathogenetic, biological, and clinical relevance of metabolic reprogramming in mesenchymal neoplasms, including GISTs [11]. Of the deregulated metabolic pathways, the de novo biosynthesis of lipids is drastically increased in rapidly proliferating cancer cells because lipids provide the building blocks of lipid rafts and various protumorigenic lipid-signaling molecules and engage in coordinating cell motility and signaling cascades [12-14]. However, the clinical and biological relevance of the lipolytic pathway, required for an immediate liberation of reserved fatty acids for metabolic and signaling demands in cancer cells, warrants further exploration [14]. Thus, considering lipid metabolic processes, we began reappraising the published transcriptome of GISTs to search for metabolic driver genes differentially expressed between high-risk and non-high-risk cases. A top-ranking upregulated gene identified through this data-mining approach was the monoglyceride lipase-encoding MGLL (also called MAGL, encoding monoacylglycerol lipase), a serine hydrolase that preferentially hydrolyzes monoglycerides into fatty acids and glycerol [14, 15].
Thus far, no study has directly evaluated the implications of altered MGLL expression in GISTs. By using two independent cohorts (Supplementary Figure 1), we validated the clinical relevance of MGLL overexpression to the full extent of transcriptional and translational characterization and clearly demonstrated that MGLL overexpression is strongly correlated with its mRNA and protein levels and adverse clinicopathological factors, and has independent prognostic utility in identifying aggressive cases. Regarding deregulated lipid metabolism, our findings provide further prognostic information as well as biological insights into the pathways regulating GIST progression.
RESULTS
Differentially upregulated MGLL expression in high-risk GISTs
Considering 559 probes covering 274 genes regulating the lipid metabolic process, we conducted unsupervised clustering on the transcriptomic dataset of 32 GISTs (GSE8167). This approach crudely segregated the samples into two clusters (Figure 1), with several notable genes differentially expressed between the high-risk and non-high-risk cases. Of these, HSD11B1, MGLL, and PLCB4 were the three top-ranking upregulated candidates exhibiting remarkable expression fold changes (log2 ratio >1 or <−1) and strong associations with the high-risk category (all p ≤ 0.0001, Supplementary Table 2). Given the novel discovery of the involvement of MGLL-driven lipolysis in promoting carcinogenesis through the modulation of the fatty acid network [14, 15], we further validated the clinical relevance and prognostic implication of MGLL in cell lines and two independent tumor cohorts.
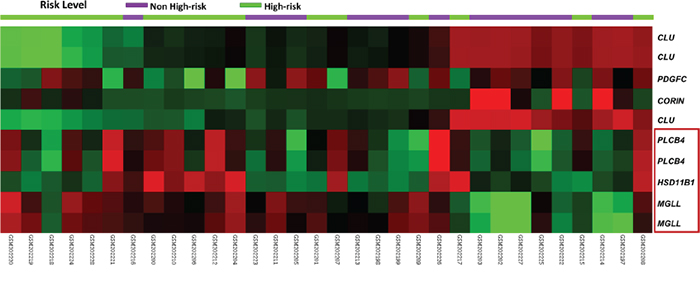
Figure 1: Heatmap of unsupervised hierarchical clustering analysis for differentially expressed genes associated with lipid metabolism. Two roughly segregated clusters were identified, which comprised more high-risk (green) GISTs on the left and more non-high-risk (purple) GISTs on the right. HSD11B1 (hydroxysteroid (11-beta) dehydrogenase 1), MGLL (monoglyceride lipase), and PLCB4 (phospholipase C beta 4) genes were top-ranking candidates highlighted in a red bracket, of which MGLL were selected for validation in this study. According to their fold changes, increased and decreased expression levels of individual genes were expressed in red and green of varying intensity, respectively.
MGLL mRNA abundance is positively associated with risk levels and protein expression
Adopting the same comparative logic in transcriptomic reappraisal, we preselected all 86 GISTs (cohort 1 in Supplementary Figure 1) for MGLL mRNA quantification to ensure congruity in the assignment of high-risk versus non-high-risk categories by using both the NCCN and NIH schemes [9, 10]. As an indicator of mRNA abundance, the luminescence-based detection of dioxetane alkaline phosphatase substrate was informative in 10 normal tissue samples and 70 primary localized tumors, whereas another 16 tumors could not be analyzed because of nucleic acid degradation. These 70 informative GISTs, 49 gastric and 21 intestinal, were classified as high-risk in 20 cases and non-high-risk in 50 (Supplementary Table 1). Compared with normal tissues, MGLL mRNA abundance was significantly higher in the GISTs in whole sections (p = 0.001, Figure 2A) and increased from normal tissues over the non-high-risk group (p = 0.030) to the high-risk group (p = 0.012), confirming the promotional role of MGLL in tumor progression. Notably, MGLL mRNA and MGLL levels were strongly and positively associated with each other (p < 0.001, r = 0.536; Figure 2B).
