
INTRODUCTION
Colorectal cancer (CRC) represents the third most commonly diagnosed malignancy and the fourth leading cause of cancer deaths [1–3]. At present, the only therapeutic strategies include surgery and adjuvant or neo-adjuvant chemotherapy; thus, there is a definite need for innovative biological targets for prognostic stratification and therapeutic interventions. Colorectal carcinogenesis implies alterations of basic cellular processes, such as differentiation, proliferation and apoptosis. These processes and the overall physiologic functions of living beings are characterized by rhythmic oscillations recurring with a periodicity of approximately 24 hours (circadian) [4–7]. The circadian rhythms are driven by molecular clockworks ticking in every cell through transcriptional-translational feedback loops (TTFLs) revolving in about 24 hours and operated by circadian genes and proteins [8, 9]. TTFLs are initiated by the transcription factors BMAL1/2 (ARNTL/ARNTL2) and CLOCK/NPAS2, which heterodimerize and activate the transcription of PERIOD (PER1-3) and CRYPTOCHROME (CRY1-2) genes. In turn, PER and CRY proteins heterodimerize and inhibit the transcriptional activity of BMAL1/2 - CLOCK/NPAS2 heterodimers [10]. BMAL1/2 activates also the expression of NR1D1/NR1D2, encoding the transcription factors Rev-Erbα/β that, in turn, negatively control the rhythmic transcription of BMAL1, hindering RORα binding to the response element in the BMAL1 promoter region [11]. In the molecular clockwork of Drosophila Melanogaster a key role is played by Timeless (Tim) [12], and precisely there are two Tim paralogs, Tim1 and Tim2 (also called Timeout). Tim1 is a canonical circadian component, whereas the role of Tim2 in the fly is presently unclear [13]. In mammals the homolog of Tim2 seems to be not essential for the ticking of the biological clock, but the encoded protein interacts with TIPIN and is crucial for replication protection and genomic stability [14], as well as for coordination of mitotic kinase activation with DNA replication termination [15], period determination and DNA damage-dependent phase advancing of the circadian clock [16]. The circadian proteins undergo also post-translational modifications, such as phosphorylation by Casein Kinase 1ε (CK1ε) and AMPK and deacetylation by SIRT1, which tag them for degradation or modify their functions, respectively [17–20].
MicroRNAs (miRNAs) are small, endogenous, single-stranded RNAs of 18–22 nucleotides (nt) in length that are emerging as important modulators of gene expression at post-transcriptional level. They promote translational repression and/or destabilization of target messenger RNAs (mRNAs) by base-pairing to partially complementary sites in the 3′ untranslated region (3′UTR), less frequently in the coding sequences (CDS), of the target mRNAs [21]. The same mRNA transcript may be targeted by different miRNAs and a single miRNA, in turn, may target several different mRNAs [22]. Derangements in the miRNA-mRNA interactions [23], as well as altered expression of clock genes [24, 25] play a role in CRC onset and development and impact colon cancer cell behavior and response to chemotherapy [26, 27].
In this study we performed an integrative analysis of miRNA-miRNA and miRNA-mRNA interactions using high-throughput mRNA and miRNA expression profiling of CRC datasets to explore miRNA-clock genes interplays searching for functional relationships that are deranged in this tumor type. We addressed this aim by investigating the miRNA correlation network built on a miRNA expression profile database, which was populated with a set of human CRC specimens and matched adjacent normal mucosa in order to identify the miRNAs that, among those targeting the core clock genes, may play a key role in this malignancy. These miRNAs populated the subset 1 of this analysis. Similarly, we investigated the miRNA correlation network built on the miRNA expression profiles obtained from the Colon adenocarcinoma (COAD) database publicly available on the web through The Cancer Genome Atlas (TCGA) Data Portal (https://tcga-data.nci.nih.gov/tcga/, downloaded March 15th, 2015). These miRNAs populated the subset 2 of this analysis. We then compared the two correlation networks in terms of connectivity and node centrality. Finally, based on the expression levels and on topological considerations of miRNAs and clock genes, we selected miR-139-5p and TIMELESS to be further validated, in vitro, in a new cohort of CRC patients and in human colon cancer cell lines.
RESULTS
A priori filtering of miRNA correlation pairs in TCGA and colon cancer microarray cohorts
We performed the Pearson Product Moment Correlation (PPMC) analysis on our CRC dataset (Table 1) and one publicly available dataset: miRNAs making significant pairs were counted and the numbers obtained were compared. The miRNAs detected in the COAD-TCGA dataset and present or not in our arrays were reported. Notably, COAD-TCGA control and tumor samples shared 501 miRNAs, while all miRNAs in control samples were shared with tumor samples. Moreover, among these latter samples, half of individual miRNAs were common to both datasets. The numbers of miRNAs that were exclusively correlated in control rather than in tumor samples were almost balanced in the microarray dataset (Supplementary Tables S1–S5).
Table 1: Clinical characteristics of the colorectal cancer patients examined for the microarray analysis
No. |
Age (years) |
Sex |
Cancer site |
Adenocarcinoma |
Histological grade |
Clinical stage |
TNM classification |
|
|---|---|---|---|---|---|---|---|---|
1 |
PS1 |
58 |
Female |
PTRCF |
Yes |
G2 |
II |
T3N0M0 |
2 |
PS3 |
81 |
Male |
PTRCF |
Yes |
G1 |
I |
T2N0M0 |
3 |
PS4 |
61 |
Male |
Sigmoid |
Yes |
G2 |
III |
T3N1M0 |
4 |
PS5 |
69 |
Female |
DTLCF |
Yes |
G1 |
IV |
T4N2M1 |
5 |
PS7 |
77 |
Male |
Sigmoid |
Yes |
G2 |
I |
T2N0M0 |
6 |
PS10 |
67 |
Male |
Sigmoid |
Yes |
G2 |
III |
T3N2M0 |
7 |
PS11 |
67 |
Male |
Sigmoid |
Yes |
G1 |
III |
T3N1M0 |
8 |
PS12 |
57 |
Male |
Sigmoid |
Yes |
G2 |
II |
T3N0M0 |
9 |
PS13 |
81 |
Female |
Sigmoid |
Yes |
G1 |
II |
T3N0M0 |
10 |
PS14 |
81 |
Female |
Rectosigmoid |
Yes |
G2 |
I |
T2N0M0 |
11 |
PS15 |
77 |
Male |
DTLCF |
Yes |
G2 |
III |
T3N1M0 |
12 |
PS17 |
73 |
Female |
Coecum |
Yes |
G3 |
III |
T3N1M0 |
13 |
PS19 |
57 |
Male |
Sigmoid |
Yes |
G2 |
II |
T3N0M0 |
14 |
PS20 |
55 |
Male |
Descendens |
Yes |
G1 |
II |
T3N0M0 |
Abbreviations: PTRCF = Proximal transverse and right colic flexure; DTLCF = Distal transverse and left colic flexure TNM (classification of malignant tumors): T, size or direct extent of the primary tumor; N, degree of spread to regional lymph nodes; M, presence of distant metastasis.
