
Introduction
Lysine Acetylation: lysine acetyltransferases (KATs) and lysine deacetylases (KDACs)
Acetylation of the ε-amino group of lysine residues has recently emerged as an important covalent post-translational modification (PTM) for regulating protein functions. Acetyltransferases catalyze the transfer of an acetyl group from acetyl-CoA to the terminal amine on the side chain of lysine residues. These enzymes are commonly called Histone acetyltransferases (HATs), because their best-known substrates are histone proteins (H2A, H2B, H3 and H4). However, the nomenclature is being changed to lysine acetyltransferases (KATs), reflecting their ability to acetylate lysine (K) on many proteins. Conversely, histone or lysine deacetylases (HDACs or KDACs), catalyze the inverse reaction by removing acetyl groups from proteins (Figure 1). The conversion of the positively charged lysine to acetyl-lysine, like the addition of negative phosphates to uncharged amino acids during phosphorylation, alters protein structure and interactions with other biomolecules.
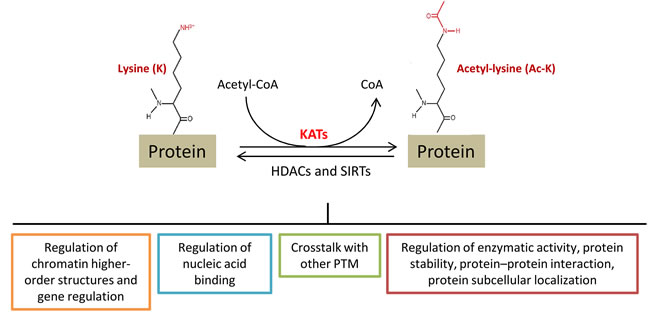
Figure 1: Lysine (K) acetylation is a reversible post-translational modification of proteins, including histones, transcription factors as well as metabolic enzymes and other nuclear and cytoplasmic proteins. Proteins can be acetylated at lysine residues (Ac-K) by specific enzymes, called lysine acetyltransferases (KATs), or deacetylated by Zn2+-dependent histone deacetylases (HDACs) and NAD+-dependent sirtuin deacetylases (SIRTs). Acetylation levels are tightly regulated by these enzymes and can impact the biological function of different proteins, some of them are here reported. PTM: post-translational modifications.
Thus far, different KATs, localized both in the nucleus and cytoplasm, have been identified in human cells [1, 2]. They can be grouped into three major families: Gcn5-related N-acetyltransferase (GNAT), p300 and CREB-binding Protein (p300/CBP) and Moz, Ybf2/Sas3, Sas2, Tip60 (MYST). Apart from these, two other KAT families exist, which belong to transcription factor-related KATs and nuclear receptor family of KATs (Table 1). The 18 KDACs that have been identified in the human genome belong to two distinct families with different catalytic mechanisms: Zn2+-dependent histone deacetylases (HDAC1-11) and NAD+-dependent sirtuin deacetylases (SIRT1-7). Zn2+-dependent deacetylases are predominantly expressed both in the nucleus and cytoplasm, whereas sirtuins are present also in the mitochondria.
In the present review, we discuss the functional implications of lysine acetylation in different cellular compartments, the changes in lysine acetylation associated with cancer biology and the potential relevance of lysine acetylation as biomarkers for cancer prognosis. Finally, we discuss the achievements and drawbacks of drugs targeting the lysine acetylation, with particular regards to the field of anti-neoplastic drug development.
Table 1: Lysine acetyltransferases (KATs) family
New nomenclature |
Former name in human |
Cellular localization |
Histone protein acetylated |
Main non-histone protein acetylated |
P300/CBP family |
||||
KAT3 |
||||
KAT3A |
CBP |
Nucleus |
H2A, H2B |
NF-kappaB, c-myb, Foxo1 |
KAT3B |
P300 |
Nucleus |
H2A, H2B |
NF-kappaB, c-myc, p53, STAT3, β-catenin, Foxo1, AR |
GNAT family |
||||
KAT1 |
HAT1 |
Nucleus |
H3, H4, H2A |
|
KAT2 |
H3, H2B |
|||
KAT2A |
GCN5 |
Nucleus |
H3, H4, H2A |
CDC6, CDK9,cyclin D1, cyclin E1 and E2F1, HDM2, PTEN, c-myc |
KAT2B |
pCAF |
Nucleus |
H3 |
p53, CDK9, c-myc, Foxo1, AR |
KAT9 |
ELP3 |
H4, H2A, H3 |
||
ATAT-1 |
MEC-17 |
Cytosol |
tubulin, cortactin |
|
AT-1 |
ER |
BACE-1, ATG9 |
||
AT-2 |
ER |
BACE-1 |
||
MYST family |
||||
KAT5 |
Tip60 |
Nucleus |
H4, H2A |
ATM, TRRAP, p53, E2F1, c-myc, DNMT1 |
KAT6 |
||||
KAT6A |
MOZ |
Nucleus |
H3 |
|
KAT6B |
MORF |
Nucleus |
||
KAT7 |
HBO1 |
Nucleus |
H3, H4 |
|
KAT8 |
MOF |
H4 |
Lysine acetyltransferases families and their link to cancer
p300/CBP family
The p300/CBP family is composed of two closely related transcriptional coactivators CBP (or KAT3A) and its paralog p300 (KAT3B). The human CBP locus resides in the chromosomal region 16p13.3 and shows homology to 22q13, where p300 is located. They have also similar structures and share an overall 63% amino acid sequence identity and around 86% sequence identity at the KAT domain. CBP and p300 have interchangeable roles during embryonic development, and in many processes they govern cellular homeostasis. Both are transcriptional co-activators of various sequence-specific transcription factors that are involved in a wide array of cellular activities, such as DNA repair, cell growth, senescence, differentiation and apoptosis [3]. p300 is also involved in the regulation of expression and function of a large number of tumor-relevant proteins, including oncoproteins c-myc [4], androgen receptor (AR) [5], tumor suppressor proteins breast cancer gene-1 (BRCA1) [6] and p53 [7].
The importance of p300/CBP is underscored by the fact that genetic alterations, as well as their functional dysregulation, are strongly linked to cancer. Germline mutations of CBP were firstly reported in Rubinstein-Taybi Syndrome, an autosomal-dominant disease characterized by mental retardation, skeletal abnormalities and a high malignancy risk. Most of the described tumor-related mutations in p300/CBP result in truncation of the p300 protein. Mutations and/or deletions of p300 and/or CBP genes have been also reported in several types of cancer, as lung, colon, breast and ovarian carcinomas [8-10], indicating a p300 role as tumor suppressor, and suggesting that it may play a role in the development of a subset of human cancers. In this context, loss of heterozygosity (LOH) at the p300 locus has been observed in numerous cancers, including hepatocellular, colorectal, oral, breast, ovarian, gastric carcinomas and glioblastomas [11]. Consistently, several studies have also shown that loss of p300 correlates with aggressive features and poor prognosis of hepatocellular carcinoma (HCC) [12, 13], breast cancer [14], cutaneous squamous cell carcinoma (SCC) [15] and nasopharyngeal carcinoma [16]. However, p300 is also found to be overexpressed in prostate cancer, where it regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth [5, 17, 18]. p300 and CBP genes are involved in various chromosomal translocation events during haematological malignancy and might contribute to aberrant growth control possibly through a gain of function mutation. For example, the chromosomal translocation events that affect CBP give rise to tumor-specific hybrid proteins [19, 20]. In particular, chromosome translocations targeting CBP have been found in acute myeloid leukemia (AML) and are associated with the development of this neoplasia following chemotherapy for other forms of cancer [21]. Recently, it was shown that the CBP gene is genetically altered in almost 15% of lung cancer cell lines and 5% of primary lung tumors. An interesting coexistence of CBP and p53 mutations was also observed in lung cancer, suggesting that CBP gene alterations might contribute to lung carcinogenesis by distorting pathways other than those engaging p53 [8].
GNAT super family
The GNAT super family includes about 12 proteins with diverse cellular functions and substrates, among them GCN5 (General Control Nonderepressible 5; KAT2A) and other proteins showing a sequence and structural similarity to GCN5, PCAF (p300/CBP Associated Factor; KAT2B), α-tubulin acetyltransferase 1 (ATAT1), the chromatin-assembly-related Hat1, the elongator complex subunit Elp3, the mediator complex subunit Nut1, and Hpa2. GNAT proteins share a domain composed of four conserved sequence motifs A-D, and unusually they also have bromodomain or chromodomain for binding acetylated or methylated lysine respectively [22].
The two main members of this family, GCN5 and PCAF are closely related proteins. The former has homologs in yeast and human, whereas the latter appears exclusively in higher eukariotes. In general, GNATs are involved in cellular growth, playing an important role in the regulation of cell cycle. For example, GCN5 specifically acetylates cell-division cycle-6 (CDC6) at three lysine residues flanking its cyclin-docking motif. This modification is crucial for the subsequent phosphorylation of the protein by cyclin A-cyclin-dependent kinase (CDKs) at a specific residue close to the acetylation site. GCN5-mediated acetylation and site-specific phosphorylation of CDC6 are both necessary for the relocalization of the protein to the cell cytoplasm in the S phase, as well as for the regulation of its stability [23]. Both GCN5 and PCAF, regulate cyclin-dependent kinase-9 (CDK9) function by specifically acetylating the catalytic core of the enzyme. This modification causes a profound inhibition of CDK9 catalytic and transcriptional activities and relocates the enzyme to the insoluble nuclear matrix compartment [24]. In non-small cell lung cancer (NSCLC), GCN5 enhances cell proliferation and G1/S transition by regulating the expression of cell cycle proteins like cyclin D1, E1 and E2F1 [25]. Under stress conditions, PCAF is required for stress-responsive histone H3 acetylation at the p21 promoter, p53-directed transcription of p21 and the resultant growth arrest [26]. Interestingly, it was also shown that PCAF possesses a ubiquitin ligase activity, that is a key determinant in controlling Hdm2 levels and, consequently, the stability and activity of p53 [27]. PCAF has been also reported to interact with the tumor suppressor PTEN and to promote its acetylation on K125 and K128 in response to growth factor stimulation [28]. Moreover, PCAF has also been shown to be a p53 target gene [29].
The involvement of GCN5, PCAF and other family members in processes that are closely linked to the hallmarks of cancer, including DNA damage repair, cell cycle regulation and post-translational regulation of both oncoproteins and tumor suppressors, suggests an important functions for these proteins in cancer progression.
GCN5 is found to be overexpressed in human glioma and NSCLC tissues, where its expression positively correlates with proliferation of cell nuclear antigen (PCNA) and tumor size [25, 30], respectively. Recently, GCN5 expression was found upregulated in human colon cancer and regulated by c-myc and E2F1 transcription factors [31].
PCAF maps to the short arm of chromosome 3, which is frequently deleted in solid tumors such HCC, ovarian cancer, gastric cancer and esophageal SCC. Accordingly, PCAF has been found to be down-regulated in HCC tissues compared with the adjacent non-tumor tissues and significantly associated with malignant portal vein invasion and poor survival of HCC patients [32]. PCAF expression has also been reported to be downregulated in HCC, gastric cancer and ovarian cancer [33]. In esophageal SCC, loss of PCAF locus correlated with advanced tumor stage and metastasis. The expression of PCAF was also found to be downregulated in esophageal SCC cell lines and this downregulation was associated with DNA hypermethylation at the PCAF promoter [34].
