
Introduction
Germline mutations occur in several cancer-related genes including, but not limited to, BRCA1, BRCA2, RB1 and TP53.[1-4] Patients with these mutations often develop tumors at a young age. Ewing sarcoma is a malignant, small, round blue-cell tumor that primarily occurs in children, adolescents and young adults. [5, 6] It is associated with a somatic translocation of chromosome 22, which typically generates a fusion protein product designated as EWS-FLI1.[7]
Approximately 50% of patients with Ewing sarcoma also express activated/phosphorylated signal transducer and activator of transcription 3 (STAT3).[8] STAT3 is known to drive pathways regulating normal cell growth, which result in tumorigenesis when dysregulated.[9] Protein tyrosine phosphatase delta (PTPRD), a tumor suppressor belonging to the protein tyrosine phosphatase (PTP) family[10], plays an essential role in dephosphorylating STAT3.[11] Genetic aberrations of PTPRD are associated with a poor prognosis in malignant tumors.[11, 12] Mutations in PTPRD have been reported in approximately 13% of head and neck squamous cell carcinoma [11], 12% of melanoma [13], and in a small subset of various other malignancies.[11] However, germline mutations of PTPRD have not been previously described.
Here we report three of eight patients (37.5%) with metastatic Ewing sarcoma who harbored germline mutations in the PTPRD gene. Using next-generation sequencing (NGS), two germline mutations were found in one patient, including one leading to truncation and subsequent loss of function of the PTPRD suppressor. Of interest, two of these three patients achieved a complete response (CR) following insulin-like growth factor 1 receptor (IGF-1R) inhibitor-based therapy. Because phosphorylated STAT3 is frequently upregulated in Ewing sarcoma [8] and PTPRD dephosphorylates STAT3, the role of germline and somatic PTPRD mutations in Ewing sarcoma as well as the implications for IGF-1R targeted therapy warrant exploration.
Results
A total of eight patients diagnosed with advanced/metastatic Ewing sarcoma with available PBMCs who had been referred to the CCTT and/or Department of Pediatrics at MD Anderson were analyzed. The median age at diagnosis was 19.5 years (range, 13 to 34 years). All patients developed metastatic disease between 0 and 7 years after initial diagnosis (Table 1).
Of the eight patients with Ewing sarcoma, three (37.5%) had germline mutations in the PTPRD gene. Patient 1 (Table 1; age 24 at diagnosis) had mutational analysis of 182 cancer-related genomic alterations in formalin-fixed paraffin-embedded tumor tissue performed using a Clinical Laboratory Improvement Amendment approved Foundation One platform. Simultaneously, DNA extracted from tumor tissue and PBMCs from the same patient were analyzed independently with next-generation whole exome sequencing in the MD Anderson Core Laboratory using the SOLiD platform. A PTPRD mutation annotated as a W775stop germline mutation was found in both the patient’s tumor and PBMCs (Figure 1). The PTPRD mutation was confirmed by polymerase chain reaction (PCR)-based Sanger sequencing in genomic DNA derived from tumor and PBMCs. The W775stop germline mutation is located in the extracellular fibronectin type III (FN3) region (Figure 1).[10] The mutation of tryptophan to a stop codon results in the truncation of all transmembrane and intracellular domains, which leads to partial loss of the dephosphorylation function of PTPRD. Because PTPRD functions as a STAT3 phosphorylation suppressor, it is plausible that partial loss of PTPRD can lead to increased STAT3 phosphorylation.[11] In addition, NGS revealed a V253I germline mutation located in the third immunoglobulin (Ig)-like domain of the receptor protein tyrosine phosphatase (RPTP)-F region, also known as LAR, within the extracellular domain (Figure 1) [10]. The impact of this mutation is, however, unclear. Patient 1 had durable CRs resulting from IGF-1R inhibitor-based therapies (Figure 2).[14, 17-20]
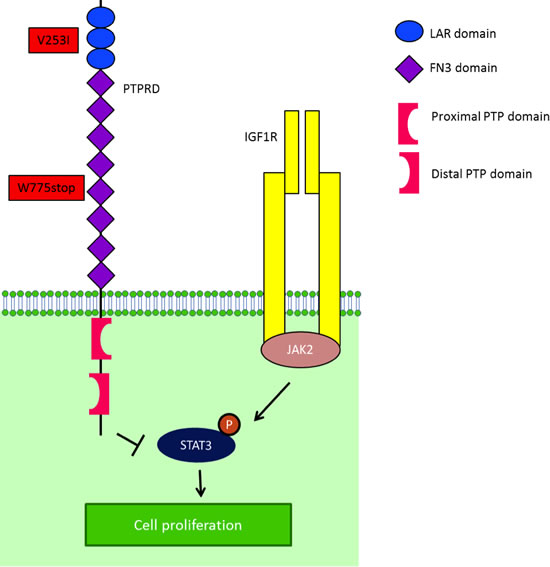
Figure 1: IGF-1R is one of the mediators of STAT3 activity. STAT3 is phosphorylated by JAK2, after being recruited to IGF-1R by RACK1. After STAT3’s phosphorylation by JAK2, PTPRD normally dephosphorylates STAT3. In the presence of a truncated PTPRD, STAT3 would remain phosphorylated.