Identification of common or unique miRNA pairs through different conditions
Weak correlations were excluded by considering only miRNA pairs with an r exceeding ± 0.8. Thus, the remaining pairs of miRNAs in tumor and control networks were compared to discover those peculiar for each disease condition. Figure 1A shows the intersections of these miRNA correlation pairs in the microarray dataset. It is worthy of note that the COAD-TCGA dataset (Figure 1B) is unbalanced in terms of number of tumor correlation pairs with respect to the number of control correlation pairs. Roughly, 10% of pairs are in common with the control ones in both datasets (Figure 1C). MiRNAs targeting clock genes were searched in both subsets with the main goal to define whether these miRNA groups had some elements in common. Indeed, a significant percentage of them was present in each subset and a further interesting part correlated (i.e., there were correlation pairs between miRNAs targeting clock genes). This finding likely indicates that pairs of correlated miRNAs may cooperate to target the core clock genes. Moreover, the strongly correlated miRNAs set was bigger in control than tumor samples, underscoring, on one hand, the important regulatory role of miRNAs in normal tissue and, on the other hand, that deregulation of almost one third of them may play a critical role in CRC development.
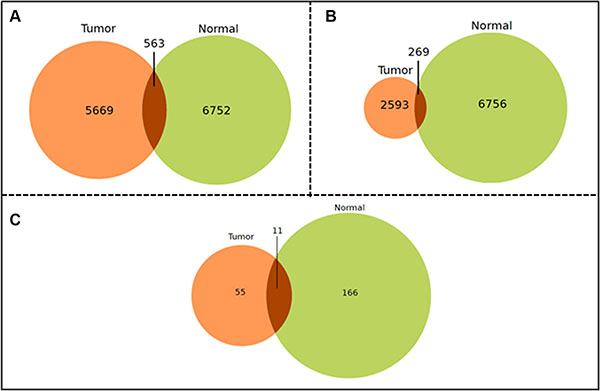
Figure 1: Venn-Diagrams of the shared miRNA-miRNA correlation pairs. Venn-Diagram of the shared miRNA-miRNA correlation pairs in the microarray (A) and the COAD-TCGA (B) datasets that are exclusive to tumor tissue (left), exclusive to non-tumor tissue (right) or common between the two tissue types (overlapping). (Filtering criteria: Pearson Product Moment Correlation score ± 0.6, absolute value, and p ≤ 0.05); (C) Venn-Diagram of the shared miRNA-miRNA correlation pairs in the microarray dataset. (Filtering criteria: Pearson Product Moment Correlation score ± 0.8, absolute value, and p ≤ 0.05).
Candidate miRNAs involved in clock genes deregulation in colorectal cancer
We drew and analyzed the networks of the following subsets: the common subset, the correlation subset exclusive for the correlation pairs in non-tumor tissue (control subset) and the correlation subset exclusive for the correlation pairs in neoplastic tissue (tumor subset). The visual representation of these networks is rendered in Figure 2. From a preliminary survey, it can be noticed that the common subset miRNAs generate a highly sparse correlation network and tend to form closed and unrelated clusters (Figure 2A). miR-20a-5p, the miRNA with the highest betweeness centrality value (0.667), reaches the maximum degree in this network that is 3. It is important to consider that, in this network, many miRNAs are also differentially expressed without a significant link between the fold-change and the r value. Moreover, many miRNAs targeting clock genes did not come out from the COAD-TCGA dataset analysis.
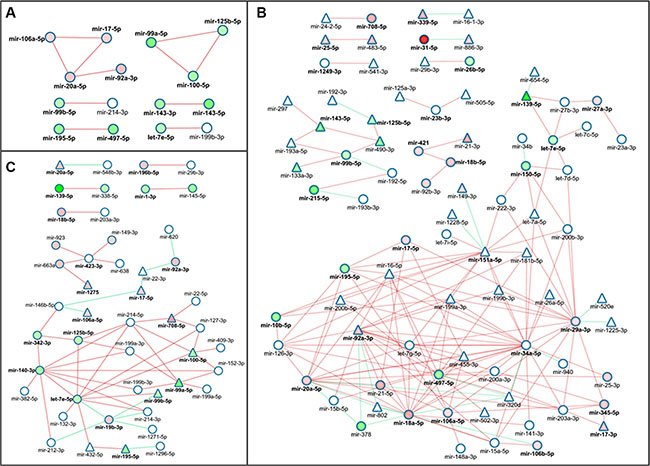
Figure 2: Network picture of colorectal cancer miRNA-ome datasets based on Pearson Product Moment Correlation network of commonly correlated miRNAs in tumor and non-tumor tissue. (A) Network representation of the common miRNA correlation pairs among neoplastic (tumor) and matched non-tumor tissue (control). (B) Network visual representation of the miRNA correlation pairs specific for the control tissue. (C) Network visual representation of the miRNA correlation pairs specific for the tumor tissue. Nodes represent miRNAs that are linked by an edge if their expression values correlate (red for positive correlations, green otherwise). Shapes and colours represent features of nodes and edges. Red nodes render up-regulated miRNAs in tumor; green nodes render down-regulated miRNAs in tumor. Bold node render miRNAs that are experimentally known to target any core clock gene. Triangle-shaped nodes render miRNAs whose expression values are cross-checked in the TCGA-COAD dataset.
Analysis of the miRNA network built on the control subset verified a strong correlation among the vast majority of the nodes, with only a few tending to form small isolated clusters (Figure 2B). Several positive correlations were present in the core network, many of which did not appear in the COAD-TCGA dataset. The node with the smallest average shortest path length is miR-34a-5p that targets CRY1, a core element of the molecular clockwork. This miRNA came out from the analysis of the COAD-TCGA dataset and displayed the highest degree centrality and the lowest average shortest path length together with a relatively low clustering coefficient. However, the expression of this miRNA did not significantly change between tumor and non-tumor tissues. On its side, the miRNA network built on the tumor subset displayed some interesting properties (Figure 2C). Apart from isolated correlation pairs, it showed an intricate correlation network and the node with the highest degree was let-7e-5p that was only slightly down-regulated in tumor tissue. This miRNA, belonging to the let family and involved in stemness, showed also a good centrality value (around 0.4), pointing it as a hub in our tumor correlation network. Moreover, this node was positively correlated with a series of other miRNAs hallmarked by low-fold change values, implying that their deregulation might trigger the reduced expression of several other miRNAs. Additionally, the fold-change and the correlation values indicated miR-19-3b as a possible negative regulator of the let-7-e-5p gene, as it was negatively correlated and up-regulated in tumor. miR-19b-3p targets TIMELESS and NR1D2 and many of its correlated neighbours, let-7e-5p, miR-99b-5p and, indirectly, miR-140-3p (separated by 1 degree and connected via miR-214-3p and miR-212-3p) are instead down-regulated, suggesting a potential regulation of miR-19b-3p on these miRNAs.