ATAT1 is the main KAT responsible for α-tubulin acetylation at K40 in higher organisms. Recent structural analyses have provided elegant insights into how ATAT1 targets and acetylates K40 of α-tubulin [35-37]. Recent reports suggest that ATAT1 plays a prominent role in many cellular processes related to cancer dissemination, including cell adhesion, migration and invasion. In breast cancer cells, ATAT1 binds and regulates cortactin acetylation levels and colocalizes with cortactin at the adherent surface of the cells. ATAT1 is required for 2D migration and invasive capability of breast cancer cells in collagen matrix [38]. Ectopic ATAT1 expression in cultured cells also increases tubulin acetylation and enhances formation of microtentacles, which are membrane protrusions in detached breast cancer cells [39]. Notably, ex vivo cultured ATAT1 -/- mouse embryonic fibroblasts displayed impaired cell adhesion and contact inhibition [40]. ATAT1 is also associated with pancreatic cancer-initiating cells [41]. A recent study on cancer cells showed that an efficient ATAT1 downregulation could impair actin architecture and induce mitotic catastrophe in cells through mechanisms partly independent of acetyl-tubulin [42]. Interestingly, in zebrafish ATAT1 appears to govern embryo development [43], conversely the viability and grossly normal development of ATAT1 -/- embryos and mice indicate that ATAT1 is not essential for basic functions such as animal survival and development, but it may be important for more advanced functions, including learning and memory [44].
MYST family
MYST family is named after its founding members MOZ, Ybf2/Sas3, Sas2, Tip60 in yeast. The family currently comprises five human KATs: Tip60 (HIV1 TAT interacting 60 kDa protein), MOF, MOZ (monocytic leukemic zinc-finger protein), MORF (MOZ related factor), HBO1 (histone acetyltransferase bound to origin recognition complex (ORC)).
MYST members are characterized by the presence of a conserved MYST domain containing an acetyl-CoA binding and a zinc finger motifs [45]. Many MYST also have other domains for recognizing other proteins and are involved in transcription control, as well as cell growth and survival [46]. Surprisingly, this protein family was poorly studied. However, an increasing number of key studies have recently shown that these chromatin modifiers are required for a diverse range of cellular processes, both in health and disease.
Tip60, a well-studied member of the MYST family, is implicated in multiple cellular pathways, such as transcription, DNA damage-induced checkpoint activation, and apoptosis. It is an important enzyme for repairing DNA and returning cellular function to normal through its regulation of ataxia telangiectasia mutant (ATM) protein kinase, which phosphorylates and therefore activates proteins involved in DNA repair. However, in order to be functional, ATM protein kinase must be acetylated by the Tip60 protein. Lack of Tip60 suppresses ATM protein kinase activity and reduces the ability of a cell to repair DNA [47]. In the DNA repair process, Tip60 serves as a cofactor for transformation/transcription domain associated protein (TRRAP), an adapter protein playing an important role in Double Strand Break repair and chromatin remodeling. TRRAP enhances DNA remodeling by binding to chromatin near broken double stranded DNA sequences, and Tip60 seems to aid this recognition [48]. Acetylation of p53 by Tip60 at K120 was also reported and it was demonstrated to be essential for p53 induction of cell death [49]. Moreover, Tip60 knockdown leads to cisplatin-sensitivity in lung cancer cell lines. During DNA damage response to cisplatin, Tip60 interacts with E2F1 and promotes the E2F1 acetylation necessary for its accumulation [50].
Given the roles of Tip60 in transcriptional activation and DNA repair, it is not surprising that this KAT has been linked to cancer. The human Tip60 locus is frequently mutated in head and neck SCC, breast carcinoma and lymphomas. Nuclear Tip60 staining on tissue microarrays is lost in a variety of tumors, and most significantly in breast carcinomas [51]. Recent studies show down-regulation of Tip60 in breast cancer, and a correlation between low Tip60 levels and p53 mutations in basal-like breast cancers, thus suggesting that Tip60 is a novel breast tumor suppressor gene whose loss results in genomic instability leading to cancer formation [52]. Tip60 can also function as a co-activator for a number of steroid hormone receptors including the AR, which is involved in the development and progression of prostate cancer. In line with this, Tip60 is functionally up-regulated in clinical prostate cancer specimens and its expression correlates with disease progression [53].
Human MOF is highly conserved from fly to human and shows the same substrate specificity [54]. It is responsible for the larger part of K16 acetylation at histone H4 in human cells, with obvious links to cancer [55, 56]. In fact, it has been well known that depletion of MOF can influence a wide range of intracellular biological functions, including chromatin stability, cell cycle, gene transcription, DNA damage repair and early embryonic development [54, 57]. MOF has also been shown to play a role in embryonic stem cell renewal. In particular, MOF is an integral component of the embryonic stem cell core transcriptional network and it primes genes for diverse developmental programs, thus playing a very important role in normal physiology and disease [58]. Except for the global reduction of histone H4 acetylated at K16, depletion of MOF in mammal cells can result in abnormal gene transcription, especially causing abnormal expression of certain tumor suppressors or oncogenes [57, 59]. For example, MOF is frequently downregulated in 41% of patients with primary breast carcinoma and in 79% of patients with medulloblastoma, and its protein expression is tightly correlated with acetylated H4 at K16 in all tested samples [60]. Notably, correlation of low expression of MOF with clinicopathological features of colorectal, gastric and renal cell carcinomas was reported, and in patients with colorectal cancer, the patterns of MOF expression were mainly associated with lymph node metastasis and tumor stage [61]. Loss of MOF levels is prognostic marker also in ovarian and hepatocellular carcinomas [62-64]. Conversely, the overexpression of MOF in NSCLC predicts poor prognosis of the disease [65].
Human MOZ and MORF form stable multisubunit complexes, which are responsible for acetylation of a substantial portion of histone H3. The KAT activity of MOZ/MORF complexes is required for normal developmental programs, including hematopoiesis and skeletogeneis, and for the regulation of various genes, especially the Hox family [46, 66-69]. Both MOZ and MORF are mutated in cancer and developmental disorders. Chromosomal aberrations involving MOZ and MORF may be a cause of AML. In AML, both MOZ and MORF fuse with multiple genes including CBP and p300 [19, 20]. The resulting proteins possess 2 catalytic KAT domains, one from the MOZ/MORF fragment and another from the p300/CBP fragment, that trigger an aberrant histone acetylation and transcriptional activation associated with overexpression of oncogenes.
The inversion inv(8)(p11;q13) generates the MOZ-TIF2 fusion protein, that is associated with AML. MOZ-TIF2-induced transformation requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP [70]. In a recent report, mutant p53 has been shown to upregulate chromatin regulatory genes expression, including MOZ, leading to alteration in global chromatin modification. This may contribute to the gain of function of mutant p53. Interestingly, the Cancer Genome Atlas shows specific upregulation of MOZ in p53 gain of function patient-derived tumors, but not in wild-type p53 or p53 null tumors [71]. Recently, the presence of homozygous deletions of MORF, and the loss of the corresponding transcript, were observed in small cell lung cancer cell lines and primary tumors. Notably, in vitro and in vivo evidence demonstrated that the depletion of MORF expression enhances cancer growth, whilst its restoration induces tumor suppressor-like features [72].
A fifth human MYST protein is HBO1, which was discovered in a two-hybrid screen on the basis of its interaction with the ORC1 subunit of the origin recognition complex ORC [73]. HBO1 appears to play an essential role in DNA replication, so it is reasonable to speculate that defects in its function should have profound consequences to the cell and lead to oncogenesis. Nevertheless, little it is known on the role of HBO1 in cancer [46].
Functional Implications of Lysine Acetylation in different cellular compartments: biological role and significance in cancer
In the last years, developments of proteomic innovations have propelled acetylation research forwards. In 2009, by using high-resolution mass spectrometry, Mann and colleagues looked in deep to the cellular acetylome [74]. They identified about 3,600 lysine acetylation sites in 1,750 proteins, and observed the presence of lysine acetylation on proteins localized in different cellular compartments. Nowadays, it is well clear that lysine acetylation is not exclusively limited to nucleus (Figure 2), but may also occur in other cellular compartments, suggesting that acetylation may also regulate cellular processes not directly related to transcriptional activation (Figure 3).
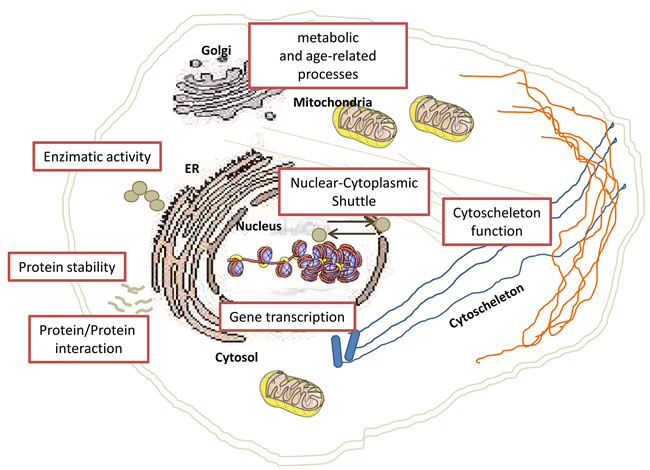
Figure 2: Schematic overview of nuclear, cytoplasmic and organelle-specific mechanisms regulated by acetylation. Lysine acetylation is well known to play a key role in regulating gene transcription and other DNA-dependent nuclear processes. Proteomics studies have identified many possible substrates of lysine acetylation and a large fraction of them resides in the cytoplasmic compartment, implicating their involvement in regulating important cellular pathways.
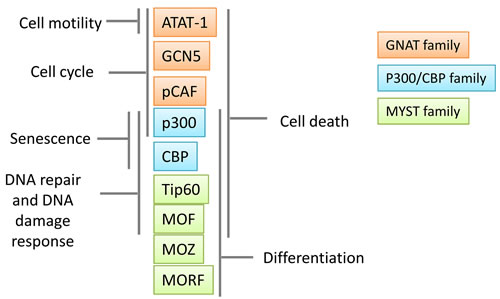
Figure 3: Role of main and well-studied lysine acetyltransferases (KATs) in cancer biology. KATs have an important role in transcription regulation and they participate in the expression of malignant phenotypes in cancer cells.
Nucleus: acetylation of histones and transcription-related factors
KATs are intimately involved in transcriptional activation. By modifying chromatin proteins and transcription-related factors, these enzymes are believed to regulate the transcription of many genes involved in cancer. In particular, chromatin architecture is strongly regulated by PTM of the N-terminal tails of the histone proteins [75]. Thus, acetylated histones represent a type of epigenetic markers within chromatin. In general, the presence of acetylated lysine in histone tails is associated with an open chromatin configuration that provides accessibility for specific transcription factors and the general transcription machinery. HDACs remove the acetyl groups, allowing formation of compacted chromatin that is associated with transcriptional gene silencing. Hence, the levels of histone acetylation play a crucial role in chromatin remodeling and in the regulation of gene transcription. Notably, histone acetylation also results in the recruitment of transcription and chromatin remodeling factors. Such factors are typically recruited through a bromodomain (BRD), functioning as acetyl-lysine binding domains.