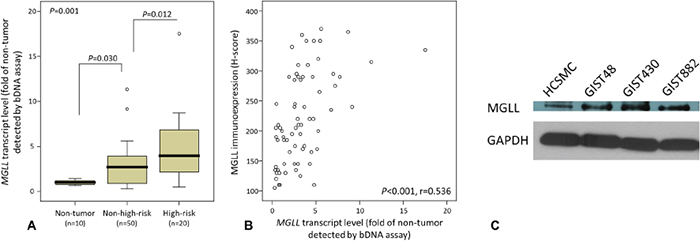
Figure 2: MGLL (monoglyceride lipase) mRNA upregulation and protein overexpression and their strong correlation validated in GIST samples and cell lines. A. Compared with the normal tissues, MGLL mRNA abundance was determined to be differentially upregulated across GISTs of various risk levels (informative n=70) and exhibit significant risk level-associated increment, indicating its role in tumor progression. B. In the same set of 70 GISTs, the scattered plot demonstrated a strong correlation between the levels of MGLL mRNA measured by Quantigene assay on the X axis and H-scores of MGLL protein immunoexpression on the Y axis (see representative images in Figure 3). C. Western blotting assay revealed increased endogenous expression of MGLL protein in all imatinib-resistant (GIST48, GIST430) and imatinib-sensitive (GIST882) cell lines, compared with the reference human colonic smooth muscle cells (HCSMC). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as the loading control.
Increased endogenous MGLL expression in GIST cell lines
We further compared the endogenous MGLL expression in GIST cell lines versus the reference human colonic smooth muscle cells (HCSMCs). Irrespective of the sensitivity or resistance to imatinib, all three GIST cell lines demonstrated increased endogenous MGLL levels in western blot analysis (Figure 2C).
MGLL overexpression is associated with unfavorable clinicopathological factors
Given the strong correlation between MGLL protein and MGLL mRNA expression levels, we next systematically analyzed the clinical relevance of MGLL immunoexpression in a large validation set of GISTs (cohort 2 in Supplementary Figure 1) through tissue microarray (TMA)-based immunohistochemistry. We included 350 informative GISTs with available follow-up data, comprising 88 no-risk or very-low-risk, 100 low-risk, 65 moderate-risk, and 97 high-risk cases defined using the NCCN scheme (Table 1) and corresponding to 127 very-low-to-low-risk, 110 intermediate-risk, and 113 high-risk cases according to the NIH scheme (Supplementary Table 3). As indicated in Table 1, MGLL overexpression was associated with nongastric location (p = 0.022) and increased size (p = 0.017) and strongly correlated with increased mitosis and risk levels defined by both the NIH and NCCN schemes (all p ≤ 0.001, Figure 3); however, the overexpression was not associated with unfavorable genotypes (p = 0.540).
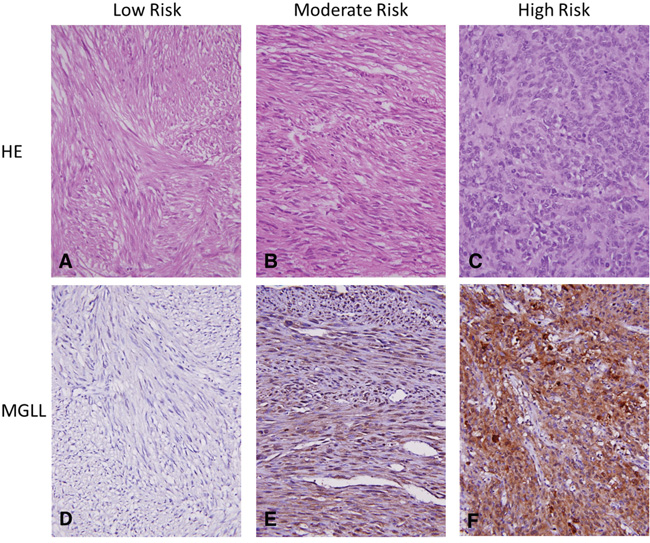
Figure 3: Representative histology and MGLL (monoglyceride lipase) immunoexpression in GISTs of various risk categories. The hematoxylin eosin stains for histological evaluation (X400) showed gradually increased cellularity from low- A., intermediate- B. to high-risk C. GISTs, which exhibited no D., weak E., and strong F. cytoplasmic immunoexpression of MGLL (monoglyceride lipase, X400), respectively.