As illustrated in Table 2 and Figure 3, miR-125b-5p, miR-140-3p, let-7e-5p and miR-99b-5p are all down-regulated in tumor as compared to non-tumor tissues and all target clock genes. Noteworthy, miR-19b-3p is weakly (−0.47) anti-correlated with miR-99b-5p in the control subset, but the anti-correlation strength becomes stronger in the tumor subset. Besides, these miRNAs are involved in the pathways controlling cancer cell proliferation and invasiveness (Supplementary Table S6)
Table 2: List of the clock genes targets of the most relevant correlated miRNAs
|
Fold Change (Tumor versus Control) |
Targets |
|---|---|---|
let-7e-5p |
Down |
PER1, CLOCK |
miR-125b-5p |
Down |
PER1, RORA, TIPIN |
miR-140-3p |
Down |
NPAS2, TIMELESS |
miR-99b-5p |
Down |
PER1, RORA, TIMELESS |
miR-19b-3p |
Up |
TIMELESS, NR1D2 |
miR-139-5p |
Down |
TIMELESS |
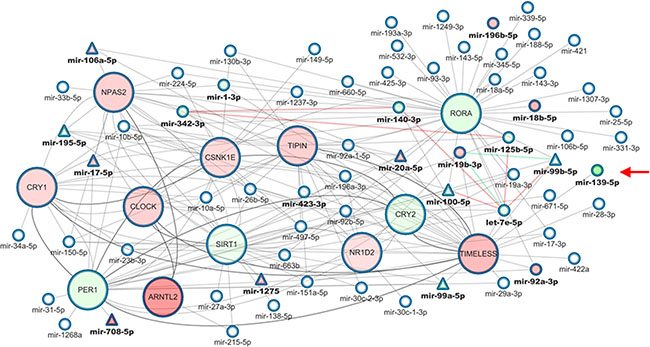
Figure 3: Core clock genes-miRNAs correlation network in colorectal cancer. Multi-layer network of clock genes and targeting miRNAs in which nodes represent clock genes or miRNAs, while edges represent correlation or interaction among miRNAs or genes. Nodes representing genes are bigger than miRNA nodes. miRNA names are bolded if any association with clock genes is known. miRNA nodes are represented as triangles if their expression values are consistent in the COAD-TCGA dataset. Red nodes are up-regulated genes/miRNAs in tumor tissue, green nodes are down-regulated genes/miRNAs in tumor tissue. Edges between genes are solid or dashed, depending on whether they represent direct or indirect interactions, as reported in BioGRID. Edges connecting miRNAs-genes render targeting interactions as from miRWalk 2.0. Arrowed edges connecting two miRNAs represent strong positive (> 0.8) or negative (≤ 0.8) correlations between them, and are colored in red or green, respectively.
In the control network, let-7e-5p is linked to miR-139-5p, a candidate tumor suppressor targeting TIMELESS and found down-regulated in many cancer types, but in the tumor network this important link was lost. Interestingly, as reported in Supplementary Table S3, miR-139-5p was down-regulated and ranked among the most deregulated miRNAs in tumor tissues (False Discovery Rate = 0.002668, Fold Change = −6.801375, p value = 0.000013). miR-139-5p recognizes two seed regions in TIMELESS mRNA and we decided to select this miRNA/gene pair for validation in an independent cohort of patients and, then, for in vitro functional studies.
TIMELESS mRNA and miR139-5p levels are altered and inversely correlated in sporadic CRCs
We quantified TIMELESS mRNA and miR-139-5p levels in 50 matched pairs of tumor tissues/non-tumor mucosa sampled from 46 well-moderately differentiated (G1-G2) and from 4 poorly differentiated-undifferentiated (G3-G4) CRCs. Median values, 25th (or first quartile, Q1) and 75th percentile (or third quartile, Q3) of GAPDH-Ct value/target gene-Ct value are shown in Figure 4. Compared to matched non-tumor tissues a statistically significant increase of TIMELESS mRNA (median = 1.25, Q1-Q3 = 0.87-2.13, p < 0.001) and a decrease of miR-139-5p levels (median = 0.15, Q1–Q3 = 0.06–0.44, p < 0.0001) was observed in tumor tissues. TIMELESS mRNA was inversely correlated with miR-139-5p expression by a Pearson’s correlation test (r = −0.320; p = 0.027) (Figure 4 upper panel). These results were corroborated by the analysis of an independent publicly available COAD-TGCA cohort comprising 210 CRC samples for which mRNA and miRNA expression profile were attainable (https://tcga-data.nci.nih.gov/tcga) (r = −0.160; p = 0.021) (Figure 4 lower panel).
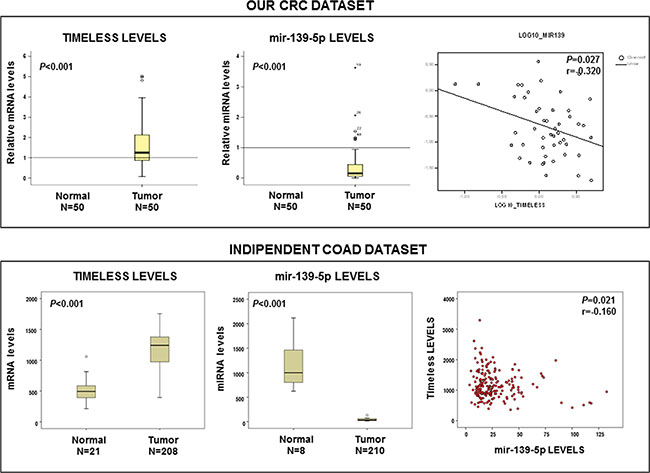
Figure 4: TIMELESS mRNA and miR-139-5p levels are inversely correlated in sporadic colorectal cancer. Box-plot analysis of TIMELESS mRNA and miR-139-5p levels in our colorectal cancer (CRC) patients validation cohort (upper panel) and in the COAD-TCGA dataset (lower panel); p value and r coefficient of correlation are reported in the panels.
Clinical-pathological features of CRC patients and association with TIMELESS mRNA and miR-139-5p levels
Genotype-phenotype associations were evaluated in our validation cohort of CRC patients. After splitting subjects at the median value of gene expression levels, the increased TIMELESS mRNA level in tumor tissue was strongly associated with disease spreading, in particular with proximal and distal lymph nodes involvement (p = 0.010) and III-IV TNM stages (p = 0.017). Lower miR-139-5p levels were associated with a non-mucinous adenocarcinoma histologic type (p = 0.035) and high microsatellite instability frequency (p = 0.035) (Table 3).
Table 3: Association of TIMELESS mRNA and miR-139.5p levels with clinical and pathological features of colorectal cancer patients included in the validation cohort
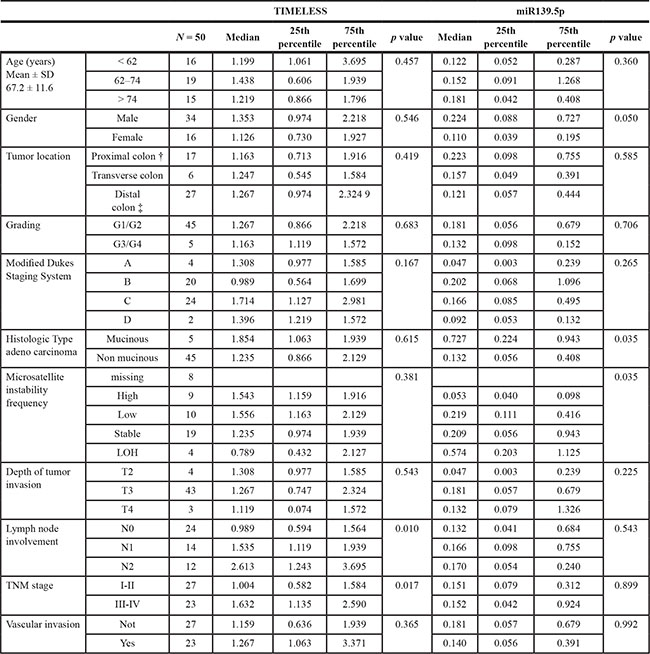
†caecum and ascending colon; ‡ descending colon, sigmoid colon and rectum; LOH = loss of heterozigosity; N0 = no lymph node involvement N1 = proximal lymph node involvement; N2 = distal lymph node involvement; TNM (classification of malignant tumors): T, size or direct extent of the primary tumor; N, degree of spread to regional lymph nodes; M, presence of distant metastasis.