Proteins containing one or more BRDs belong to BET (Bromodomain and extraterminal domain) family, an evolutionarily conserved family of proteins associated with chromatin and present in nearly all nuclear KATs. These proteins perform transcription regulatory functions under normal conditions, while in cancer they regulate transcription of several oncogenes, such as c-Myc and Bcl-2 [76]. Notably, different PTMs may reside on the same histone molecule. For example, PTMs neighboring acetylated lysines can strongly alter the affinity of BRD—and likewise, the enzymatic of KATs and HDACs may be modulated by close-by PTMs [77]. Recently in an elegant paper Feller and coworkers have applied a mass spectrometry-based strategy to generate a comprehensive catalog of combinatorial histone acetylation and other PTM motifs in Drosophila cells. In particular, they have described the histone acetylation changes in response to ablation of known or suspected KATs and HDACs [78]. Surprisingly, they have found that depletion of single KAT activities leads to complex alterations of the epigenome including, the reduction of primary substrates, the global redistribution of acetyl groups to secondary sites, and changes to methylation of histones.
Many studies have suggested alterations in histone acetylation as potential diagnostic or prognostic biomarkers in human diseases such as cancer [75]. Recently, there has been a significant growth in our knowledge about the involvement of aberrant patterns of histone modifications in cancer development. Analysis of global histone modification in a large cohort of solid cancer has instead revealed differential levels of bulk histone acetylation across tumors, and the occurrence of some of these histone marks correlates with tumor morphology and biological subtype. In general, tumors with adverse traditional prognostic or phenotypic characteristics were found to have reduced levels of detectable H3 and H4 acetylation. Loss of selected histone acetylation and methylation marks has been shown to predict patient outcome in human carcinoma [79]. For example, acetylation of H3 at K56 is often increased in multiple types of cancer and undifferentiated cells [80]. Barlesi and collaborators showed that acetylation of histone H2A at K5 and H3 at K9, as well as histone H3 di-methylation at K4, influenced overall and disease-free survival of NSCLC patients who had undergone resection, suggesting the role of epigenetic modifications in lung carcinogenesis [81]. In another study, cancer cells compared with normal lung displayed an aberrant pattern of histone H4 modifications with hyperacetylation at K5 and K8, hypoacetylation at K12 and K16, and loss of K20 trimethylation [82]. A recent study on immunostaining analyzed 408 NSCLC tissues, showing that global histone H3 and H4 modification patterns are potential markers of tumor recurrence and disease-free survival [83]. Breast cancer patients analysis reveals that the loss of acetylated H4 at K16 may serve as an early sign of cancer, and low levels of H3 acetylated at K9 and K14 and H4 at K12 are prognostic of poor outcomes [84]. Independent studies have demonstrated the prognostic significance of histone acetylation in esophageal SCC [85, 86]. In particular, abnormal levels of H3 and H4 acetylation correlate with the severity and histological differentiation of this tumor histotype.
In general, acetylation can lead to increase in DNA binding affinity of transcription factors such as c-myb, STAT3, E2F1 [87, 88], this in turn could lead to increase in transactivation and gene expression by these proteins. Stat3 is acetylated by p300 and the major Stat3 acetylation site is located at K685. The acetylation on Stat3 enhances its DNA binding and transactivation activities, and increases its nuclear localization [88]. In the case of c-myb, acetylation by CBP increases the transactivating capacity of c-myb by enhancing its association with CBP [87]. pCAF acetylates one of the cell-cycle regulator E2F family member, E2F1, at the DNA binding domain, which increases DNA binding ability of the protein and thus E2F mediated transcriptional activation. Other acetylases (GCN5, CBP and p300) can also acetylate E2F1, but much less efficiently [89]. Conversely, in the case of FoxO1, three acetylation sites (K242, K245, and K262) located in the protein domain that directly participates in DNA recognition and/or stabilization of the FoxO-DNA complex, were identified and their acetylation attenuates FoxO1 transcriptional activity. In fact, the positive charge of these lysines in FoxO1 contributes to its DNA-binding, and acetylation at these residues by CBP attenuates its ability to bind cognate DNA sequence [90].
Acetylation of transcription factors can alter their activity, but this is dependent on the functional domains that are acetylated. For example, the tumor suppressor p53 contains a lysine-rich basic domain near its C-terminus. Six different lysine residues, spanning sites 370-386 on human p53, can be modified by acetylation and other PTM that facilitates p53 activation. However, acetylation may serve multiple roles for p53, including stabilizing the protein, altering association with other proteins or other p53 monomers, enhancing its binding to DNA, and regulating transcription [91].
Cytosol: acetylation of cytoskeleton-associated proteins, nuclear-cytoplasmic shuttle, and other cellular factors
Lysine acetylation plays a crucial role in the regulation of cytoskeleton-associated proteins including actin and tubulin. There are three major isoforms of actin. β and γ actins form the stress fibers and are important for cell shape and cell movement. α-actin forms the core of the thin filament of the sarcomere where it interacts with a variety of proteins to produce the force for muscle contraction. Using proteomic approaches it has been shown that all three actin isoforms can be acetylated [74, 92] and acetylation of K61 in γ-actin may result in stabilization of actin stress fibers [92]. Several regulatory proteins of the actin cytoskeleton are also modified by acetylation. In particular, cortactin is regulated through acetylation by histone acetyltransferase p300, and deacetylation by HDAC6 and SIRT1. Acetylation abrogates binding of cortactin to filamentous actin and leads to diminished actin dynamics and altered cell motility [93, 94].
Like actin, the cytoskeletal protein α-tubulin is also acetylated, with the K40 residue on α-tubulin identified as the acetylation site. Acetylation of α-subunit tubulin occurs in a context of tubulin assembly, which is conserved among species. In mammals, the responsible enzymes have been identified and characterized as ATAT1 and HDAC6 [95]. Using a proteomic approach for acetylated proteins, several other acetylated residues in different tubulin isoforms (α as well as β subunits) were detected, but their role and significance have still to be elucidated [74]. There is growing evidence that acetylation of tubulin globally impacts on cellular functions and several reviews have recently been devoted to that subject [95, 96]. Briefly, studies have shown that tubulin acetylation contributes to microtubule stabilization and allows for more efficient bundling of acetylation-stabilized microtubules. It is also implicated in other cell functions, including intracellular endoplasmic reticulum (ER) localization and ER-mitochondria interactions [97]. Moreover, elevated tubulin acetylation promotes adhesion and invasion of breast cancer cells, and a relationship between high level of α-tubulin acetylation and metastatic behaviour of basal-like breast cancers, has also been recently reported [39]. High level of acetylated tubulin have been also found correlated with a higher tumor grade in SCC of the head and neck [98], and its expression has been suggested as a prognostic marker in epithelial malignancies and as a marker for sensitivity to chemotherapy [99].
Subcellular distribution of many proteins can be also influenced by acetylation. Interestingly, for some proteins, acetylation favors localization to the cytoplasm, whereas for others, acetylation enhances the retention of proteins in the nucleus. For example, c-Abl acetylation on K730 leads it from nuclear to cytoplasmic delocalization and promotes myogenic differentiation [100]. Similarly, an increased acetylation on β-catenin at K49 resulted in increased membrane localization in human induced pluripotent stem cells derived from neuronal cells [101]. Conversely, acetylated form of eIF5A was primarily enriched in the nucleus, whereas unacetylated eIF5A was primarily cytoplasmic; this distinction was even more pronounced in the presence of HDAC inhibitors [102]. Acetylation of one nuclear localization signal sequence of Net1A, a Rho guanine-nucleotide-exchange factor, regulates its subcellular localization to impact RhoA activity and actin cytoskeletal organization [103].
In some cases, acetylation also competes with other modifications [104]. For some transcription factors such as p53, FoxO, and c-myc, it has been observed that protein stability can be increased by acetylation, through blocking ubiquitination of the same residues, which will target the protein for proteasomal degradation. Acetylation and ubiquitination were found to modify the same residues at the C-terminal domain of p53 (K residues number, 370, 372, 373, 381, 382). Thus, acetylation blocks the export and degradation of p53 preventing ubiquitination [91]. Ubiquitination and acetylation can occur on the same lysine residues as well on FoxO. Using acetylation-defective and acetylation-mimicking mutants, Kitamura et al. have found that these mutual effects play an important role in the FoxO1-dependent oxidative damage response in pancreatic β cells [105]. Also acetylation of c-myc by either GCN5/PCAF or Tip60 results in a dramatic increase in protein stability [106]. Alternatively, acetylation can also decrease protein stability, for example DNA methyltransferase 1 (DNMT1), the primary enzyme that maintains DNA methylation, is destabilized by Tip60 acetylation, which targets DNMT1 for proteasomal degradation [107].
Acetylation can also alter the enzymatic activity of certain enzymes or their ability to interact with other proteins. Lu and coworkers have demonstrated that MOF not only acetylates histone H4 at K16, but it also acetylates itself to regulate its recruitment and activities on the chromatin [108]. ATM kinase activity is tightly regulated by Tip60-dependent acetylation at K3016, which is located in the highly conserved C-terminal FAT domain adjacent to the kinase domain. Mutation of K3016 does not affect unstimulated ATM kinase activity but inhibits the conversion of inactive ATM dimers to active ATM monomers, and prevents the ATM-dependent phosphorylation of the p53 and CHK2 proteins [109].
Functional IMPLICATIONS of organelle-specific acetylation
For a long time, lysine acetylation was thought to be limited to the cytosol and nucleus, where both the donor of the acetyl group and KAT were available. On the contrary, over the last few years it has been proved that a large number of organelle-resident and/or organelle-transiting proteins undergo lysine acetylation in the lumen of the organelle, as mitochondrial and ER.
In 2006, an extensive proteomic survey of cellular proteins have revealed that a large number of mitochondrial proteins are subject to reversible lysine acetylation [92]. In this proteomic analysis of the acetylated peptides obtained from purified mouse liver mitochondria, 277 lysine acetylation sites were identified in 133 mitochondrial proteins. Thus revealing that at least 20% of all mitochondrial proteins are lysine acetylated and suggesting that lysine acetylation is an abundant PTM in the mitochondrion. A more recent proteomic analysis of lysine-acetylated mitochondrial proteins has shown that as much as 50% of all mitochondrial proteins are acetylated, and most of them are involved in energy metabolism. These include proteins involved in the tricarboxylic acid (TCA) cycle, oxidative phosphorylation, β-oxidation of lipids, amino acid metabolism, carbohydrate metabolism, nucleotide metabolism, and the urea cycle [110, 111]. It is well known that the three deacetylases (SIRT3, SIRT4, and SIRT5) located in the mitochondria are poised to mediate mitochondrial protein acetylation levels. Among them SIRT3 is localized in the mitochondrial matrix and its expression is selectively activated during fasting and calorie restriction. Activated SIRT3 deacetylates several key metabolic enzymes (e.g. acetyl-coenzyme A synthetase, long-chain acyl-coenzyme A (acyl-CoA) dehydrogenase (LCAD), and 3-hydroxy-3-methylglutaryl CoA synthase 2) and enhances their enzymatic activity [112]. Interestingly, mitochondrial acetyltransferases (MATs) have not been identified, raising the question as to how mitochondrial proteins become acetylated. One explanation is that high acetyl-CoA levels in the mitochondria could facilitate a similar non-enzymatic acetylation mechanism. Alternatively, MATs could mediate the acetylation reaction or mitochondrial proteins could be acetylated by a novel mitochondrial enzyme, not yet recognizable as a KATs. A multitude of mitochondrial proteins are now known to be acetylated. Some of them are modified in a reversible and nutrient-responsive manner. Accordingly, alterations in mitochondrial acetylation states, and, hence, alterations in carbon substrate utilization, may contribute to the unusual preference for aerobic glycolysis and glutaminolysis often observed in numerous forms of cancer [113].