Table 1: Clinicopathological and KIT/PDGFA genotypic correlations with MGLL immunoexpression in cohort 2 of primary GISTs in in tissue microarrays
MGLL Expression |
p-value |
||
|---|---|---|---|
Low |
High |
||
Sex |
0.915 |
||
Male |
86 |
87 |
|
Female |
89 |
88 |
|
Age (years) |
60.14±13.026 |
59.59±12.527 |
0.749 |
Location |
0.022* |
||
Gastric |
116 |
95 |
|
Non-gastric |
59 |
80 |
|
Histologic Type |
0.080 |
||
Spindle |
140 |
126 |
|
Epithelioid & Mixed |
35 |
49 |
|
Tumor Size (cm)& |
5.895+/-4.0668 |
6.914+/-4.3204 |
0.017* |
Mitotic Count (50HPFs)& |
6.75+/-18.687 |
11.71+/-27.174 |
<0.001* |
NIH Risk |
0.001* |
||
Low/Very low |
73 |
54 |
|
Intermediate |
62 |
48 |
|
High |
40 |
73 |
|
NCCN Guideline |
<0.001* |
||
None/Very low |
57 |
31 |
|
Low |
55 |
45 |
|
Moderate |
32 |
33 |
|
High |
31 |
66 |
|
Mutation Types |
0.540 |
||
Favorable Types |
51 |
55 |
|
Unfavorable Types |
47 |
60 |
|
NIH, National Institutes of Health; NCCN, National Comprehensive Cancer Network; MGLL, monoglyceride lipase; *: Statistically significant; &: Wilcoxon rank-sum test.
MGLL overexpression independently predicts worse outcomes
Unlike overall survival (OS), the disease-free survival (DFS) of patients receiving primary surgery is not confounded by imatinib therapy administered for relapsed and disseminated diseases. Therefore, we primarily detailed disease-free DFS-related findings in this article. Univariately, MGLL overexpression was strongly predictive of worse outcomes (Figure 4A, p < 0.0001), with the median DFS being 42.7 months in the MGLL-overexpressing GISTs and 61.0 months in their MGLL-underexpressing counterparts. The nongastric location (p = 0.0023) and unfavorable mutated KIT/PDGFRA genotypes (p = 0.0005) significantly predicted inferior DSS. Moreover, significant poor prognosticators of DFS included the presence of epithelioid histology, increased tumor size and mitosis, and higher GIST risk defined by both the NIH and NCCN schemes (all p < 0.0001; Figure 4B, Table 2, and Supplementary Table 3). Regarding OS at the univariate level (Supplementary Tables 4 and 5), age older than 70 years (p = 0.0048) was a significant poor prognosticator, whereas nongastric location (p = 0.0807) showed only a marginal trend toward poorer OS. Notably, MGLL overexpression (p = 0.0007, Figure 4C) still strongly indicated poorer OS, which was also significantly associated with increased tumor size and mitosis and higher GIST risk defined by both the NCCN and NIH schemes (Figure 4D; all p < 0.0001). Although epithelioid histology was a significant prognosticator (p = 0.002), it was not as powerful as that seen in DFS.
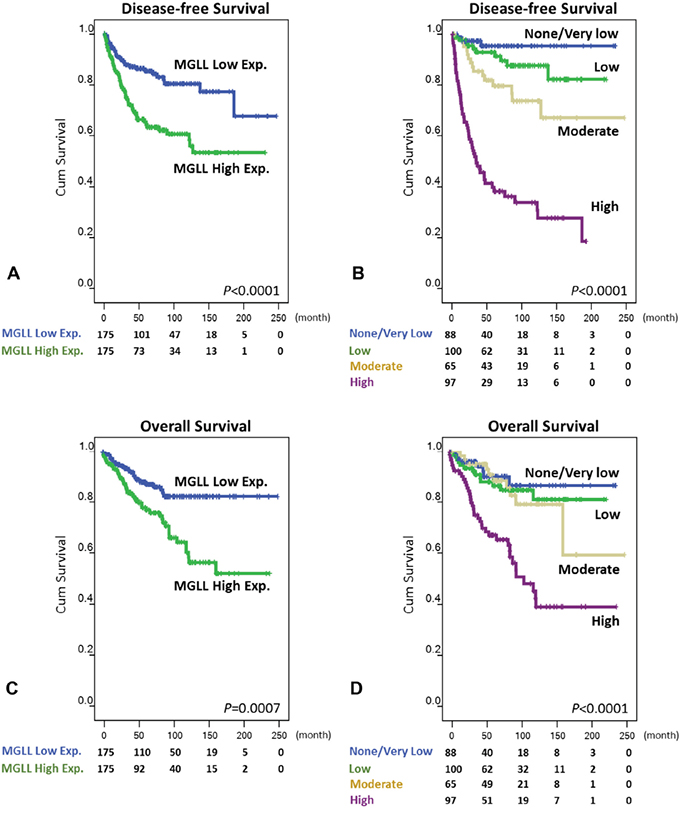
Figure 4: Kaplan-Meier analyses of univariate disease-free survival A, B. and overall survival C, D. in patients with primary GISTs according to MGLL (monoglyceride lipase) immunoexpression status (A, C) and risk levels defined by National Comprehensive Cancer Network scheme (B, D).