No statistically significant difference was found for survival rates in a Kaplan–Meier analysis of censored data after stratifying patients according to the median expression value of TIMELESS mRNA (Log Rank–Mantel-Cox test p = 0.721) and miR139-5p (Log Rank-Mantel-Cox test p = 0.515) (Supplementary Figure S1).
TIMELESS and miR-139-5p levels are inversely related in human colon cancer cell lines
TIMELESS expression at the mRNA and protein levels was evaluated in CaCo2, HCT116, HT29, SW480 and SW620 cells: TIMELESS mRNA expression was higher in tumor than non-tumor mucosa and mainly in CaCo2 and HT29 cells with respect to HCT116, SW480 and SW620 cells (Figure 5A). Western blot analysis on extracts from the same cells showed higher levels of the protein in CaCo2, HCT116 and in particular SW620 cells, suggesting a key role played by miR-139-5p in TIMELESS post-transcriptional regulation (Figure 5B). Indeed, miR-139-5p expression displayed an opposite pattern to TIMELESS, with lower levels in all the examined cell lines than non-tumor mucosa; lower levels were detected in CaCo2 and HT29 with respect to HCT116, SW480 and SW620 cells (Figure 5A). Bioinformatic analysis identified two seed regions for miR-139-5p in the CDS and 3′UTR of TIMELESS mRNA, respectively (Figure 5C). Thus, we investigated the miR-139-5p-dependent TIMELESS regulation in vitro. Modulation of miR-139-5p expression through mimic or antisense RNAs induced remarkable and inverse variations of TIMELESS in CRC cells. In detail, miR-139-5p inhibition resulted in TIMELESS overexpression in HCT116 and HT29 with respect to SW480 and SW620, while no statistically significant variation was observed in CaCo2 cells. In contrast, transfection of a miR-139-5p mimic down-regulated TIMELESS in all cells lines under investigation (Figure 5D). These data confirm that miR-139-5p inversely correlates with TIMELESS regulating its protein levels in human colon cancer cells.
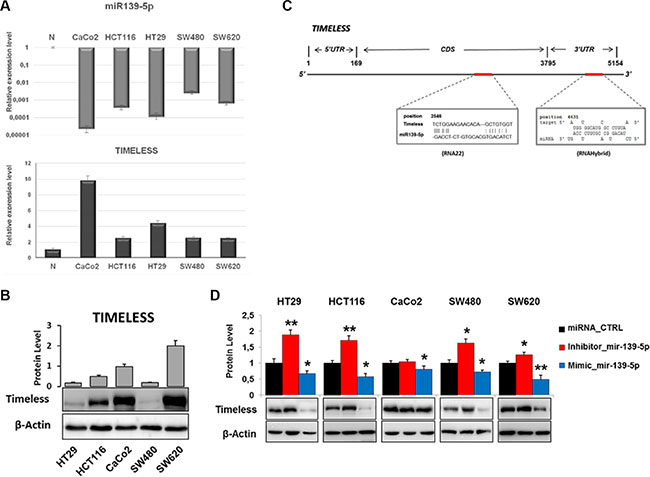
Figure 5: miR-139-5p negatively regulates TIMELESS expression in human colon cancer cell lines. (A) The bar plot reports miR-139-5p and TIMELESS mRNA levels evaluated by qRT-PCR analysis in the indicated cell lines with respect to the average expression detected in non-tumor tissues. (B) Immunoblot detection of TIMELESS protein in five human colon cancer cell lines; relative fold-change, obtained by densitometric analysis and normalization to β-actin, is reported in the corresponding histograms. (C) Schematic representation of human TIMELESS mRNA: two specific highly conserved miR-139-5p binding sites are located in the coding region (CDS; RNA22 prediction) and in 3′ UTR (RNAhybrid prediction). (D) Western blot analysis of TIMELESS in cells transfected with a miR-139-5p mimic or inhibitor, or scrambled controls; the histogram reports the protein fold-change with respect to scrambled-transfected cells. Samples were analyzed in triplicate and data are reported as mean ± S.D. and representative of three independent experiments. *p ≤ 0.05; **p ≤ 0.01.
FISH comparative analysis of TIMELESS copy numbers in human colon cancer cell lines
We hypothesized that the different TIMELESS levels found in the colon cancer cell lines could be related to the occurrence of copy number gains involving the encoding gene. To address this issue, FISH co-hybridization experiments were performed in all five colorectal cancer cell lines by using a probe set encompassing the whole gene region. The overall results showed low copy number gains of TIMELESS in all cell lines except HCT116, showing signals only on the two normal chromosome 12 homologs (Figure 6B). Specifically, HT29 displayed a whole-chromosome 12 trisomy (Figure 6D); in SW480, two markers showed positivity for all the three probes, as well as one normal chromosome 12 (Figure 6C); CaCo2, in addition to trisomy 12, showed positivity for a marker chromosome of unknown origin (Figure 6E). Finally, SW620 cells showed the most complex karyotype, displaying multiple signals to be ascribed to a whole chromosome 12 tetrasomy and two marker chromosomes positive for TIMELESS signals (Figure 6F).
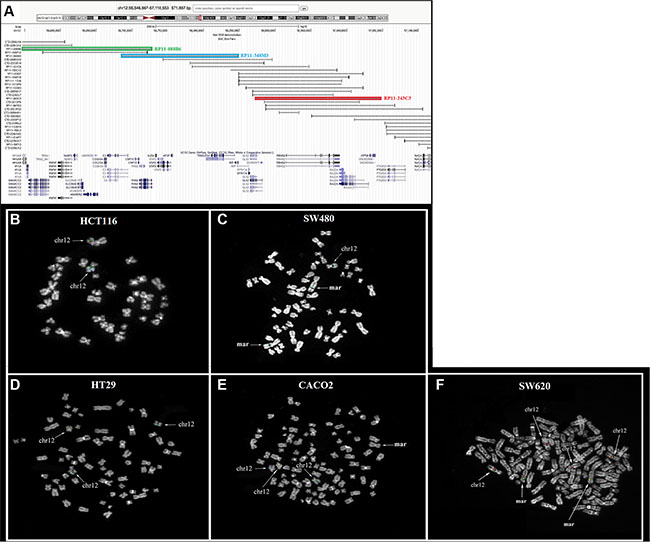
Figure 6: Comparative analysis of TIMELESS copy numbers performed by FISH in human colon cancer cell lines. (A) The BAC probe set used for the screening of TIMELESS copy number gains. The colored rectangles (blue, red, and green) correspond to the FISH results obtained respectively for probes RP11-348M3, RP11-243C3, and RP11-888B6. Genes are reported in blue at the bottom of the figure. (B–F) FISH analysis results. In HCT116 the three probes gave signals on the two normal chromosome 12 homologs (B); in SW480, two markers showed positivity for all the probes used, as well as one normal chromosome 12 (C); HT29 displayed a whole chromosome 12 trisomy (D); CACO2 showed the positivity of a marker chromosome of unknown origin, in addition to chromosome 12 trisomy (E); SW620 displayed a whole-chromosome 12 tetrasomy and the positivity of two additional marker chromosomes (F).