In 2007, Costantini et al. reported transient dual enzymatic machinery acting both in the ER and Golgi apparatus. In particular, they have reported that membrane protein β-site APP cleaving enzyme 1 (BACE1), an aspartic-acid protease that processes amyloid precursor protein involved in the pathogenesis of Alzheimer’s disease, is acetylated and deacetylated in seven different residues both in the ER and Golgi apparatus, respectively [114]. Subsequent proteomic studies have assessed the ER acetylome, and predicted wide-ranging biological implications of this pathway [115]. The list of ER-resident proteins includes chaperones and enzymes involved with PTM and folding. After that, many membrane and secreted proteins were reported to undergo transient lysine acetylation in the ER lumen [115-117]. This implies not only the presence of KATs in this cellular compartment but also an ER import system for acetyl-CoA. To date, ER acetylation has been found to be catalyzed by two different ER-based KATs, which were named AT-1 (also known as camello-like 2 and N-acetyltransferase 8B) and AT-2 (also known as camello-like 1 and N-acetyltransferase 8), both members of the camello family, which belongs to the GNAT superfamily [118]. Recently, AT-1 and consequently ER acetylation were genetically modified in vivo [119]. Importantly, the overexpression of AT-1 per se does not cause acetylation of proteins that are normally non-acetylated [117]. Targeting AT-1 function in mice leads to the appearance of neurodegenerative features, inflammation and cancer [119]. In conclusion, what was once a cytosolic and nuclear event now appears to be an essential component of mitochondria and ER functions as well.
Small molecule modulators of KATs as cancer therapeutics
The discovery of protein acetylation has led to the identification of many novel epigenetic drug targets. The first Food and Drug Administration-approved acetylation-modifying agent was the HDAC inhibitor vorinostat (also known as suberanilohydroxamic acid (SAHA)), initially identified as an agent inducing the differentiation of tumor cells in vitro. To date, other 3 HDACi (romidepsin, belinostat, and panobinostat) have been approved for cancer treatment and many other HDACi are currently used in clinical trials [120]. By contrast, the area of identifying KAT inhibitors (KATi) is not as well explored as HDACi (Table 2).
Table 2: Involvement of Lysine acetyltransferases (KATs) in cancer
KATs |
Cancer Type |
Alteration |
Oncogene or tumor suppressor |
Ref. |
p300/CBP |
Lung, colon, breast and ovarian cancers |
mutation |
tumor suppressor |
|
Hepato, colorectal, oral, breast, ovarian, gastric cancers and glioblastomas |
loss of heterozygosity or deletion |
tumor suppressor |
||
Prostate cancers |
overexpression |
oncogene |
||
Haematological malignancies |
chromosome translocations |
oncogene |
||
GCN5 |
Glioma, colon and lung cancers |
overexpression |
oncogene |
|
PCAF |
Hepato, ovarian, gastric and esophageal cancers |
deletion |
tumor suppressor |
[33] |
Tip60 |
Head and neck, breast cancers and lymphomas |
mutation |
tumor suppressor |
[51] |
Prostate cancer |
overexpression |
oncogene |
[53] |
|
MOF |
Breast, renal cell colorectal, gastric, ovarian, and hepato cancers, medulloblastoma |
deletion |
tumor suppressor |
[60] |
Lung cancer |
overexpression |
oncogene |
[65] |
|
MOZ/MORF |
Haematological malignancy |
chromosome translocations |
oncogene |
|
Acute leukemia |
chromosome translocations |
oncogene |
[70] |
|
MORF |
Lung cancer |
deletion |
tumor suppressor |
[72] |
The first KATi specific for p300 (Lys-CoA) and for PCAF (H3-CoA-20) were reported in 2000 [121]. The major drawback of these compounds is that they are cell impermeable, thus greatly restricting their utility. In 2005, Zheng and collaborators synthesized the first potent and selective KATi which is effective in live cells, by linking Lys-CoA and H3-CoA-20 to a cell permeabilizing ‘tat’ peptide [122].
In addition to bi-substrate analogues, several natural products have been reported as KATi. Curcumin, a polyphenol responsible for the yellow color of the spice turmeric, is the only KATi in clinical trials (https://clinicaltrials.gov/) and exhibits great promise as a therapeutic agent for its anti-inflammatory and antitumor properties. It possesses KAT inhibitory activity with specificity for the p300/CBP. In cells, curcumin promotes proteasome-dependent degradation of p300 and the closely related CBP protein without affecting PCAF or GCN5. Moreover, curcumin inhibits the KAT activity of purified p300 as assessed using histone H3 substrate [123]. Interestingly, it could also inhibit the acetylation of p53 [124]. The anticancer potential of curcumin stems from its ability to suppress cell proliferation of a wide variety of tumor cells. In vitro studies have demonstrated that curcumin is an efficient inducer of apoptosis and some degree of selectivity for cancer cells has been observed. Curcumin may also alter the effectiveness of radiotherapy and chemotherapy [125]. To date, about 57 clinical studies are ongoing with curcumin and its different formulations alone or in combination with conventional chemotherapeutic agents.
Anacardic acid, isolated from cashew nut shell liquid, has been identified as a potent inhibitor of p300 and PCAF, Tip60 KAT activity in vitro [126]. It has been shown that anacardic acid blocks Tip60-dependent activation of ATM and DNA-PK protein kinases by DNA damage in vivo [127]. Recently it was also found to inhibit MOF. Based on this evidence, Wapenaar and coworkers have designed a small collection of anacardic acid derived inhibitors and calculated their respective binding constants for MOF. This study will be the starting point for developing of selectively MOF inhibitors, which will ultimately enable the exploitation of this KAT as a novel drug target in disease [128].
Starting from the leading natural product KATi anacardic acid, a series of analogues was synthesized and investigated for KAT inhibitory properties and effects on cancer cell growth. The compounds have an improved inhibitory potency for PCAF/p300 KAT activity, and there is a clear correlation between their inhibitory potency and cytotoxicity toward a broad panel of cancer cells [129, 130]. MG153, a small molecule derived from anacardic acid, is a potent inhibitor of p300/PCAF, that decreases cell proliferation and induces apoptosis and resistance to DNA damage in BCR-ABL-expressing cells [131]. Interestingly, Pentadecylidenemalonate 1b, another analogue of anacardic acid, was identified as the first mixed activator/inhibitor of KATs. It potentiates PCAF KAT activity while inhibiting those of p300/CBP and recombinant CBP [132].
Garcinol is a polyisoprenylated benzophenone derivative isolated from Garcinia indica, and it is a potent inhibitor of PCAF and p300 enzymes. It was found to induce apoptosis and alter global gene expression in cancer cells. However, cells were found to be poorly permeable to this compound, thus limiting its practical application [124].
Epigallocatechin-3-gallate (EGCG) is the most abundant catechin in tea, and it has been found as a KATi in natural compound screening. EGCG can block p300-mediated acetylation of p65, impairing its translocation to the nucleus, thus inhibiting NF-κB activity and decreasing NF-κB target genes expression. EGCG impairs B cell transformation by Epstein-Barr Virus (EBV) and completely blocks EBV infection-induced cytokine expression Thus, EGCG could represent a potential therapy for B cell malignancies [133]. Plumbagin (RTK1), is a natural compound isolated from Plumbago rosea root extract, which inhibits p300 KAT activity potently in vitro and in vivo. Interestingly, RTK1 specifically inhibits the p300-mediated acetylation of p53. RTK1 also induces a distinct modification profile, involving transcriptional activation marks like histone H3 trimethylated at K4 and phosphorylated at S10 in the context of histone acetylation brought about by KAT3B/ p300 [134-136].
Recently, oridonin, a natural diterpenoid isolated from the Chinese medicinal herb Rabdosia rubescens was found to possess acetyltransferase-inhibitory effects on multiple KAT including p300, GCN5, Tip60, and PCAF. In gastric cancer cells, oridonin treatment inhibited cell proliferation and induced cell death by engaging the mitochondrial pathway of apoptosis, at least partially through p53-and caspase-3-mediated mechanisms [137].
In silico docking of commercial small molecule library has identified C646 as a competitive inhibitor of p300. It is a cell-permeable pyrazolone-containing small molecule with potent p300/CBP inhibitory activity with an IC50 value in the nanomolar range [138]. To date, C646 is the best characterized p300 KATi compound, which suppresses histone acetylation in human cancer cells in vitro. A recent study has been proposed C646 as potential candidate for treating AML1-ETO (AE) fusion protein -positive AML. C646 inhibits cellular proliferation, reduces colony formation, evokes partial cell cycle arrest in G1 phase, and induces apoptosis in AE-positive AML cell lines and primary blasts isolated from leukemic mice and AML patients. Notably, AE-positive AML cells are more sensitive to lower C646 doses than AE-negative ones. Finally C646 induces growth inhibition on AE-positive AML cells, and this event is associated with reduced global histone H3 acetylation and declined c-kit and bcl-2 levels [139]. C646 antitumor activity has been also show in different model of solid tumors. In prostate cancer cell lines, C646-mediated inhibition of p300 leads to induction of caspase-dependent apoptosis in several androgen-dependent and castration-resistant prostate cancer cells [140]. In NSCLC, Oike et al. have reported that C646 sensitizes cancer cells to radiotherapy, and enhances mitotic catastrophe induced by radiation [141]. C646 also inhibits the growth and promotes chemo-responsiveness of human melanoma cells. Mechanistically, p300 transcriptome has identified functional roles of p300 in promoting cell cycle progression, chromatin assembly, and activation of DNA repair pathways through direct transcriptional regulatory mechanisms [142]. Recently, a functional screening of synthetic-lethal genes in CBP-deficient cancers has proposed p300 inhibition as a promising therapeutic target for treatment of CBP-deficient cancers. In fact, ablation of p300 in CBP-knockout and -deficient cancer cells induces G1/S cell-cycle arrest, followed by apoptosis, and C646 specifically suppresses the growth of CBP-deficient lung and hematopoietic cancer cells in vitro and in vivo [143]. However, a recent chemical proteomic study has revealed that thiol reactivity of C646 may limit its ability to antagonize acetylation in cells [144]. In another recent study, a mass spectrometry analysis has shown a slight increase in acetylation of histone H3 in cells treated with high dose of C646. Indeed, they have also found an inhibition of HDACs at high concentrations, thus discovering a lack of selectivity of C646 at higher concentrations that needs to be taken into account for further development of C646 applications [145].
Recently, a novel cell-permeable p300/CBP-selective inhibitor, HATi II, has been described. HATi II inhibited the proliferation of a panel of glioma cells in a dose-dependent manner, by inducing cell cycle arrest at the G2/M phase, and apoptosis. In addition, microarray analysis and quantitative real-time PCR indicated that HATi II activates the p53 signaling pathway in glioma cells [146].