Table 1: Patients with Ewing sarcoma tested for germline mutations in PTPRD and outcomes with IGF-1R-based therapy.
Age at diagnosis |
Mutation |
Time from diagnosis metastasis |
Best RECIST response to IGF-1R inhibitor monotherapy (PFS) |
Best RECIST response to IGF-1R+mTOR inhibitor (PFS) |
|||
1 |
24 |
V253I, W775stop |
6 months |
CR, -100% (3 years) |
CR, -100% (2 years) |
||
2 |
22 |
T781A |
0 |
Not treated |
PD, +21% |
||
3 |
33 |
Wild-type |
7 years |
Not treated |
SD, -27% (16+ months) |
||
4 |
18 |
Wild-type |
2 years |
Not treated |
SD, -23% (20 months) |
||
5 |
13 |
R995C |
1 years |
Not treated |
CR, -100% (28 months) |
||
6 |
21 |
Wild-type |
0 |
Not treated |
PD, -42%, new lesion |
||
7 |
13 |
Wild-type |
0 |
Not treated |
SD, -14% (4.5 months) |
||
8 |
13 |
Wild-type |
0 |
Not treated |
Not treated |
||
Abbreviations: RECIST, response evaluation criteria in solid tumors; IGF-1R, insulin-like growth factor receptor 1; CR, complete response; PD, progressive disease; SD, stable disease
PBMCs from an additional seven patients (2 to 8, Table 1) with advanced Ewing sarcoma were analyzed for PTPRD germline mutations in all 35 exons (exons 11-45, ENST00000381196) using Sanger sequencing, as previously described.[11] Among these seven patients, five had wild-type PTPRD and two had PTPRD germline mutations: T781A and R995C. The tumor tissues from these two patients were not available for testing. A second patient with a PTPRD mutation (Patient 5, Table 1) also attained a CR on IGF-1R inhibitor-based therapy.
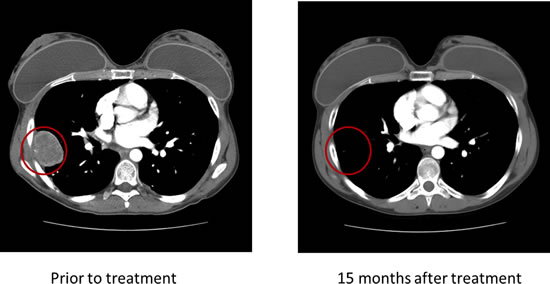
Figure 2: Patient 1 demonstrated a durable complete response to therapy with an IGF-1R inhibitor.
Discussion
We identified germline PTPRD mutations in three (37.5%) of eight patients with advanced/metastatic Ewing sarcoma. The first patient had two mutations (Table 1). The PTPRD W775stop germline mutation is expected to express truncated PTPRD protein, leading to loss of function; however, it has not previously been reported in the Exome Sequencing Project (ESP) [21] and Catalogue of Somatic Mutations in Cancer (COSMIC) databases [22] in the general population or in cancer patients (Figure 1). The W775stop germline mutation can lead to the truncation of all transmembrane and intracellular domains, which can putatively result in partial loss of the dephosphorylation function of PTPRD. Because PTPRD serves to suppress STAT3 phosphorylation [11], partial loss of PTPRD can possibly lead to increased STAT3 phosphorylation, which has been reported in approximately 50% of patients with Ewing sarcoma.[8] Patient 1 also had a PTPRD V253I germline mutation located within the extracellular domain coding region; its functional consequences are unclear (Figure 1).[10] In another two patients, we identified germline mutations T781A and R995C, which are single nucleotide polymorphisms of the PTPRD suppressor that have been reported in a small subset (2.2% and 7.4%, respectively) of the general population [21], but whose functional impact is unclear.