miR-139-5p regulates proliferation, migration, colony forming efficiency and apoptosis in human colon cancer cell lines
A potential impact of TIMELESS expression changes on cancer cell phenotype modifications was suggested by data coming from the analysis of CRC patient specimens. To investigate this possibility we assessed the effects of TIMELESS down-regulation upon miR-139-5p ectopic expression on colon cancer cell proliferation, apoptosis, migration and colony forming efficiency. Cell proliferation was evaluated by automatic cell counting at different time points (24, 48 and 72 hrs) from transfection. A time-dependent reduction of proliferation was observed in all cell lines upon miR-139-5p mimic transfection (p < 0.05); the reduction trend was less evident in HT29 cells (Figure 7A). In contrast, due to the low basal miR-139-5p levels in all cell lines, a miR-139-5p antisense increased the proliferation rate only in HT29 cells (p < 0.05). Consistently, miR-139-5p overexpression or inhibition induced higher or lower pre-apoptosis and apoptosis than basal cells, respectively (Figure 7B). Colony forming efficiency and migration assays were severely reduced in all CRC cell lines examined (p < 0.05 and p < 0.01) upon miR139-5p mimic ectopic expression. In contrast, the miR-139-5p antisense induced higher migration in HT29 and SW480 cells and, in these latter ones, also larger colonies (Figure 8A and 8B).
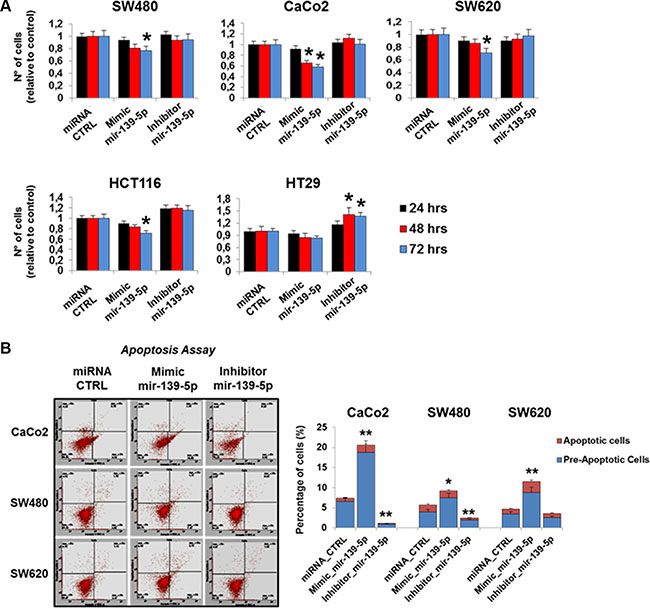
Figure 7: miR-139-5p inhibits cell proliferation and apoptosis in human colon cancer cells lines. (A) Graphical representation of cell numbers assessed by automatic cell counting at different time points (24, 48 and 72 hrs) from transfection with the miR-139-5p mimic, miR-139-5p inhibitor, or the appropriate scrambled controls. (B) Flow cytometry analysis of pre-apoptotic and apoptotic cells (Caco-2, SW480 and SW620) transfected with the miR-139-5p mimic, inhibitor or the appropriate scrambled controls. One representative experiment out of three is shown. Samples were analyzed in triplicate and data are reported as mean ± S.D. and representative of three independent experiments; *p ≤ 0.05; **p ≤ 0.01.
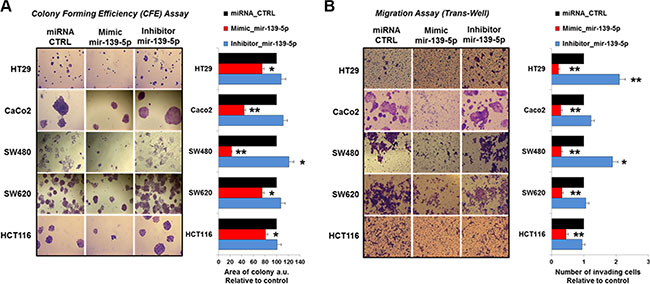
Figure 8: miR-139-5p reduces the colony forming area and cell invasion in human colon cancer cells lines. (A) Plate colony-forming assay performed in the indicated cell lines transfected with miR-139-5p mimic or inhibitor or relative miRNA controls and stained with crystal violet (left panel); the histogram reports the value of colonies areas (right panel). (B) Number of invading cells measured in at least five fields of the trans-well migration assay in all five human colon cancer cell lines transfected as in (A). Data are reported as mean ± SD of at least four independent experiments; *p ≤ 0.05; **p ≤ 0.01.
DISCUSSION
The definition of higher-order mRNA-miRNA interaction networks allows tumor microarray data interpretation and hypothesis generation. This, in turn, may help in discovering biological pathways deregulated in tumorigenesis and their possible roles in cancer onset and progression [28]. This method of analysis takes advantage of network-based models and, in the present study, was conducted pinpointing core clock genes and their targeting miRNAs. The data from our series of tumor and matched non-tumor tissues of CRC patients were validated with those obtained from high-throughput publicly available molecular profiling of matched colorectal tumor and non-tumor specimens.
MiRNAs play an important role in gene regulatory mechanisms negatively modulating gene expression at the post-transcriptional level. Approximately 30% of all mRNAs in mammals are regulated by miRNAs [29] and recognition occurs via base-pairing interactions with seed motifs located in the CDS or the 3′-UTR of target mRNAs. Generally, a given miRNA is expected to fine-tune a large set of mRNAs within a cell [30]. Consistently, miRNAs have been implicated in the control of genes driving proliferation, development, and differentiation [31]. Besides, miRNA expression is regulated in a circadian pattern in living beings starting from D. melanogaster [32] and miRNAs, in turn, modulate the biological clock [33, 34].
Interestingly, our analysis in CRC tissues shows a profound subversion of the appropriate relationships among miRNAs with respect to those detected in non-tumor tissue, with loss of connections with the main nodes and acquisition of abnormal ones with new partners that acquire key roles in the network. Among miRNAs regulating clock genes, miR-34a-5p is particularly important as it targets CRY1 and has recently been identified as a valuable prognostic biomarker in CRC. It is, in fact, down-regulated in tumor tissues and positively correlated with disease-free survival; it is able to hinder CRC recurrence, restraining cell growth, migration and invasion, triggering cell apoptosis and cell cycle arrest in a p53-dependent manner [35]. In the non-tumor tissue network, miR-34a-5p shows the highest degree centrality as well as the lowest average shortest path length with a relatively low clustering coefficient, while in the tumor network it looses the hub function that is acquired by let-7e-5p.