Table 2: Univariate and multivariate disease-free survival analyses according to MGLL expression status, NCCN guidelines, and other prognostic factors
Parameter |
Univariate analysis |
Multivariate analysis |
||||
|---|---|---|---|---|---|---|
No. Case |
No. Event |
p-value |
HR |
95% CI |
p-value |
|
Sex |
0.4667 |
|||||
Male |
177 |
43 |
||||
Female |
173 |
44 |
||||
Age (years) |
0.0584 |
|||||
<70 |
259 |
59 |
||||
>=70 |
91 |
28 |
||||
Location |
0.0023* |
0.875 |
||||
Gastric |
211 |
40 |
1 |
- |
||
Non-gastric |
139 |
47 |
0.961 |
0.585-1.578 |
||
Histologic Type |
<0.0001* |
0.001* |
||||
Spindle |
266 |
51 |
1 |
- |
||
Mixed/Epithelioid |
84 |
36 |
2.266 |
1.374-3.736 |
||
Tumor Size (cm)# |
<0.0001* |
|||||
=<5 cm |
161 |
16 |
||||
>5; =<10 cm |
131 |
38 |
||||
>10 cm |
58 |
33 |
||||
Mitotic Count (50HPFs)# |
<0.0001* |
|||||
0-5 |
249 |
33 |
||||
6-10 |
43 |
14 |
||||
>10 |
58 |
40 |
||||
NCCN Guideline |
<0.0001* |
<0.0001* |
||||
None/Very low |
88 |
3 |
1 |
- |
||
Low |
100 |
10 |
3.436 |
2.967-52.632 |
||
Moderate |
65 |
15 |
3.460 |
1.601-7.463 |
||
High |
97 |
59 |
12.658 |
1.736-6.803 |
||
Mutation Type |
0.0005* |
0.061 |
||||
Favorable type |
106 |
22 |
1 |
- |
||
Unfavorable type |
107 |
45 |
1.668 |
0.977-2.845 |
||
MGLL expression# |
<0.0001* |
0.031* |
||||
Low Exp. |
175 |
28 |
1 |
- |
||
High Exp. |
175 |
59 |
1.869 |
1.058-3.300 |
||
NCCN, National Comprehensive Cancer Network; MGLL, monoglyceride lipase; #, Tumor size and mitotic activity were not introduced in multivariate analysis, since these two parameters were component factors of NCCN guidelines; *, Statistically significant. HR, hazard ratio.
When adopting only the NCCN guidelines in the multivariate analysis for DFS (Table 2), MGLL overexpression (p = 0.031, hazard ratio [HR] = 1.869) remained an independent prognosticator of poorer outcomes, as did higher GIST risk (p < 0.0001) and the presence of epithelioid histology (p = 0.001). However, the GISTs harboring mutated KIT/PDGFRA of unfavorable genotypes exhibited only a trend toward independent prediction of poorer DFS (p = 0.061). We further examined the effect of the NIH scheme (Supplementary Table 3) on the multivariate survival analyses regarding the independence of MGLL overexpression on DFS, which demonstrated generally similar results and statistical power to those defined using the NCCN scheme. MGLL overexpression (p = 0.008, HR = 2.116) again remained the independent prognosticator of poorer outcomes, as did higher NIH risk levels (p < 0.001), the presence of epithelioid histology (p = 0.003), and unfavorable genotypes (p = 0.034). Regarding OS, MGLL overexpression still independently predicted adverse events (Supplementary Tables 4 and 5), together with increased risk levels and unfavorable genotypes (coanalyzed using the NCCN guidelines: p = 0.032, HR = 2.024; coanalyzed using the NIH scheme: p = 0.034, HR = 1.984). Taken together, MGLL overexpression and higher risk levels represent independent poor prognosticators, regardless of the risk criteria and endpoints being analyzed.
DISCUSSION
Among the deregulated metabolic events in carcinogenesis, increasing attention is being focused on the development of lipogenic phenotypes, which lead to tumor growth in various cancer types by providing energy substrates, building blocks for lipid rafts, and oncogenic lipid products [11-13]. In human cancers, oncogenic lipogenesis is prototypically exemplified through increased fatty acid synthase levels, resulting in enhanced de novo fatty acid synthesis and poor prognosis [12]. Fatty acid synthase, predominantly overexpressed in high-risk and metastatic GISTs, has a proproliferative oncogenic attribute through the positive regulation of cyclin A1. In GISTs, we recently reported the negative prognostic effect of aberrantly increased levels of alpha-methylacyl-CoA racemase (AMACR), which functions as a gatekeeper for fueling the β-oxidation of branched-chain fatty acids [8]. Partly driven by gene amplification, the overexpressed AMACR promotes cell proliferation and represents an exploitable target of AMACR chemical inactivators [8]. In this series, we began with transcriptomic reappraisal of aberrantly expressed genes regulating lipid metabolism and systematically validated the clinical relevance of increased MGLL and MGLL mRNA levels in GISTs.