Two of three patients with germline PTPRD mutations attained a CR in response to therapies with a IGF-1R monoclonal antibody and or a combination of an IGF-1R monoclonal antibody and the mTOR inhibitor temsirolimus. IGF-1R is one of the mediators of STAT3 activity. STAT3 is phosphorylated by JAK2, after being recruited to the IGF-1R by RACK1 (Figure 1).[23] After STAT3 phosphorylation by JAK2, PTPRD normally serves to dephosphorylate STAT3. In the presence of truncated PTPRD, STAT3 would remain phosphorylated. However, based on the biology described, an IGF-1R inhibitor might interfere with the initial phosphorylation of STAT3, perhaps explaining the response to IGF-1R inhibition in these patients.
Somatic PTPRD mutations have been identified in a small subset of patients with cancer types such as glioblastoma multiforme, melanoma, squamous cell carcinoma of the head and neck, and others.[11, 13] To date, germline mutations of PTPRD have not been described in Ewing sarcoma. Our sample size is small, and the functional consequences of the mutations have not been fully elucidated; therefore, our findings should be interpreted with caution. However, the role of germline PTPRD mutations in predisposition to Ewing sarcoma, a disease of children, adolescents, and young adults, and the implications of these mutations for therapy with IGF-1R and STAT3 inhibitors warrants further investigation..
Patients and methods
Patients
We reviewed the electronic medical records of eight patients with advanced/metastatic Ewing sarcoma referred to the Clinical Center for Targeted Therapy (CCTT) at The University of Texas MD Anderson Cancer Center (MD Anderson) who had available peripheral blood mononuclear cells (PBMCs) and, in some cases, tumor tissue. Data were collected from transcribed notes, radiology films and reports in the electronic medical record and other source documentation. Registering patients in the database, clinical, pathologic, laboratory and pathology assessment were performed at MD Anderson. The study and all treatments were conducted in accordance with the guidelines of the MD Anderson Institutional Review Board.
Molecular analysis
Targeted NGS of tumor tissue was performed in a Clinical Laboratory Improvement Amendment-approved laboratory at Foundation Medicine (Cambridge, MA). Genomic libraries were captured for 3,230 exons in 182 cancer-related genes plus 37 introns from 14 genes that are often rearranged in cancer and sequenced to an average median depth of 734X with 99% of bases covered >100X.
Whole exome sequencing of tumor and PBMC samples was performed using an Applied Biosystems 5500XL SOLiD Next-Generation Sequencing platform (Life Technologies, Carlsbad, CA) in the MD Anderson Core laboratory. The 5500XL SOLiD system generates 300 GB of mappable sequence tags in a single run. This level of data throughput enables 30X coverage of the human genome with high accuracy and depth, facilitating detection of rare somatic mutations, insertions and deletions in patient samples.
Sanger sequencing of tumor tissue and PBMCs used to identify PTPRD mutations in all 35 exon (exons 11-45, ENST00000381196) was done as previously described.[11]
Treatment
Patients were enrolled in clinical trials with a single-agent IGF-1R monoclonal antibody or a combination of a monoclonal IGF-1R monoclonal antibody and the mTOR inhibitor temsirolimus.[14, 15] Treatment was carried out according to the specific requisites in these treatment protocols and continued until disease progression or unacceptable toxicity occurred.
Assessments, including history, physical examination, and laboratory evaluations, were performed as specified in each protocol, typically before the initiation of therapy, weekly during the first cycle, and then, at a minimum, at the beginning of each new treatment cycle. Response was assessed from computed tomography (CT) scans and/or magnetic resonance imaging (MRI) at baseline before treatment initiation and then every 2 cycles (8 weeks). All radiographs were read in the Department of Radiology at MD Anderson and reviewed in the Department of Investigational Cancer Therapeutics tumor measurement clinic. Responses were categorized per RECIST criteria.[16] In brief, a CR was defined as the disappearance of all measurable and non-measurable disease; partial response (PR) was defined as at least a 30% decrease in the sum of the longest diameter of measurable target lesions; progressive disease (PD) was defined as at least a 20% increase in the sum of the longest diameter of measurable target lesions, or unequivocal progression of a non-target lesion, or the appearance of a new lesion; and stable disease (SD) was defined as neither sufficient shrinkage to qualify as a PR nor an increase sufficient to qualify as PD.