This latter miRNA is implicated in colorectal carcinogenesis [36] and targets PER1 and CLOCK mRNAs and in our analysis it gains a link to miR-19b-3p, which targets TIMELESS and NR1D2 mRNAs. It is included in the paralogous miRNA gene cluster originating the miR-17 family miRNAs (miR-17~92, miR-106a~363 and miR-106b ~ 25) involved in the control of cell cycle, proliferation, apoptosis, development and referred to as “oncomir” as frequently deregulated in hematopoietic and solid cancers [37, 38]. Finally let-7e-5p lost the link with miR-139-5p, an important tumor suppressor found deregulated in different cancer types [39–43].
miR-19b-3p is correlated with miR-99b-5p, targeting PER1, RORA and TIMELESS mRNAs and, indirectly, with miR-140-3p, targeting NPAS2 and TIMELESS mRNAs. Furthermore, bioinformatic analysis identified miR-139-5p seed regions in the CDS and 3′UTR of TIMELESS mRNA, which plays a key role in DNA damage response, telomere length and integrity maintenance, cell cycle progression as well as processes coordinated at the DNA replication fork such as DNA synthesis, S-phase-dependent checkpoint activation and chromosome cohesion [44–47].
The potential relationship between TIMELESS and miR-139-5p was validated in our cohort of sporadic CRC patients, confirming the inverse expression pattern and suggesting that deregulation of this axis may play a role in disease progression. This was further corroborated in a series of human colon cancer cell lines in which miR-139-5p induced TIMELESS down-modulation, determining the cell phenotype. In fact, restoration of miR-139-5p significantly inhibited cell proliferation and apoptosis as evidenced by cell count and FACS analysis. In contrast, re-expression of miR-139-5p in in vitro assays inhibited cell migration in all five cell lines examined.
In conclusion, miRNA-miRNA interactions add complexity to the functionality and enhance the robustness of coding/noncoding RNA regulatory networks. Alteration of this multifaceted interaction plays a crucial role in carcinogenesis. Our integrative analysis of the miRNA-mRNA interplay as well as of the expression patterns of potential miRNA-miRNA pairs based on the expression profiles in CRC tissues and matched adjacent non-tumor mucosa underscores the pivotal role that the miRNA-clock genes axis plays in colorectal tumorigenesis and improves our understanding of the miRNA-mRNA regulatory networks deregulated in this common malignancy.
MATERIALS AND METHODS
Microarray data (subset 1)
The transcriptome and miRNAome of 14 pairs of matched tumor tissues and adjacent non-tumor mucosa obtained at surgery from CRC patients was analyzed by using the GeneChip Human Exon 1.0 ST array and the GeneChip miRNA 2.0 array (Affymetrix, Santa Clara, CA, USA), respectively [48, 49]. The clinical-pathological characteristics of the patients investigated in the current study are listed in Table 1.
COAD-TCGA data (subset 2)
Expression profiles of COAD and healthy tissues were downloaded from the TCGA data portal, as preprocessed and normalized probeset intensity values (level 3 data).
Selection of miRNAs targeting clock genes and miRNA symbols disambiguation
We picked up specific experimentally validated miRNAs targeting the core clock genes by the miRWalk 2.0 web service [50]. Several miRNA symbols in both the microarray and TCGA data resulted to be not updated and thus, for these, it was not possible to distinguish between the 5p from the 3p forms. We have then disambiguated and updated these symbols by a custom Python script operating on the latest CDF file of the miRNA 1.0 chip and on the list of miRNA aliases available through the miRBase web site. The only two miRNAs, i.e. miR-378 and miR-34b, which were not possible to disambiguate were kept with their original names.
Subset and miRNA-Gene target analyses
The expression values of two miRNAs were considered coherently correlated in the tumor and normal networks if their r values were significant and both were either positive or negative. Any miRNA pair was tagged as clock-controller if at least one miRNA of the pair controlled one or more clock genes. MiRNAs were further highlighted according to their differential expression, when compared to control tissues.
Network analysis
Networks were drawn and analyzed by Cytoscape ver. 3.2.1 (http://www.cytoscape.org/). Nodes represent miRNAs and edges represent significant correlations between two miRNAs. Attributes were added to the nodes and edges according to the following features. Nodes were colored according to their fold-changes: red for up-regulated miRNAs, green for down-regulated miRNAs and white for miRNAs with no significant or missing fold-change values. Moreover, shapes of nodes were thought to represent the co-occurrence of a miRNA in our microarray data (rounded nodes) or in the COAD-TCGA data (triangular nodes). Moreover, miRNAs targeting clock genes were marked with bold labels. Edges were colored according to their correlation levels: red for positive correlations and green for negative correlations. For each node, we calculated the following topological indices: Degree, Closeness, Centrality, Betweeness, Clustering Coefficient, Radiality and Average Shortest Path Length, as already explained elsewhere [51, 52]. In Figure 7 bigger sized nodes represented clock genes, while correlated miRNAs were linked with dashed edges.
Validation cohort of CRC patients
The expression levels of TIMELESS mRNA and miR-139-5p were evaluated in the tumor tissues and adjacent non-tumor mucosa of an independent cohort of 50 CRC patients (34 men and 16 women, mean age ± SD 67.2 ± 11.6 years), surgically treated for colon and rectum primary tumors at our Hospital. The study was approved by the Ethical Board of our Institute, and all patients gave written informed consent. Patient characteristics are shown in Table 3. All tumor and non-tumor tissue specimens were collected between 9:00 a.m. and 17:00 p.m. of the same day (8 h), dissected immediately and snap-frozen in liquid nitrogen. All tissue samples were derived from surgical specimens. The frozen sections were evaluated by the pathologist to confirm the diagnosis of both tumor and non-tumor tissue. The non-tumor mucosa was collected in a part of the bowel close to the resection margins. The first and the last section of each sample were stained with haematoxylin and eosin and were analyzed by the pathologist to evaluate the tumor cell percentage. Tissue samples having at least 80% of tumor cell content were frozen in liquid nitrogen until the molecular analysis. These data were corroborated by the examination of TIMELESS and miR-139-5p expressions values of COAD and healthy tissues downloaded from the TCGA data portal, as preprocessed and normalized probeset intensity values (level 3).
RNA extraction from freshly frozen tissue and colon cancer cell lines and first-strand cDNA synthesis
Preparation of total RNA from about 150–200 mg of freshly frozen tissue specimens was done using the TRIzol reagent (Invitrogen Corporation, Carlsbad, CA, USA). The amount of total extracted RNA was determined by using the Nano Drop Spectrophotometer (Nanodrop Technology,Wilmington, DE, USA), and RNA integrity was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) after subsequent digestion by DNaseI. Next, 1.0 μg of total RNA was reversed transcribed using the High-Capacity cDNA Archive Kit following the manufacturer’s instructions (Applied Biosystems, Applera, Foster City, CA, USA).