Small-molecule inhibitors of p300, such as L002 and isothiazolones, have been also identified by high-throughput screening as potent anticancer agents. L002 and its analogs have been identified from a library of 622,079 compounds. They inhibit the activity of p300 and related acetyltransferases (PCAF, and GCN5), but do not affect the activity of other KATs or HDACs or histone methyltransferases. Notably, in vitro leukemia, lymphoma and breast cancer cell lines are extremely sensitive to L002, whereas it potently suppresses tumor growth and histone acetylation of breast cancer xenografts [147].
Isothiazolones are antagonists of both p300 and PCAF [148]. Recently, two selected pyridoisothiazolone compound PU139, a pan-KATi , and PU141, a KAT3 selective KATi, were analyzed for their KAT inhibitory profile and for their anti-proliferative properties on a panel of human cancer cell lines. Interestingly, PU139 triggers caspase-independent cell death in cell culture, whereas both inhibitors block growth of neuroblastoma xenografts. Moreover, PU139 was shown to synergize with doxorubicin in vivo, and to reduce histone lysine acetylation in vivo at concentrations that block neoplastic xenograft growth [149]. Among isothiozolone-based inhibitors, 5-chloroisothiazolone has been identified as a PCAF specific inhibitor [150]. The thiazole-based synthetic compound, BF1 (1-(4-(4- chlorophenyl) thiazol-2-yl)-2-(propan-2-ylidene) hydrazine) can inhibit p300 and GCN5 in vitro, and when tested in both neuroblastoma and glioblastoma cell lines, it shows a global reduction in histone H3 and specific acetylation at K18 [151].
A series of cycloalkylidene-(4-phenylthiazol-yl)-hydrazone (CPTH) derivatives have been recently synthesized. The lead compound CPTH2 has been discovered through a phenotypic screening in yeast, then validated in vitro as a KATi. In vivo, it also decreases acetylation of bulk histone H3 at the specific H3 acetylated at K14 [152]. Among the first series of CPTH derivatives, our group has found that the compound named CPTH6 shows a time-dependent inhibition of KAT activity and acetylation of H3 and H4 histones in both AML and several solid tumors cell lines. We have also demonstrated that CPTH6 significantly reduces cell proliferation and induces cell cycle perturbation, apoptosis and monocytic-differentiation in U937 AML cells [153]. Interestingly, in a panel of cancer cell lines CPTH6 and analogs are able to modulate the autophagic flux inhibiting autophagosome maturation. Notably, CPTH6 treatment decreases α-tubulin acetylation and fails to increase autophagic markers in cells in which acetyltransferase ATAT1 expression was silenced, indicating a possible role of α-tubulin acetylation in CPTH6-induced alteration in autophagy [154]. More recently, the efficacy of CPTH6 has been tested on a panel of Lung Cancer Stem-like Cells (LCSC) derived from NSCLC patients. Notably, LCSCs exhibit greater growth inhibition than established NSCLC cells. Growth inhibitory effect of CPTH6 in LCSC lines is mainly due to apoptosis induction. Moreover, differentiated progeny of LCSC lines is more resistant to CPTH6 in terms of loss of cell viability and reduction of protein acetylation, when compared to their undifferentiated counterparts. Interestingly, CPTH6 is able to inhibit the growth of LCSC-derived xenografts and to reduce cancer stem cell content in treated tumors [155].
With the aim of identifying KATi more potent than the lead compounds CPTH2 and CPTH6, Carradori and collaborators have synthesized several new (thiazol-2-yl)hydrazones including some related thiazolidines and pyrimidin-4(3H)-ones, and tested the whole library against human p300 and PCAF KAT enzymes. Five compounds are more efficient than CPTH2 and CPTH6 in inhibiting the p300 enzyme, and when tested in human leukemia cells, they are more potent than CPTH6 in inducing apoptosis and cytodifferentiation [156].
Beyond anacardic acid and garcinol, only a small number of Tip60 inhibitors have been reported. Other Tip60 inhibitors include 6-alkylsalicylates [157], NU9056 an isothiazole-based KATi, which is effective against prostate cancer cells [158], Pentamidine a drug used for treating parasitic protozoan [159], and some small inhibitors reported by Wu and co-workers [160]. Recently a high-throughput screening has led to the identification of TH1834 as Tip60-selective inhibitor by using computational tools to design drugs based on the binding pocket of Tip60 TH1834 inhibits Tip60 in vitro and can sensitize breast cancer cells to ionizing radiation [161].
Table 3: Lysine acetyltransferase inhibitors (KATi) in cancer therapy
Category |
Drug |
Targeted KATs |
Mechanism and main cellular effect |
Ref. |
|
Natural Compound |
|||||
|
Curcumin |
CBP, p300, PCAF |
Promotes CBP/p300 degradation and inhibits KAT activity. Inhibits cell proliferation of a wide variety of tumor cells, and sensitizes to radiotherapy and chemotherapy. Affects various signaling pathways in cancer cells independently by its KAT inhibitory activity |
||
Anacardic acid (AA) |
Tip60 |
Blocks the Tip60-dependent activation of ATM by DNA damage. Sensitizes human tumor cells to the cytotoxic effects of ionizing radiation |
[127] |
||
Plumbagin (RTK1) |
p300 |
Inhibits Ac-CoA and histone binding. Induces cell death and affects various signaling pathways in cancer cells |
|||
Garcinol |
PCAF, p300 |
Inhibits Ac-CoA and histone binding. Induces apoptosis and alters global gene expression in cancer cells |
[124] |
||
Epigallocatechin- 3-gallate (EPGG) |
p300 |
Blocks p300-mediated acetylation of p65, impairing its translocation to the nucleus, and inhibiting NF-κB activity and target genes expression |
[133] |
||
Oridonin |
p300, GCN5, Tip60, PCAF |
Inhibits cell proliferation and induces apoptosis |
[137] |
||
Natural Compound analogs |
|||||
AA derivatives |
1° series anacardic acid derivatives |
p300, PCAF |
Are cytotoxic toward a broad panel of cancer cells |
||
6-Alkylsalicylates |
Tip60 |
Are competitive with Ac-CoA and non-competitive with the histone substrate |
[157] |
||
Pentadecylidenemalonate 1b |
PCAF, p300/CBP |
Activates PCAF and inhibits p300/CBP, and induces apoptosis in cancer cells |
[132] |
||
MG153 |
PCAF |
induces apoptosis and resistance to DNA damage in BCR-ABL-expressing cells |
[131] |
||
Bisubstrate inhibitor |
|||||
Lys-CoA |
p300 |
Inhibits Ac-CoA and substrate binding |
[122] |
||
H3-CoA-20 |
PCAF |
Inhibits Ac-CoA and substrate binding |
[122] |
||
Synthetic compound |
|||||
L002 and its analogs |
p300,PCAF, GCN5 |
Suppress tumor growth and histone acetylation of breast cancer xenografts |
[147] |
||
HATi II |
p300, CBP |
Induces cell cycle arrest and apoptosis in cancer cells |
[146] |
||
C646 |
p300 |
Inhibits Ac-CoA and substrate binding. Suppress histone acetylation in human cells in vitro Inhibits cell proliferation and induces cell cycle arrest and apoptosis in cancer cells |
|||
Bisbenzamidine derivative |
Pentamidine |
Tip60 |
Inhibits acetylation of histone H2A, inhibits radiation-induced focus formation and homologous recombination repair pathway |
[159] |
|
Thiazole derivatives |
CPTH2, CPTH6 |
GCN5/PCAF |
Compete with substrates. Induce differentiation and apoptosis in cancer cells. Modulate autophagic flux in human cancer cells Selectively kill lung cancer stem cells |
||
CPTH derivatives |
p300, PCAF |
Induce apoptosis and cytodifferentiation in leukemia cells |
[156] |
||
BF1 |
GCN5, p300 |
Competes with substrates. Induces histone hypoacetylation and apoptosis in human cancer cells |
[151] |
||
Isothiazolones derivatives
|
NU9056 |
Tip60 |
Is effective against prostate cancer cells |
[158] |
|
TH1834 |
Tip60 |
Inhibits AcCoA binding. Sensitizes breast cancer cells to ionizing radiation |
[161]. |
||
PU141 |
KAT3 |
Exerts anti-proliferative properties on a panel of human cancer cell lines |
[149] |
||
PU139 |
p300,CBP, PCAF, GCN5 |
Synergizes with doxorubicin and to reduces histone lysine acetylation in vivo at concentrations that block neoplastic xenograft growth |
[149] |
||
5-Cloroisothiazolone |
PCAF |
not determined |
[150] |
||
Limitations of KAT INHIBITORS and potential ALTERNATIVE strategies
Almost all studies on KATi are currently still in a preclinical phase. Better agents, with greater specificity and better pharmokinetic properties are sorely needed. Nowadays, a compelling alternative to the direct targeting of KATs is coming forward based on the inhibition of BET family proteins, enzymes recognizing lysine acetylated residues [162, 163]. These inhibitors act as acetyl-lysine mimics that occupy the acetylated lysine-binding site in bromodomain-containing proteins. Pharmacological inhibition of BET proteins shows therapeutic activity in a variety of different pathologies, particularly in models of hematologic malignancies, cancer and inflammation [164-166]. Such effects have been attributed to a specific set of downstream target genes, whose expression is modulated by pharmacological targeting of BET proteins. Currently, there have been five registered active clinical trials investigating the targeting of BET family proteins, such as RVX-208, I-BET 762, OTX 015, CPI-0610 and TEN-010, which reported encouraging results in treating hematologic malignancies (https://clinicaltrials.gov/).
Concluding remarks and future perspectives
Lysine acetylation occurs not only at the histone tails but also in non-histone proteins. Importantly, aberrant lysine acetylation in cancer cells affects virtually all cellular pathways that have been associated to tumorigenesis. Furthermore, alterations in the function of histone-modifying complexes deregulate the control of chromatin-based processes, ultimately leading to oncogenic transformation and the development of cancer. Consistent with this notion, aberrant patterns of histone modifications and KATs mutations have been associated with a large number of human malignancies, and could provide cancer biomarkers for predicting prognosis and for determining better treatment options for cancer patients. Hence, the diverse role of KATs, the increasing numbers of KAT subtypes and the increasing evidence that the deregulation of epigenetic processes plays a key role in several diseases hold promise for future therapeutic strategies focused on KAT inhibition.
Extensive studies have explored small-molecule inhibitors of KAT family proteins for cancer therapy. However, they have not entered clinical trials. The non-specificity, pleiotropic effects, toxicity profiles and doses in the micromolar range are the major drawbacks in the KATi advancement. Curcumin, administered alone or with conventional drugs, is the only KATi tested in clinical trials against breast, prostate, colorectal and other solid cancers. However, it is important to note that curcumin shows enzymatic inhibitory activity on other proteins differently from KATs, which may explain the observed anticancer activity. Hence, the potential of using KATi for cancer therapy is still in its infancy.