In normal adipocytes, MGLL serves as a lipolytic enzyme with serine-hydrolyzing capacity and enables the liberation of free fatty acids by catalyzing the final step of lipolysis to supply fuel for energy consumption [14, 17]. The current knowledge regarding the regulation and biological functions of MGLL in neoplastic diseases remains limited, with the exact role of MGLL in various aspects of cancer biological processes being rarely characterized [13-15]. To complement the swift incorporation of nascent fatty acids into the cellular lipid stores, a molecular mechanism responsible for lipolysis is probably required for liberating fatty acid moieties from this oil depot for fulfilling the requirements of rapidly proliferating cancers [14]. Nomura et al. reported that aggressive cancer lines of various cellular lineages and primary ovarian carcinomas and melanomas may hijack overexpressed MGLL to drive tumorigenesis by remodeling fatty acids to yield a signaling network enriched with pro-oncogenic lipid metabolites such as lipophosphatidic acid and prostaglandins [14]. By contrast, the reduced expression or even absence of MGLL has been reported in various common carcinomas, with MGLL proposed as a potential tumor suppressor [18, 19] because of its growth-suppressive effect on cell colony formation [19]. Notably, contradictory results regarding the oncogenic versus tumor suppressive functions of MGLL have been reported for the same tumor type, such as colorectal cancers [19-21]. These contentions on the functional role of MGLL suggest that its complex disparity in cancer biology may be tumor context-dependent.
Given the strong positive association between MGLL mRNA and immunoexpression levels, increased MGLL levels were attributable to the increased mRNA levels in GISTs, at least in part. Compared with the adjacent nontumoral tissue, the MGLL mRNA abundance was significantly higher in the entire GIST group, and it increased stepwise in parallel with the increase in the risk. Furthermore, MGLL overexpression was more frequent in GISTs characterized by the nongastric location, increased tumor size and mitosis, and higher risk levels defined using both the NIH and NCCN schemes. Regardless of whichever risk scheme being introduced, MGLL overexpression was notably identified as an independent poor prognosticator of DFS and OS in the large TMA cohort of GISTs, with an approximately 2-fold increased risk of adverse outcome in multivariate analysis. Taken together, these features clearly indicate that MGLL represents an oncogenic lipid-metabolizing enzyme that confers growth advantages to and aggravates the progression of GISTs.
The concomitantly increased MGLL mRNA and MGLL levels in GISTs imply transcriptional control as a mechanism regulating MGLL expression, which appears inferable from the transcriptional derepression of MGLL promoter through the loss of tumor-suppressive PRDM5 transcription factor in intestinal carcinogenesis [19]. However, the regulation of MGLL expression may operate at multiple levels. For instance, MGLL was reported as a potential tumor suppressor in hepatocellular carcinomas, associated with its loss of protein expression, which was posttranslationally dictated by SND1-promoted ubiquitination for proteasome-mediated proteolysis [18]. As revealed in the datasets of cancer genomic projects [22], MGLL amplification is common in various tumor types such as prostatic carcinomas with neuroendocrine phenotypes [23] and pancreatic ductal adenocarcinomas [24]. Further understanding of the mechanisms underlying deregulated lipid metabolism may provide novel therapeutic strategies for imatinib-refractory GISTs, for which MGLL may represent a promising target metabolic driver in light of the rapid emergence of several novel MGLL-targeting chemical inhibitors [17, 25].
In summary, MGLL was substantiated as a critical lipid-metabolizing enzyme contributing to aggressiveness in GISTs, given its risk increment-associated mRNA upregulation and protein overexpression. MGLL overexpression is associated with adverse clincopathological factors and is independently predictive of unfavorable prognosis, suggesting its causative role in conferring aggressive phenotypes to primary localized, imatinib-naïve GISTs. However, the expression status of MGLL is associated with neither KIT/PDGFRA genotypes nor imatinib resistance or sensitivity. Future studies may further elucidate the molecular underpinning of MGLL overexpression in GISTs and the therapeutic relevance of MGLL inhibitors to facilitate the development of alternative targeted therapy for imatinib-resistant GISTs with high-risk aggressiveness.