Quantitative Real-Time reverse transcription-PCR assay
Quantitative real-time PCR (qRT-PCR) assay was used to assess the differential expression of the TIMELESS gene and miR-139-5p in CRC specimens matched to normal mucosa. Human QuantiTect Primers Assay (SYBR Green QuantiTect Primers Assay; QIAGEN, Hamburg, Germany) was used for TIMELESS (QT00019789) and qRT-PCRs were performed in a 25-μl final volume, in three replicates per sample, by using QuantiFast SYBR Green PCR kit (QIAGEN, Hamburg, Germany) and run in an ABI PRISM® 7700 Sequence Detection System (Applied Biosystems, Applera, Foster City, CA, USA) according to the following conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles at 95°C for 15 s and at 60°C for 1 min. Data were acquired as threshold cycle (CT) value. Expression levels of target gene were normalized using the GAPDH housekeeping control gene and the relative amount of mRNA in each target gene to GAPDH was calculated as the average 2−ΔΔCT method. Since the amplification efficiencies of target genes and internal control were equal, the relative change of target gene expression in tumor cells compared with normal colon mucosa (ΔCt calibrator value) was calculated using the equation 2−ΔΔCt, where ΔΔCt = ΔCt(calibrator) −ΔCt(target). The ΔCt values were defined by subtracting the average GAPDH Ct value from the average target gene Ct value. The SD of the difference was calculated from the SDs of target gene and GAPDH values. After each qRT-PCR, a melting profile as well as agarose gel electrophoresis of each sample was done to rule out nonspecific PCR products and primer dimers. For miRNA detection, 10 ng of RNA were reverse transcribed using the Taq-Man miRNA reverse transcription kit, including miR-139-5p (Assay ID: 478312_mir) and endogenous control RNU6B (Assay ID: 001093) primers in the reaction. Reverse transcription was followed by qRT-PCR assay with a TaqMan PCR Universal master mix and the appropriate miRNA specific TaqMan probe (Life Technologies Corporation). Expression levels of miR-139-5p were normalized to RNU6B and relative expression levels were calculated within each independent experiment using the formula 2−ΔΔCT.
Microsatellite instability
The Microsatellite instability (MSI) analysis was performed using the Bethesda panel of microsatellite (BAT25, BAT26, D5S346, D17S250 and D2S123) evaluated by means of a multiplex-PCR and PAGE analysis. Tumors showing instability in four or more markers were classified as high MSI (MSI-H), those showing instability in two markers as low MSI (MSI-L), and those showing no instability as microsatellite-stable (MSS).
Cell culture
CaCo2, HCT116, HT29, SW480 and SW620 cells were acquired from ATCC (American Type Cell Culture) and cultured as appropriate at 37°C in 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) and Minimum Essential Medium (MEM) Alpha media, supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 ng/ml streptomycin (Invitrogen Life Technologies, Milan Italy).
Immunoblot detection
CaCo2, HCT116, HT 29, SW480 and SW620 cells were lysed in a 2× Laemmli Sample buffer [250 mM Tris-HCl, pH 6.8; 500 mM DTT; 10% sodium dodecyl sulfate (SDS), 0.5% bromophenol blue and 50% glycerol) supplemented with protease inhibitor cocktail (1 mM phenylmethanesulphonylfluoride and 1 mM sodium orthovanadate). After boiling at 100°C for 3 min, equal amount of proteins were loaded on 10% polyacrylamide gels and separated by electrophoresis. Protein transfer was performed on PVDF membrane (Millipore S.p.A. Milan Italy). Membranes were blocked with 5% skim milk in wash buffer (20 mM Tris-HCl, pH 7.6, 140 mM NaCl, 0.1% Tween 20) and incubated with the specific primary antibodies diluted in blocking solution, at appropriate dilutions. Following three washes, membranes were incubated with a secondary goat anti-mouse or goat anti-rabbit horseradish peroxydase-conjugated antibody (BioRad, Hercules, CA, USA) diluted 1:3.000 in wash buffer. After three further washes, proteins were revealed by chemiluminescence (ECL, Amersham Biosciences AB, Uppsala, Sweden) and the signal detected on an X-ray film (Amersham Biosciences AB, Uppsala, Sweden). Rabbit polyclonal anti-TIMELESS antibody (ab72458) was purchased from ABCAM (Cambridge, UK). Mouse anti-β-Actin antibody (F-3022) was purchased from Sigma-Aldrich (Milan, Italy).
Fluorescent in situ hybridization analysis
Each colorectal cancer cell line was tested by fluorescent in situ hybridization (FISH) to detect copy number gains of the chromosomal region (12q13.3) harboring the TIMELESS gene (chr12:56,810,157-56,843,200). BAC (Bacterial Artificial Chromosomes) clones encompassing both the ~200 kb upstream and downstream regions to TIMELESS were selected according to the latest release (February 2009, GRCh37/hg19) of the University of California Santa Cruz (UCSC) Human Genome Browser (http://genome.ucsc.edu). The probes used were RP11-888B6 (chr12:56,517,568-56,727,528), RP11-348M3 (chr12:56,684,981-56,848,839) and RP11-243C3 (chr12: 56,872,287-57,047,110) (Figure 6A). Metaphase spreads were prepared as previously described [53] and FISH experiments were carried out as reported elsewhere [54]. Briefly, 600 ng of each probe were directly labeled with Cy3-, Cy5-, or FITC-conjugated dUTP by nick translation. Hybridization was performed at 37°C in 2X saline-sodium citrate (SSC), 50% (v/v) formamide, 10% (wt/vol) dextran sulfate, 5 μg Cot-1 DNA (Bethesda Research Laboratories, Gaithersburg, MD, USA), and 3 μg sonicated salmon sperm DNA in a volume of 10 μl. Three post-hybridization washes were performed at 60°C in 0.1XSSC. Chromosomes were identified by 4′,6-diamidino-2-phenylindole (DAPI) staining. Digital images were obtained using a Leica DMRXA epifluorescence microscope equipped with a cooled CCD camera (Princeton Instruments, Boston, MA, USA). Cy3 (red; New England Nuclear, Boston, MA, USA), FITC (green; Fermentas Life Sciences, Milan, Italy), Cy5 (IR; New England Nuclear), and DAPI (blue) fluorescence signals, were detected using specific filters, and recorded separately as gray-scale images. Pseudocoloring and images merging were performed by use of Adobe Photoshop software (Adobe Systems, Seattle, WA).
Computational prediction of miR-139-5p seed regions on TIMELESS mRNA
Putative miR-139-5p binding sites in TIMELESS mRNA were predicted by the RNAHybrid (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/) [55] and RNA22 algorithms (https://cm.jefferson.edu/rna22/) [56] using the mature miRNA sequence reported in miRbase v.21 and the TIMELESS mature mRNA sequence found on RefSeq ad using default settings in both cases.
Cell transfection assays
CaCo2, HCT116, HT29, SW480 and SW620 cells at 70–80% confluence were transiently transfected with synthetic miR-139–5p mimic (MSY0000250, QIAGEN) or miR-139-5p inhibitor (MIN0000250, QIAGEN) or the appropriate scrambled controls (AllStar or mirScript Inhibitor-Negative Control) using HiPerfect Transfection Reagent (QIAGEN; Hilden, Germany), following the manufacturer’s instructions. Transfection efficiency was estimated by qRT-PCR evaluating miR-139-5p vs RNU6B expression levels.