Statistical analysis
There was no formal statistical analysis; however, progression-free survival (PFS) was defined as the time interval from the start of therapy to the first observation of disease progression or death, whichever occurred first.
Acknowledgements
Dr. Anderson wishes to acknowledge the Sam Butler Callahan Ewing sarcoma fund. We thank Ms. Joann Aaron for scientific review and editing of this article.
References
1. Claus EB, Schildkraut JM, Thompson WD and Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer. 1996; 77(11):2318-2324.
2. Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JL, Fan I, Wong B and Narod SA. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001; 68(3):700-710.
3. Harbour JW. Molecular basis of low-penetrance retinoblastoma. Arch Ophthalmol. 2001; 119(11):1699-1704.
4. Malkin D, Li FP, Strong LC, Fraumeni JF, Jr., Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA and et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990; 250(4985):1233-1238.
5. Burchill SA. Molecular abnormalities in Ewing’s sarcoma. Expert Rev Anticancer Ther. 2008; 8(10):1675-1687.
6. Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA and Juergens H. Ewing’s sarcoma family of tumors: current management. Oncologist. 2006; 11(5):503-519.
7. Janknecht R. EWS-ETS oncoproteins: the linchpins of Ewing tumors. Gene. 2005; 363:1-14.
8. Lai R, Navid F, Rodriguez-Galindo C, Liu T, Fuller CE, Ganti R, Dien J, Dalton J, Billups C and Khoury JD. STAT3 is activated in a subset of the Ewing sarcoma family of tumours. J Pathol. 2006; 208(5):624-632.
9. Calo V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N and Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol. 2003; 197(2):157-168.
10. Julien SG, Dube N, Hardy S and Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011; 11(1):35-49.
11. Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, Gerald WL, Liau L, Cloughesy TC, Mischel PS, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009; 106(23):9435-9440.
12. Chan TA, Glockner S, Yi JM, Chen W, Van Neste L, Cope L, Herman JG, Velculescu V, Schuebel KE, Ahuja N and Baylin SB. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS Med. 2008; 5(5):e114.
13. Solomon DA, Kim JS, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, Ressom H, Jean W, Bigner D, Yan H, Samuels Y and Waldman T. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008; 68(24):10300-10306.
14. Kurzrock R, Patnaik A, Aisner J, Warren T, Leong S, Benjamin R, Eckhardt SG, Eid JE, Greig G, Habben K, McCarthy CD and Gore L. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin Cancer Res. 2010; 16(8):2458-2465.
15. Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, Ludwig J, Chen HX, Doyle LA and Kurzrock R. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(9):2625-2631.
16. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D and Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45(2):228-247.
17. Subbiah V, Naing A, Brown RE, Chen H, Doyle L, LoRusso P, Benjamin R, Anderson P and Kurzrock R. Targeted morphoproteomic profiling of Ewing’s sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) inhibitors: response/resistance signatures. PLoS One. 2011; 6(4):e18424.
18. Naing A, Lorusso P, Fu S, Hong DS, Anderson P, Benjamin RS, Ludwig J, Chen HX, Doyle LA and Kurzrock R. Insulin Growth Factor-Receptor (IGF-1R) Antibody Cixutumumab Combined with the mTOR Inhibitor Temsirolimus in Patients with Refractory Ewing’s Sarcoma Family Tumors. Clin Cancer Res. 2012;18(9):2625-31.
19. Subbiah V and Kurzrock R. Ewing’s sarcoma: overcoming the therapeutic plateau. Discov Med. 2012; 13(73):405-415.
20. Naing A, Kurzrock R, Burger A, Gupta S, Lei X, Busaidy N, Hong D, Chen HX, Doyle LA, Heilbrun LK, Rohren E, Ng C, Chandhasin C and LoRusso P. Phase I trial of cixutumumab combined with temsirolimus in patients with advanced cancer. Clin Cancer Res. 2011; 17(18):6052-6060.
21. Exome Variant Servier [cited 2012 09/19]; Available from: http://evs.gs.washington.edu/EVS/.
22. Catalogue of Somatic Mutations in Cancer [cited 2012 09/19]; Available from: http://www.sanger.ac.uk/genetics/CGP/cosmic/
23. Zhang W, Zong CS, Hermanto U, Lopez-Bergami P, Ronai Z and Wang LH. RACK1 recruits STAT3 specifically to insulin and insulin-like growth factor 1 receptors for activation, which is important for regulating anchorage-independent growth. Mol Cell Biol. 2006; 26(2):413-424.