MATERIALS AND METHODS
Reappraisal of published transcriptomic datasets
Focusing on driver(s) deregulated in lipid metabolism, we reappraised transcriptomic dataset of imatinib-naïve gastric and intestinal stromal tumors at various risk levels to search for aberrantly expressed genes critical in tumor progression. These samples were deposited in Gene Expression Omnibus (GSE8167) and profiled for global mRNA expression by using GeneChip Human Genome U133 Plus 2.0 arrays. The raw CEL files were imported into the Nexus Expression 3 software (BioDiscovery Hawthorne, CA, USA) to analyze all probe sets without preselection or filtering. Unsupervised comparative analysis was performed to identify significant genes differentially expressed between the high-risk and non-high-risk samples, with special attention paid to the lipid metabolic process in Gene Ontology (GO: 0006629). The ranking in the expression fold change (at least >1-fold in the log2-transformed ratio) and the power of statistical significance (p ≤ 0.0001 according to the Student t test) were considered for prioritizing potential candidate genes for further validation.
Tumor cohorts
The institutional review board of Chang Gung Memorial Hospital approved this study (102-3911B). To validate the transcriptomic reappraisal results, we exploited the first cohort of 86 primary localized GISTs with formalin-fixed tissues for assessing MGLL mRNA expression level by using QuantiGene assays and MGLL protein immunoexpression on whole sections from 70 cases with informative data of MGLL mRNA quantitation (cohort 1 in Supplementary Figure 1). The second cohort comprised 370 primary tumors resected before 2009 (cohort 2 in Supplementary Figure 1), from which triplicate representative cores for each case had been assembled into TMAs used in a previous publication [8]. TMA sections were recut for MGLL immunostaining, yielding 350 informative cases, including 213 successfully determined for KIT/PDGFRA genotypes as described previously [8]. All cases were imatinib-naïve before disease relapse in both cohorts, the clinicopathological characteristics of which are listed in Table 1 and Supplementary Table 1.
QuantiGene branched-chain DNA assay
This novel assay employed a sandwich nucleic acid hybridization technique to quantitatively measure the relative mRNA abundance of housekeeping and target transcripts in tissue homogenates obtained from formalin-fixed tumor tissues [26]. In brief, custom probes specifically targeting the MGLL transcript were designed for detection through the QuantiGene Multiplex 2.0 assay system (Affymetrix/Panomics Inc., Santa Clara, CA), according to the manufacturer’s instructions. Oligonucleotides of the probe set were mixed with the lysed paraffin sections, and the mixture was then added to a 96-well plate coated with capture probe oligonucleotide. Target RNA was captured during overnight incubation at 55°C. Unbound material was removed by three-run washes with 300 μL of wash buffer, followed by the hybridization of DNA amplifier molecules and three additional washes after incubation every time. After the final wash, the dioxetane alkaline phosphatase substrate Lumiphos Plus (Lumingen Inc., Southfield, MI, USA) was added to the reaction wells for detection using a Luminex 100 microplate luminometer (Luminex, Austin, TX, USA). The detected readout of MGLL mRNA abundance was further normalized through the expression level of reference glyceraldehyde-3-phosphate dehydrogenase transcript.
Cell culture
GIST882, GIST48, and GIST430 cell lines were kindly provided by Professor Jonathan Fletcher and cultured by following published methods [8, 16]. In brief, cell lines were maintained in Iscove’s modified Dulbecco’s medium (Invitrogen, Carlsbad, CA, USA) supplemented with 15% fetal bovine serum (FBS), 100 U/mL penicillin/streptomycin, and 4 mM L-glutamine (Invitrogen) at 37°C in 5% CO2. GIST882 was established from an untreated GIST with an imatinib-sensitive K642E mutation in KIT exon13. GIST48 and GIST430 were derived from progressing GISTs on imatinib therapy. GIST48 harbored primary homozygous V560D mutation in KIT exon11 and secondary heterozygous D820A mutation in KIT exon17. GIST430 exhibited primary heterozygous exon 11 in-frame deletion and secondary heterozygous exon13 missense mutation. Primary HCSMCs (ScienCell, Carlsbad, CA, USA) were cultured at 37°C in smooth muscle medium containing 500 mL of basal medium, 10 mL of FBS, 5 mL of growth supplement, and 5 mL of penicillin–streptomycin solution until 90% confluence.
Western blot analysis
To evaluate the endogenous MGLL expression, equal amounts of total protein (25 μg) extracted from GIST cell lines and primary HCSMCs were separated on 4%–12% gradient NuPAGE gels (Invitrogen), transferred onto PVDF membranes (Amersham, Arlington Heights, IL, USA), and blocked with 5% skim milk in Tris-buffered saline with Tween 20 at room temperature for 1 h. The membranes were then probed with antibodies against MGLL (1:1000, Epitomics, Burlingame, CA, USA) and GADPH (1:3000, Chemicon, Temecula, CA, USA) as the loading control. After incubation with the secondary antibody for 1.5 h, MGLL protein was visualized using an enhanced chemiluminescence system (Amersham) and semiquantitatively measured through densitometry.