A recent study carried out in Drosophila cells by Feller and collaborators [78], has elegantly demonstrated the role of different KATs in the acetylation of specific histone residues and highlighted as an increased knowledge of chromatin modification network is required for a more targeted utilization of epigenetic inhibitors in a clinical setting. Therefore, additional studies characterizing the mechanistic consequences of KATs overexpression or mutation in various cancers, as well as studies in animal models of disease, would be beneficial in understanding the role of several KATs in tumorigenesis. The identification of selective KATi would allow better design of combination therapies using these inhibitors in combination with other epigenetic agents, such as bromodomain inhibitors or conventional cancer therapy.
Acknowledgments
We are grateful to Dr. Adele Petricca for secretarial assistance and Dr. Simone Bonacelli for English revision.
Conflicts of Interest
No potential conflicts of interest were disclosed.
FUNDING
Dr. Daniela Trisciuoglio is recipient of a fellowship from the Umberto Veronesi Foundation. This work was supported by grants from the Italian Association for Cancer Research (DDB, IG 14100).
REFERENCES
1. Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annual Review of Biochemistry. 2001; 70: 81-120.
2. Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nature reviews.Molecular cell biology. 2007; 8: 284-95.
3. Kalkhoven E. CBP and p300: HATs for different occasions. Biochemical pharmacology. 2004; 68: 1145-55.
4. Vervoorts J, Lüscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Lüscher B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO reports. 2003; 4: 484-90.
5. Zhong J, Ding L, Bohrer LR, Pan Y, Liu P, Zhang J, Sebo TJ, Karnes RJ, Tindall DJ, van Deursen J, Huang H. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer research. 2014; 74: 1870-80.
6. Pao GM, Janknecht R, Ruffner H, Hunter T, Verma IM. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proceedings of the National Academy of Sciences of the United States of America (PNAS). 2000; 97: 1020-5.
7. Teufel DP, Freund SM, Bycroft M, Fersht AR. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proceedings of the National Academy of Sciences of the United States of America (PNAS). 2007; 104: 7009-14.
8. Kishimoto M, Kohno T, Okudela K, Otsuka A, Sasaki H, Tanabe C, Sakiyama T, Hirama C, Kitabayashi I, Minna JD, Takenoshita S, Yokota J. Mutations and deletions of the CBP gene in human lung cancer. Clinical Cancer Research. 2005; 11: 512-9.
9. Gayther SA, Batley SJ, Linger L, Bannister A, Thorpe K, Chin SF, Daigo Y, Russell P, Wilson A, Sowter HM, Delhanty JD, Ponder BA, Kouzarides T, et al. Mutations truncating the EP300 acetylase in human cancers. Nature genetics. 2000; 24: 300-3.
10. Bryan EJ, Jokubaitis VJ, Chamberlain NL, Baxter SW, Dawson E, Choong DY, Campbell IG. Mutation analysis of EP300 in colon, breast and ovarian carcinomas. International Journal of Cancer. 2002; 102: 137-41.
11. Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004; 23: 4225-31.
12. Yokomizo C, Yamaguchi K, Itoh Y, Nishimura T, Umemura A, Minami M, Yasui K, Mitsuyoshi H, Fujii H, Tochiki N, Nakajima T, Okanoue T, Yoshikawa T. High expression of p300 in HCC predicts shortened overall survival in association with enhanced epithelial mesenchymal transition of HCC cells. Cancer letters. 2011; 310: 140-7.
13. Li M, Luo RZ, Chen JW, Cao Y, Lu JB, He JH, Wu QL, Cai MY. High expression of transcriptional coactivator p300 correlates with aggressive features and poor prognosis of hepatocellular carcinoma. Journal of translational medicine. 2011; 9.
14. Xiao XS, Cai MY, Chen JW, Guan XY, Kung HF, Zeng YX, Xie D. High Expression of p300 in Human Breast Cancer Correlates with Tumor Recurrence and Predicts Adverse Prognosis. Chinese Journal of Cancer Research. 2011; 23: 201-7.
15. Chen MK, Cai MY, Luo RZ, Tian X, Liao QM, Zhang XY, Han JD. Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. British Journal of Dermatology. 2015; 172: 111-9.
16. Liao ZW, Zhou TC, Tan XJ, Song XL, Liu Y, Shi XY, Huang WJ, Du LL, Tu BJ, Lin XD. High expression of p300 is linked to aggressive features and poor prognosis of nasopharyngeal carcinoma. Journal of translational medicine. 2012; 10.
17. Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, Tindall DJ. p300 in prostate cancer proliferation and progression. Cancer research. 2003; 63: 7638-40.
18. Gang X, Yang Y, Zhong J, Jiang K, Pan Y, Karnes RJ, Zhang J, Xu W, Wang G, Huang H. P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget. 2016; 7:15135-49. doi: 10.18632/oncotarget.7715.
19. Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dubé I, Frischauf AM, Horsman D, Mitelman F, Volinia S, et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nature genetics. 1996; 14: 33-41.
20. Chaffanet M, Gressin L, Preudhomme C, Soenen-Cornu V, Birnbaum D, Pébusque MJ. MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes, Chromosomes and Cancer. 2000; 28: 138-44.
21. Sun XJ, Man N, Tan Y, Nimer SD, Wang L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Frontiers in Oncology. 2015; 5.
22. Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: a structural overview. Annual Review of Biophysics and Biomolecular Structure. 2000; 29: 81-103.
23. Paolinelli R, Mendoza-Maldonado R, Cereseto A, Giacca M. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nature Structural and Molecular Biology. 2009; 16: 412-20.
24. Sabò A, Lusic M, Cereseto A, Giacca M. Acetylation of conserved lysines in the catalytic core of cyclin-dependent kinase 9 inhibits kinase activity and regulates transcription. Molecular and cellular biology. 2008; 28: 2201-12.
25. Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, Deng A, Chen J, Wang G, Zhu S, Kang J. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. The Journal of Biological Chemistry. 2013; 288: 14510-21.
26. Love IM, Sekaric P, Shi D, Grossman SR, Androphy EJ. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle. 2012; 11: 2458-66.
27. Linares LK, Kiernan R, Triboulet R, Chable-Bessia C, Latreille D, Cuvier O, Lacroix M, Le Cam L, Coux O, Benkirane M. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nature cell biology. 2007; 9: 331-8.
28. Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. The Journal of Biological Chemistry. 2006; 281: 26562-8.
29. Watts GS, Oshiro MM, Junk DJ, Wozniak RJ, Watterson S, Domann FE, Futscher BW. The acetyltransferase p300/CBP-associated factor is a p53 target gene in breast tumor cells. Neoplasia. 2004; 6: 187-94.
30. Liu K, Zhang Q, Lan H, Wang L, Mou P, Shao W, Liu D, Yang W, Lin Z, Lin Q, Ji T. GCN5 Potentiates Glioma Proliferation and Invasion via STAT3 and AKT Signaling Pathways. International journal of molecular sciences. 2015; 16: 21897-910.
31. Yin YW, Jin HJ, Zhao W, Gao B, Fang J, Wei J, Zhang DD, Zhang J, Fang D. The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene expression. 2015; 16: 187-96.
32. Li Q, Liu Z, Xu M, Xue Y, Yao B, Dou C, Jia Y, Wang Y, Tu K, Zheng X, Yao Y. PCAF inhibits hepatocellular carcinoma metastasis by inhibition of epithelial-mesenchymal transition by targeting Gli-1. Cancer letters. 2016; 375: 190-8.
33. Ying MZ, Wang JJ, Li DW, Yu G, Wang X, Pan J, Chen Y, He MX. The p300/CBP associated factor is frequently downregulated in intestinal-type gastric carcinoma and constitutes a biomarker for clinical outcome. Cancer biology & therapy. 2010; 9: 312-20.
34. Zhu C, Qin YR, Xie D, Chua DT, Fung JM, Chen L, Fu L, Hu L, Guan XY. Characterization of tumor suppressive function of P300/CBP-associated factor at frequently deleted region 3p24 in esophageal squamous cell carcinoma. Oncogene. 2009; 28: 2821-8.
35. Kormendi V, Szyk A, Piszczek G, Roll-Mecak A. Crystal structures of tubulin acetyltransferase reveal a conserved catalytic core and the plasticity of the essential N terminus. The Journal of Biological Chemistry. 2012; 287: 41569-75.
36. Taschner M, Vetter M, Lorentzen E. Atomic resolution structure of human -tubulin acetyltransferase bound to acetyl-CoA. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109: 19649-54.
37. Friedmann DR, Aguilar A, Fan J, Nachury MV, Marmorstein R. Structure of the -tubulin acetyltransferase, TAT1, and implications for tubulin-specific acetylation. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109: 19655-60.
38. Castro-Castro A, Janke C, Montagnac G, Paul-Gilloteaux P, Chavrier P. ATAT1/MEC-17 acetyltransferase and HDAC6 deacetylase control a balance of acetylation of alpha-tubulin and cortactin and regulate MT1-MMP trafficking and breast tumor cell invasion. European journal of cell biology. 2012; 91: 950-60.
39. Boggs AE, Vitolo MI, Whipple RA, Charpentier MS, Goloubeva OG, Ioffe OB, Tuttle KC, Slovic J, Lu Y, Mills GB, Martin SS. -Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer research. 2015; 75: 203-15.
40. Aguilar A, Becker L, Tedeschi T, Heller S, Iomini C, Nachury MV. -tubulin K40 acetylation is required for contact inhibition of proliferation and cell-substrate adhesion. Molecular biology of the cell. 2014; 25: 1854-66.
41. Bailey JM, Alsina J, Rasheed ZA, McAllister FM, Fu YY, Plentz R, Zhang H, Pasricha PJ, Bardeesy N, Matsui W, Maitra A, Leach SD. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology. 2014; 146: 245-56.
42. Chien JY, Tsen SD, Chien CC, Liu HW, Tung CY, Lin CH. TAT1 downregulation induces mitotic catastrophe in HeLa and A549 cells. Cell Death Discov. 16006 (2016); 2.
43. Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J. MEC-17 is an alpha-tubulin acetyltransferase. Nature. 2010; 467: 218-22.
44. Kim GW, Li L, Gorbani M, You L, Yang XJ. Mice lacking -tubulin acetyltransferase 1 are viable but display -tubulin acetylation deficiency and dentate gyrus distortion. The Journal of Biological Chemistry. 2013; 288: 20334-50.
45. Sapountzi V, Côté J. MYST-family histone acetyltransferases: beyond chromatin. Cellular and Molecular Life Sciences. 2011; 68: 1147-56.
46. Avvakumov N, Côté J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene. 2007; 26: 5395-407.
47. Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 13182-7.
48. Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nature cell biology. 2006; 8: 91-9.
49. Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular cell. 2006; 24: 841-51.
50. Van Den Broeck A, Nissou D, Brambilla E, Eymin B, Gazzeri S. Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis. 2012; 33: 320-5.
51. Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, Confalonieri S, Cesaroni M, Marchesi F, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007; 448: 1063-7.
52. Bassi C, Li YT, Khu K, Mateo F, Baniasadi PS, Elia A, Mason J, Stambolic V, Pujana MA, Mak TW, Gorrini C. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell death and differentiation. 2016.
53. Halkidou K, Gnanapragasam VJ, Mehta PB, Logan IR, Brady ME, Cook S, Leung HY, Neal DE, Robson CN. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene. 2003; 22: 2466-77.