Immunohistochemistry
Whole blocks and TMA sections were microwave-heated to retrieve tissue antigen and incubated with the primary antibody against MGLL (1:100; Epitomics), followed by detection with ChemMate EnVision kit (Dako, Glostrup, Denmark). Blinded to patient outcomes and molecular testing results, one pathologists (I.C.C.) independently assessed cytoplasmic MGLL expression through H-scoring [27], defined by the equation ΣPi (i + 1), where i is the intensity of stained tumor cells (0–3+) and Pi is the percentage of stained tumor cells (0%–100%). Specifically for the TMA cohort, MGLL overexpression was defined for cases when their means of triplicate H-scores were higher the median value of the 350 informative cases.
KIT/PDGFRA mutation analysis
The methods of direct sequencing of KIT exon 11 and denatured high-performance liquid chromatography screening for KIT exons 9, 13, and 17 and PDGFRA exons 12 and 18 with confirmatory sequencing have been described previously [8, 28].
Statistical analysis
The Mann–Whitney U test was performed on the full-sectioned samples to determine the difference in MGLL mRNA abundance between adjacent normal tissue and GISTs and between the high-risk and non-high-risk groups. Pearson correlation analysis was used to evaluate the association between MGLL mRNA abundance and MGLL immunoexpression. In the TMA validation set, we evaluated the associations of MGLL immunoexpression with clinicopathological factors by using the Chi-square and Wilcoxon rank-sum tests for categorical and continuous variables, respectively. Follow-up data were available for 350 cases as of April 2009 (median, 49.9 months; range, 1–247 months). The endpoints were DFS and OS. KIT/PDGFRA genotypes were dichotomized into two groups according to prognosis, as reported previously [8, 28]. In brief, the favorable genotypes included PDGFRA mutation involving exons 12 or 18, 3′ tandem insertion of KIT exon 11 with or without point mutation, and a single point mutation of KIT exon 11. The unfavorable genotypes were Ala502-Tyr503 insertion of KIT exon 9, wild-type for both KIT and PDGFRA, and 5′ deletion of KIT exon 11 with or without a point mutation. We used the log-rank test to compare univariate prognostic analyses. Significant prognosticators with univariate p < 0.05 were generally included in the multivariate Cox regression analysis. As component factors of the NIH risk scheme and NCCN guidelines, tumor size and mitotic activity were not introduced in the multivariate comparisons.
Abbreviations
GIST, gastrointestinal stromal tumor; KIT, KIT protooncogene receptor tyrosine kinase: MGLL, monoglyceride lipase; NIH, National Institutes of Health; NCCN, National Comprehensive Cancer Network; PDGFRA, platelet-derived growth factor receptor alpha.
ACKNOWLEDGMENTS
The authors thank the genomics (CMRPG880251) and tissue bank (CLRPG8B0033 and CLRPG8E0161) core laboratories of Kaohsiung Chang Gung Memorial Hospital for their technical assistance.
CONFLICTS OF INTEREST
No conflicts of interest were declared.
GRANT SUPPORT
This work was sponsored by the Taiwan National Science Council (NSC99-2628-B-182A-064-MY3, 102-2628-B-182A-002-MY3 to HYH, NSC99-2320-B-384-001-MY2, 104-2314-B-384-009-MY3, and MOHW104-TDU-M-212-133004 to CFL) and Chang Gung Memorial Hospital (CMRPG8C0983 to HYH, CMRPG8C0993 to LTT).
Author contributions
HYH, CFL, and CIC conceived the experiments, analyzed the data, and prepared the manuscript. TTL, JL, CKC, YYC, FMF, SHL, and TJC collected and analyzed the data. TTL, CIC, and SCY performed the experiments. All authors were involved in writing the paper and had final approval of the submitted manuscript.
REFERENCES
1. Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004; 22: 3813-3825.
2. Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, Leversha MA, Jeffrey PD, Desantis D, Singer S, Brennan MF, Maki RG, DeMatteo RP. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005; 11: 4182-4190.
3. Antonescu CR, Sommer G, Sarran L, Tschernyavsky SJ, Riedel E, Woodruff JM, Robson M, Maki R, Brennan MF, Ladanyi M, DeMatteo RP, Besmer P. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003; 9: 3329-3337.
4. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006; 24: 4764-4774.
5. Martín J, Poveda A, Llombart-Bosch A, Ramos R, López-Guerrero JA, García del Muro J, Maurel J, Calabuig S, Gutierrez A, González de Sande JL,Martínez J, De Juan A, Laínez N, et al. Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol. 2005; 23: 6190-6198.