54. Rea S, Xouri G, Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007; 26: 5385-94.
55. Su J, Wang F, Cai Y, Jin J. The Functional Analysis of Histone Acetyltransferase MOF in Tumorigenesis. International journal of molecular sciences. 2016; 17: 10.3390/ijms17010099.
56. Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Molecular and cellular biology. 2005; 25: 6798-6810.
57. Gupta A, Guerin-Peyrou TG, Sharma GG, Park C, Agarwal M, Ganju RK, Pandita S, Choi K, Sukumar S, Pandita RK, Ludwig T, Pandita TK. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Molecular and cellular biology. 2008; 28: 397-409.
58. Li X, Li L, Pandey R, Byun JS, Gardner K, Qin Z, Dou Y. The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network. Cell stem cell. 2012; 11: 163-178.
59. Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, Cote J, Pandita TK. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Molecular and cellular biology. 2010; 30: 3582-3595.
60. Pfister S, Rea S, Taipale M, Mendrzyk F, Straub B, Ittrich C, Thuerigen O, Sinn HP, Akhtar A, Lichter P. The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. International journal of cancer. 2008; 122: 1207-1213.
61. Cao L, Zhu L, Yang J, Su J, Ni J, Du Y, Liu D, Wang Y, Wang F, Jin J, Cai Y. Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. International journal of oncology. 2014; 44: 1207-1214.
62. Liu N, Zhang R, Zhao X, Su J, Bian X, Ni J, Yue Y, Cai Y, Jin J. A potential diagnostic marker for ovarian cancer: Involvement of the histone acetyltransferase, human males absent on the first. Oncology letters. 2013; 6: 393-400.
63. Wang Y, Zhang R, Wu D, Lu Z, Sun W, Cai Y, Wang C, Jin J. Epigenetic change in kidney tumor: downregulation of histone acetyltransferase MYST1 in human renal cell carcinoma. Journal of experimental & clinical cancer research. 2013; 32: 8-9966-32-8.
64. Cai M, Hu Z, Liu J, Gao J, Tan M, Zhang D, Zhu L, Liu S, Hou R, Lin B. Expression of hMOF in different ovarian tissues and its effects on ovarian cancer prognosis. Oncology reports. 2015; 33: 685-692.
65. Chen Z, Ye X, Tang N, Shen S, Li Z, Niu X, Lu S, Xu L. The histone acetylranseferase hMOF acetylates Nrf2 and regulates anti-drug responses in human non-small cell lung cancer. British journal of pharmacology. 2014; 171: 3196-3211.
66. Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene. 2007; 26: 5408-5419.
67. Perez-Campo FM, Costa G, Lie-a-Ling M, Kouskoff V, Lacaud G. The MYSTerious MOZ, a histone acetyltransferase with a key role in haematopoiesis. Immunology. 2013; 139: 161-165.
68. Perez-Campo FM, Borrow J, Kouskoff V, Lacaud G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood. 2009; 113: 4866-4874.
69. Crump JG, Swartz ME, Eberhart JK, Kimmel CB. Moz-dependent Hox expression controls segment-specific fate maps of skeletal precursors in the face. Development. 2006; 133: 2661-2669.
70. Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML, Gilliland DG. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer cell. 2003; 3: 259-271.
71. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, Huang J, Hua X, Arrowsmith CH, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015; 525: 206-211.
72. Simo-Riudalbas L, Perez-Salvia M, Setien F, Villanueva A, Moutinho C, Martinez-Cardus A, Moran S, Berdasco M, Gomez A, Vidal E, Soler M, Heyn H, Vaquero A, et al. KAT6B Is a Tumor Suppressor Histone H3 Lysine 23 Acetyltransferase Undergoing Genomic Loss in Small Cell Lung Cancer. Cancer research. 2015; 75: 3936-3945.
73. Iizuka M, Stillman B. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. The Journal of biological chemistry. 1999; 274: 23027-23034.
74. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325: 834-840.
75. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nature reviews.Genetics. 2007; 8: 286-298.
76. Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochimica et biophysica acta. 2014; 1839: 676-685.
77. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149: 214-231.
78. Feller C, Forne I, Imhof A, Becker PB. Global and specific responses of the histone acetylome to systematic perturbation. Molecular cell. 2015; 57: 559-571.
79. Esteller M. Epigenetics in cancer. The New England journal of medicine. 2008; 358: 1148-1159.
80. Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009; 459: 113-117.
81. Barlesi F, Giaccone G, Gallegos-Ruiz MI, Loundou A, Span SW, Lefesvre P, Kruyt FA, Rodriguez JA. Global histone modifications predict prognosis of resected non small-cell lung cancer. Journal of clinical oncology. 2007; 25: 4358-4364.
82. Van Den Broeck A, Brambilla E, Moro-Sibilot D, Lantuejoul S, Brambilla C, Eymin B, Khochbin S, Gazzeri S. Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clinical cancer research. 2008; 14: 7237-7245.
83. Song JS, Kim YS, Kim DK, Park SI, Jang SJ. Global histone modification pattern associated with recurrence and disease-free survival in non-small cell lung cancer patients. Pathology international. 2012; 62: 182-190.
84. Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, Grainge MJ, Ball GR, Abdelghany MK, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer research. 2009; 69: 3802-3809.
85. Tzao C, Tung HJ, Jin JS, Sun GH, Hsu HS, Chen BH, Yu CP, Lee SC. Prognostic significance of global histone modifications in resected squamous cell carcinoma of the esophagus. Modern pathology. 2009; 22: 252-260.
86. I H, Ko E, Kim Y, Cho EY, Han J, Park J, Kim K, Kim DH, Shim YM. Association of global levels of histone modifications with recurrence-free survival in stage IIB and III esophageal squamous cell carcinomas. Cancer epidemiology, biomarkers & prevention. 2010; 19: 566-573.
87. Sano Y, Ishii S. Increased affinity of c-Myb for CREB-binding protein (CBP) after CBP-induced acetylation. The Journal of biological chemistry. 2001; 276: 3674-3682.
88. Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. The Journal of biological chemistry. 2005; 280: 11528-11534.
89. Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. The EMBO journal. 2000; 19: 662-671.
90. Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 11278-11283.
91. Reed SM, Quelle DE. p53 Acetylation: Regulation and Consequences. Cancers. 2014; 7: 30-69.
92. Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, Grishin NV, White M, Yang XJ, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Molecular cell. 2006; 23: 607-618.
93. Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Molecular cell. 2007; 27: 197-213.
94. Zhang Y, Zhang M, Dong H, Yong S, Li X, Olashaw N, Kruk PA, Cheng JQ, Bai W, Chen J, Nicosia SV, Zhang X. Deacetylation of cortactin by SIRT1 promotes cell migration. Oncogene. 2009; 28: 445-460.
95. Li L, Yang XJ. Tubulin acetylation: responsible enzymes, biological functions and human diseases. Cellular and molecular life sciences. 2015; 72: 4237-4255.
96. Yu I, Garnham CP, Roll-Mecak A. Writing and Reading the Tubulin Code. The Journal of biological chemistry. 2015; 290: 17163-17172.
97. Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. The Journal of cell biology. 2010; 190: 363-375.
98. Saba NF, Magliocca KR, Kim S, Muller S, Chen Z, Owonikoko TK, Sarlis NJ, Eggers C, Phelan V, Grist WJ, Chen AY, Ramalingam SS, Chen ZG, et al. Acetylated tubulin (AT) as a prognostic marker in squamous cell carcinoma of the head and neck. Head and neck pathology. 2014; 8: 66-72.
99. Marcus AI, Zhou J, O’Brate A, Hamel E, Wong J, Nivens M, El-Naggar A, Yao TP, Khuri FR, Giannakakou P. The synergistic combination of the farnesyl transferase inhibitor lonafarnib and paclitaxel enhances tubulin acetylation and requires a functional tubulin deacetylase. Cancer research. 2005; 65: 3883-3893.
100. di Bari MG, Ciuffini L, Mingardi M, Testi R, Soddu S, Barila D. c-Abl acetylation by histone acetyltransferases regulates its nuclear-cytoplasmic localization. EMBO reports. 2006; 7: 727-733.
101. Iaconelli J, Huang JH, Berkovitch SS, Chattopadhyay S, Mazitschek R, Schreiber SL, Haggarty SJ, Karmacharya R. HDAC6 inhibitors modulate Lys49 acetylation and membrane localization of beta-catenin in human iPSC-derived neuronal cells. ACS chemical biology. 2015; 10: 883-890.
102. Ishfaq M, Maeta K, Maeda S, Natsume T, Ito A, Yoshida M. Acetylation regulates subcellular localization of eukaryotic translation initiation factor 5A (eIF5A). FEBS letters. 2012; 586: 3236-3241.
103. Song EH, Oh W, Ulu A, Carr HS, Zuo Y, Frost JA. Acetylation of the RhoA GEF Net1A controls its subcellular localization and activity. Journal of cell science. 2015; 128: 913-922.
104. Mellert HS, McMahon SB. Biochemical pathways that regulate acetyltransferase and deacetylase activity in mammalian cells. Trends in biochemical sciences. 2009; 34: 571-578.
105. Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell metabolism. 2005; 2: 153-163.
106. Patel JH, Du Y, Ard PG, Phillips C, Carella B, Chen CJ, Rakowski C, Chatterjee C, Lieberman PM, Lane WS, Blobel GA, McMahon SB. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Molecular and cellular biology. 2004; 24: 10826-10834.
107. Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, Sedwick D, Ewing RM, Wang Z. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Science signaling. 2010; 3: ra80.
108. Lu L, Li L, Lv X, Wu XS, Liu DP, Liang CC. Modulations of hMOF autoacetylation by SIRT1 regulate hMOF recruitment and activities on the chromatin. Cell research. 2011; 21: 1182-1195.
109. Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Molecular and cellular biology. 2007; 27: 8502-8509.
110. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010; 327: 1000-1004.
111. Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010; 327: 1004-1007.
112. Hirschey MD, Shimazu T, Huang JY, Schwer B, Verdin E. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harbor symposia on quantitative biology. 2011; 76: 267-277.
113. Wagner GR, Payne RM. Mitochondrial acetylation and diseases of aging. Journal of aging research. 2011; 2011: 234875.
114. Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. The Biochemical journal. 2007; 407: 383-395.
115. Pehar M, Lehnus M, Karst A, Puglielli L. Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by N(epsilon)-lysine acetylation in the lumen of the organelle and predicts broad biological impact. The Journal of biological chemistry. 2012; 287: 22436-22440.
116. Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO reports. 2008; 9: 916-922.
117. Jonas MC, Pehar M, Puglielli L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. Journal of cell science. 2010; 123: 3378-3388.
118. Ko MH, Puglielli L. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. The Journal of biological chemistry. 2009; 284: 2482-2492.
119. Peng Y, Li M, Clarkson BD, Pehar M, Lao PJ, Hillmer AT, Barnhart TE, Christian BT, Mitchell HA, Bendlin BB, Sandor M, Puglielli L. Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. The Journal of neuroscience. 2014; 34: 6772-6789.
120. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. The Journal of clinical investigation. 2014; 124: 30-39.
121. Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, Wolffe AP, Nakatani Y, Roeder RG, Cole PA. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Molecular cell. 2000; 5: 589-595.
122. Zheng Y, Balasubramanyam K, Cebrat M, Buck D, Guidez F, Zelent A, Alani RM, Cole PA. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. Journal of the American Chemical Society. 2005; 127: 17182-17183.
123. Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, Neckers L. Curcumin is an inhibitor of p300 histone acetylatransferase. Medicinal chemistry (Shariqah (United Arab Emirates)). 2006; 2: 169-174.
124. Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PP, Kundu TK. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. The Journal of biological chemistry. 2004; 279: 33716-33726.
125. Lopez-Lazaro M. Anticancer and carcinogenic properties of curcumin: considerations for its clinical development as a cancer chemopreventive and chemotherapeutic agent. Molecular nutrition & food research. 2008; 52 Suppl 1: S103-27.
126. Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. The Journal of biological chemistry. 2003; 278: 19134-19140.
127. Sun Y, Jiang X, Chen S, Price BD. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS letters. 2006; 580: 4353-4356.
128. Wapenaar H, van der Wouden PE, Groves MR, Rotili D, Mai A, Dekker FJ. Enzyme kinetics and inhibition of histone acetyltransferase KAT8. European journal of medicinal chemistry. 2015; 105: 289-296.
129. Eliseeva ED, Valkov V, Jung M, Jung MO. Characterization of novel inhibitors of histone acetyltransferases. Molecular cancer therapeutics. 2007; 6: 2391-2398.
130. Ghizzoni M, Boltjes A, Graaf C, Haisma HJ, Dekker FJ. Improved inhibition of the histone acetyltransferase PCAF by an anacardic acid derivative. Bioorganic & medicinal chemistry. 2010; 18: 5826-5834.
131. Kusio-Kobialka M, Dudka-Ruszkowska W, Ghizzoni M, Dekker FJ, Piwocka K. Inhibition of PCAF by anacardic acid derivative leads to apoptosis and breaks resistance to DNA damage in BCR-ABL-expressing cells. Anti-cancer agents in medicinal chemistry. 2013; 13: 762-767.
132. Sbardella G, Castellano S, Vicidomini C, Rotili D, Nebbioso A, Miceli M, Altucci L, Mai A. Identification of long chain alkylidenemalonates as novel small molecule modulators of histone acetyltransferases. Bioorganic & medicinal chemistry letters. 2008; 18: 2788-2792.
133. Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, Kim MJ, Cha JH, Kim YJ, Jun WJ, Lee JM, Yoon HG. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer research. 2009; 69: 583-592.
134. Ravindra KC, Selvi BR, Arif M, Reddy BA, Thanuja GR, Agrawal S, Pradhan SK, Nagashayana N, Dasgupta D, Kundu TK. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. The Journal of biological chemistry. 2009; 284: 24453-24464.
135. Dalvoy Vasudevarao M, Dhanasekaran K, Selvi RB, Kundu TK. Inhibition of acetyltransferase alters different histone modifications: probed by small molecule inhibitor plumbagin. Journal of Biochemistry. 2012; 152: 453-462.
136. Raghu D, Karunagaran D. Plumbagin downregulates Wnt signaling independent of p53 in human colorectal cancer cells. Journal of natural products. 2014; 77: 1130-1134.
137. Shi M, Lu XJ, Zhang J, Diao H, Li G, Xu L, Wang T, Wei J, Meng W, Ma JL, Yu H, Wang YG. Oridonin, a novel lysine acetyltransferases inhibitor, inhibits proliferation and induces apoptosis in gastric cancer cells through p53- and caspase-3-mediated mechanisms. Oncotarget. 2016. Mar 10. doi: 10.18632/oncotarget.8033. [Epub ahead of print]
138. Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chemistry & biology. 2010; 17: 471-482.
139. Gao XN, Lin J, Ning QY, Gao L, Yao YS, Zhou JH, Li YH, Wang LL, Yu L. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PloS one. 2013; 8: e55481.
140. Santer FR, Hoschele PP, Oh SJ, Erb HH, Bouchal J, Cavarretta IT, Parson W, Meyers DJ, Cole PA, Culig Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Molecular cancer therapeutics. 2011; 10: 1644-1655.
141. Oike T, Komachi M, Ogiwara H, Amornwichet N, Saitoh Y, Torikai K, Kubo N, Nakano T, Kohno T. C646, a selective small molecule inhibitor of histone acetyltransferase p300, radiosensitizes lung cancer cells by enhancing mitotic catastrophe. Radiotherapy and oncology. 2014; 111: 222-227.
142. Yan G, Eller MS, Elm C, Larocca CA, Ryu B, Panova IP, Dancy BM, Bowers EM, Meyers D, Lareau L, Cole PA, Taverna SD, Alani RM. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. The Journal of investigative dermatology. 2013; 133: 2444-2452.
143. Ogiwara H, Sasaki M, Mitachi T, Oike T, Higuchi S, Tominaga Y, Kohno T. Targeting p300 Addiction in CBP-Deficient Cancers Causes Synthetic Lethality by Apoptotic Cell Death due to Abrogation of MYC Expression. Cancer discovery. 2016; 6: 430-445.
144. Shrimp JH, Sorum AW, Garlick JM, Guasch L, Nicklaus MC, Meier JL. Characterizing the Covalent Targets of a Small Molecule Inhibitor of the Lysine Acetyltransferase P300. ACS medicinal chemistry letters. 2015; 7: 151-155.
145. van den Bosch T, Boichenko A, Leus NG, Ourailidou ME, Wapenaar H, Rotili D, Mai A, Imhof A, Bischoff R, Haisma HJ, Dekker FJ. The histone acetyltransferase p300 inhibitor C646 reduces pro-inflammatory gene expression and inhibits histone deacetylases. Biochemical pharmacology. 2016; 102: 130-140.
146. Xu LX, Li ZH, Tao YF, Li RH, Fang F, Zhao H, Li G, Li YH, Wang J, Feng X, Pan J. Histone acetyltransferase inhibitor II induces apoptosis in glioma cell lines via the p53 signaling pathway. Journal of experimental & clinical cancer research. 2014; 33: 108-014-0108-3.
147. Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, Jahn SC, Saldanha SA, Chase P, Planck J, Geary KR, Ma H, Law BK, et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Molecular cancer therapeutics. 2013; 12: 610-620.
148. Stimson L, Rowlands MG, Newbatt YM, Smith NF, Raynaud FI, Rogers P, Bavetsias V, Gorsuch S, Jarman M, Bannister A, Kouzarides T, McDonald E, Workman P, et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Molecular cancer therapeutics. 2005; 4: 1521-1532.
149. Gajer JM, Furdas SD, Grunder A, Gothwal M, Heinicke U, Keller K, Colland F, Fulda S, Pahl HL, Fichtner I, Sippl W, Jung M. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis. 2015; 4: e137.
150. Ghizzoni M, Haisma HJ, Dekker FJ. Reactivity of isothiazolones and isothiazolone-1-oxides in the inhibition of the PCAF histone acetyltransferase. European journal of medicinal chemistry. 2009; 44: 4855-4861.
151. Secci D, Carradori S, Bizzarri B, Bolasco A, Ballario P, Patramani Z, Fragapane P, Vernarecci S, Canzonetta C, Filetici P. Synthesis of a novel series of thiazole-based histone acetyltransferase inhibitors. Bioorganic & medicinal chemistry. 2014; 22: 1680-1689.
152. Chimenti F, Bizzarri B, Maccioni E, Secci D, Bolasco A, Chimenti P, Fioravanti R, Granese A, Carradori S, Tosi F, Ballario P, Vernarecci S, Filetici P. A novel histone acetyltransferase inhibitor modulating Gcn5 network: cyclopentylidene- [4-(4’-chlorophenyl)thiazol-2-yl)hydrazone. Journal of medicinal chemistry. 2009; 52: 530-536.
153. Trisciuoglio D, Ragazzoni Y, Pelosi A, Desideri M, Carradori S, Gabellini C, Maresca G, Nescatelli R, Secci D, Bolasco A, Bizzarri B, Cavaliere C, D’Agnano I, et al. CPTH6, a thiazole derivative, induces histone hypoacetylation and apoptosis in human leukemia cells. Clinical cancer research. 2012; 18: 475-486.
154. Ragazzoni Y, Desideri M, Gabellini C, De Luca T, Carradori S, Secci D, Nescatelli R, Candiloro A, Condello M, Meschini S, Del Bufalo D, Trisciuoglio D. The thiazole derivative CPTH6 impairs autophagy. Cell death & disease. 2013; 4: e524.
155. Di Martile M, Desideri M, De Luca T, Gabellini C, Buglioni S, Eramo A, Sette G, Milella M, Rotili D, Mai A, Carradori S, Secci D, De Maria R, et al. Histone acetyltransferase inhibitor CPTH6 preferentially targets lung cancer stem-like cells. Oncotarget. 2016; 7: 11332-11348. doi: 10.18632/oncotarget.7238.
156. Carradori S, Rotili D, De Monte C, Lenoci A, D’Ascenzio M, Rodriguez V, Filetici P, Miceli M, Nebbioso A, Altucci L, Secci D, Mai A. Evaluation of a large library of (thiazol-2-yl)hydrazones and analogues as histone acetyltransferase inhibitors: enzyme and cellular studies. European journal of medicinal chemistry. 2014; 80: 569-578.
157. Ghizzoni M, Wu J, Gao T, Haisma HJ, Dekker FJ, George Zheng Y. 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. European journal of medicinal chemistry. 2012; 47: 337-344.
158. Coffey K, Blackburn TJ, Cook S, Golding BT, Griffin RJ, Hardcastle IR, Hewitt L, Huberman K, McNeill HV, Newell DR, Roche C, Ryan-Munden CA, Watson A, et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PloS one. 2012; 7: e45539.
159. Kobayashi J, Kato A, Ota Y, Ohba R, Komatsu K. Bisbenzamidine derivative, pentamidine represses DNA damage response through inhibition of histone H2A acetylation. Molecular cancer. 2010; 9: 34-4598-9-34.
160. Wu J, Wang J, Li M, Yang Y, Wang B, Zheng YG. Small molecule inhibitors of histone acetyltransferase Tip60. Bioorganic chemistry. 2011; 39: 53-58.
161. Gao C, Bourke E, Scobie M, Famme MA, Koolmeister T, Helleday T, Eriksson LA, Lowndes NF, Brown JA. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Scientific reports. 2014; 4: 5372.
162. Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L, Zhang Y, Liu B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015; 6: 5501-5516. doi: 10.18632/oncotarget.3551.
163. Romero FA, Taylor AM, Crawford TD, Tsui V, Cote A, Magnuson S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. Journal of medicinal chemistry. 2016; 59: 1271-1298.
164. Wyce A, Ganji G, Smitheman KN, Chung CW, Korenchuk S, Bai Y, Barbash O, Le B, Craggs PD, McCabe MT, Kennedy-Wilson KM, Sanchez LV, Gosmini RL, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PloS one. 2013; 8: e72967.
165. Coude MM, Braun T, Berrou J, Dupont M, Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME, Herait P, Baruchel A, Dombret H, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015; 6: 17698-17712. doi: 10.18632/oncotarget.4131.
166. Hytti M, Tokarz P, Maatta E, Piippo N, Korhonen E, Suuronen T, Honkakoski P, Kaarniranta K, Lahtela-Kakkonen M, Kauppinen A. Inhibition of BET bromodomains alleviates inflammation in human RPE cells. Biochemical pharmacology. 2016.