6. Bachet JB, Hostein I, Le Cesne A, Brahimi S, Beauchet A, Tabone-Eglinger S, Subra F, Bui B, Duffaud F, Terrier P, Coindre JM, Blay JY, Emile JF. Prognosis and predictive value of KIT exon 11 deletion in GISTs. Br J Cancer. 2009; 101: 7-11.
7. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, Nilsson B, Braconi C, Bordoni A, Magnusson MK, Sufliarsky J, Federico M, Jonasson JG, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015; 33: 634-642.
8. Li CF, Chen LT, Lan J, Chou FF, Lin CY, Chen YY, Chen TJ, Li SH, Yu SC, Fang FM, Tai HC, Huang HY. AMACR amplification and overexpression in primary imatinib-naive gastrointestinal stromal tumors: a driver of cell proliferation indicating adverse prognosis. Oncotarget. 2014; 5: 11588-11603. doi: 10.18632/oncotarget.2597.
9. Demetri GD, von Mehren M, Antonescu CR, DeMatteo RP, Ganjoo KN, Maki RG, Pisters PW, Raut CP, Riedel RF, Schuetze S, Sundar HM, Trent JC,Wayne JD. NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Netw. 2010; 8 Suppl 2: S1-41; quiz S42-44.
10. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, Miettinen M, O'Leary TJ, Remotti H, Rubin BP, Shmookler B, Sobin LH, Weiss SW. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002; 33: 459-465.
11. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011; 11: 85-95.
12. Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010; 6: 551-562.
13. Liu R, Huang Y. Lipid Signaling in Tumorigenesis. Mol Cell Pharmacol. 2014; 6: 1-9.
14. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010; 140: 49-61.
15. Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, Hoover HH, Cravatt BF. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol. 2011; 18: 846-856.
16. Rossi S, Ou W, Tang D, Bhattacharya N, Dei Tos AP, Fletcher JA, Loda M. Gastrointestinal stromal tumors overexpress fatty acid synthase. J Pathol. 2006; 209: 369-375.
17. Scalvini L, Piomelli D, Mor M. Monoglyceride lipase: Structure and inhibitors. Chem Phys Lipids. 2016; 197: 13-24.
18. Rajasekaran D, Jariwala N, Mendoza RG, Robertson CL, Akiel MA, Dozmorov M, Fisher PB, Sarkar D. Staphylococcal nuclease and tudor domain containing 1 (SND1) promotes hepatocarcinogenesis by inhibiting monoglyceride lipase (MGLL). J Biol Chem. 2016; 291:10736-46. doi: 10.1074/jbc.M116.715359.
19. Sun H, Jiang L, Luo X, Jin W, He Q, An J, Lui K, Shi J, Rong R, Su W, Lucchesi C, Liu Y, Sheikh MS, et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene. 2013; 32: 234-241.
20. Ye L, Zhang B, Seviour EG, Tao KX, Liu XH, Ling Y, Chen JY, Wang GB. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011; 307: 6-17.
21. Galli GG, Multhaupt HA, Carrara M, de Lichtenberg KH, Christensen IB, Linnemann D, Santoni-Rugiu E, Calogero RA, Lund AH. Prdm5 suppresses Apc(Min)-driven intestinal adenomas and regulates monoacylglycerol lipase expression. Oncogene. 2014; 33: 3342-3350.
22. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2: 401-404.
23. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016; 22: 298-305.
24. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, Choti MA, Yeo CJ, McCue P, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015; 6: 6744.
25. Tuccinardi T, Granchi C, Rizzolio F, Caligiuri I, Battistello V, Toffoli G, Minutolo F, Macchia M, Martinelli A. Identification and characterization of a new reversible MAGL inhibitor. Bioorg Med Chem. 2014; 22: 3285-3291.
26. Knudsen BS, Allen AN, McLerran DF, Vessella RL, Karademos J, Davies JE, Maqsodi B, McMaster GK, Kristal AR. Evaluation of the branched-chain DNA assay for measurement of RNA in formalin-fixed tissues. J Mol Diagn. 2008; 10: 169-176.
27. Ma LJ, Lee SW, Lin LC, Chen TJ, Chang IW, Hsu HP, Chang KY, Huang HY, Li CF. Fibronectin overexpression is associated with latent membrane protein 1 expression and has independent prognostic value for nasopharyngeal carcinoma. Tumor Biol. 2014; 35: 1703-1712.
28. Li CF, Huang WW, Wu JM, Yu SC, Hu TH, Uen YH, Tian YF, Lin CN, Lu D, Fang FM, Huang HY. Heat shock protein 90 overexpression independently predicts inferior disease-free survival with differential expression of the alpha and beta isoforms in gastrointestinal stromal tumors. Clin Cancer Res. 2008; 14: 7822-7831.