Cell proliferation assay
Cell number and density of viable cells were determined using CAsy Cell Counter (Innovatis, Sittingbourne, UK) as previously reported [57]. Briefly, each sample (cell suspension) was prepared three times in CAsyTon (Innovatis) buffer, followed by triplicate measurements of 200 μl sample volume. Viable cells were measured by Casy Cell Counter by excluding all counts that were of a size smaller that 10 μm (dead cells and debris). Three biological replicates were prepared and each assayed in triplicate and the results expressed as mean ± standard deviation (SD).
Apoptosis assay
Apoptosis assay was performed as previously reported [58] CaCo2, SW480 and SW620 cells were transfected with miR-139-5p mimic or inhibitor or Controls 48 h at 37°C. Apoptosis was evaluated by AnnexinV-FITC and Propidium Iodide stained cells using Apoptosis Detection Kit (BD Biosciences), according to the manufacturer’s protocols. All flow cytometry results were analyzed with FACSuite Software v.1.0.5.3841 (BD Biosciences). Three biological replicates were prepared and each assayed in triplicate and the results expressed as mean ± standard deviation (SD).
Cell migration and colony forming assay
Cell migration and colony forming assay were performed as previously reported [59]. Briefly, to perform the plate colony–forming assay, 24 hours after transfection, cells were trypsinized and seeded in six-well plates (5 × 103 cells); after 7 days, cells were fixed, stained and photographed under microscope. The area of single colonies was then measured by ImageJ software (National Institutes of Health Image, http://rsbweb.nih.gov/nih-image). We also performed Transwell cell migration assay. Briefly, cells after 24 h transfection were trypsinized and adjusted to 8 × 105 cells/ml of cell suspension. Cells in a 200-μl volume were seeded into the upper chamber of Transwell, and added 800 μl medium with 20% FBS in the lower chamber. Cells were then cultured at 37°C for 48 h and the cells on the surface of the up chamber were swapped with cotton swap and the cells under the surface of the low chamber were stained with crystal violet (0.1%). Cells were then photographed under an inverted microscope for capturing pictures and counted for migrated cell numbers. Three biological replicates were prepared and the results expressed as mean ± standard deviation (SD).
Statistical analysis
Raw signals of probes were background corrected, log-transformed to achieve normal distribution and then normalized using the RMA algorithm. Differential expression between samples was assessed by paired t-test. A miRNA was considered differentially expressed if its fold-change of expression was significantly greater than 1.5 or lower than −1.5. Correlation of expression was measured by Pearson Product Moment Correlation (PPMC) and the Pearson correlation coefficient r for all the probesets of tumor and non-tumor tissue. Only the pairs of human probesets with r significantly greater than 0.8 or lower than −0.8, were taken into account for the subsequent analysis steps. Associations between gene expression level and phenotypic characteristics were evaluated by the Pearson’s chi-squared test. Survival rates were calculated by the Kaplan–Meier method for analysis of censored data. Subgroup analyses were performed after splitting the subjects into high/low expressing, based on the median value of the gene of interest. A p-value < 0.05 was considered statistically significant. All analyses were performed using the SPSS v17 Statistical Package (SPSS, Chicago, IL, USA), the R framework and Partek Genomics Suite 6.6 (Partek Inc., St. Louis, MO, USA).
ACKNOWLEDGMENTS
We would like to thank Stefano Castellana for providing excellent technical assistance.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest with respect to the authorship and/or publication of this article.
FUNDING
The study was supported by the “5x1000” voluntary contribution and by a grant to GM from the Italian Ministry of Health through Department of Medical Sciences, Division of Internal Medicine and Chronobiology Unit, IRCCS Scientific Institute and Regional General Hospital “Casa Sollievo della Sofferenza”, Opera di Padre Pio da Pietrelcina, San Giovanni Rotondo (FG), Italy (RC1203ME46, RC1302ME31, RC1403ME50, RC1504ME53, RC1603ME43) and to CTS from AIRC (IG15413).
Authors’ contributions
GM conceived the study; TM, MRV and DC performed statistical and bioinformatic analysis; GM, FT, AdP, LS, GB, VC contributed biopsies, plasmid vectors, cell lines and performed cell culture experiments; AP, AG, OP, MC, RR, CT, TC performed microarrays, qRT-PCR, immunoblotting and in vitro experiments; DT, CTS and GeM performed FISH experiments; GM, AdP, ADC, TC, TM, DC, LS and VC analyzed the data and wrote the manuscript. All authors have read and approved the final manuscript. All authors have read and approved the journal’s authorship agreement.
REFERENCES
1. Rozen P, Liphshitz I, Barchana M. The changing epidemiology of colorectal cancer and its relevance for adapting screening guidelines and methods. Eur J Cancer Prev. 2011; 20:46–53.
2. Sasieni PD, Shelton J, Ormiston-Smith N, Thomson CS, Silcocks PB. What is the lifetime risk of developing cancer: the effect of adjusting for multiple primaries. Br J Cancer. 2011; 105:460–465.
3. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
4. Bass J, Takahashi JS. Circadian Integration of metabolism and energetics Science 2010; 330:1349–1354.
5. Bass J. Circadian topology of metabolism. Nature. 2012; 491:348–356.
6. Mazzoccoli G, Pazienza V, Vinciguerra M. Clock genes and clock-controlled genes in the regulation of metabolic rhythms. Chronobiol Int. 2012; 29:227–251.
7. Cermakian N, Lange T, Golombek D, Sarkar D, Nakao A, Shibata S, Mazzoccoli G. Crosstalk between the circadian clock circuitry and the immune system. Chronobiol Int. 2013; 30:870–888.
8. Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet. 2011; 74:175–230.
9. Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. 2012; 74:246–260.
10. Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010; 72:517–549.
11. Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, Liddle C, Auwerx J, Downes M, et al. Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature. 2012; 485:123–127.
12. Scully AL, Kay SA: Time flies for Drosophila. Cell. 2000, 100:297–300.
13. Benna C, Bonaccorsi S, Wülbeck C, Helfrich-Förster C, Gatti M, Kyriacou CP, Costa R, Sandrelli F. Drosophila timeless2 is required for chromosome stability and circadian photoreception. Curr Biol. 2010; 20:346–352.
14. Cho WH, Kang YH, An YY, Tappin I, Hurwitz J, Lee JK. Human Tim-Tipin complex affects the biochemical properties of the replicative DNA helicase and DNA polymerases. 2013; 110:2523–2527.
15. Dheekollu J, Wiedmer A, Hayden J, Speicher D, Gotter AL, Yen T, Lieberman PM. Timeless links replication termination to mitotic kinase activation. PLoS One. 2011; 6:e19596.
16. Engelen E, Janssens RC, Yagita K, Smits VA, van der Horst GT, Tamanini F. Mammalian TIMELESS is involved in period determination and DNA damage-dependent phase advancing of the circadian clock. PLoS One. 2013; 8:e56623.
17. Eide EJ, Vielhaber EL, Hinz WA, Virshup DM. The Circadian Regulatory Proteins BMAL1 and Cryptochromes Are Substrates of Casein Kinase Iε. J. Biol. Chem. 2002; 277:17248–17254.
18. Lamia KA, Sachdeva UM, Ditacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, Thompson CB, Evans RM. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009; 326:437–440.
19. Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008; 134:329–340.
20. Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008; 134:317–328.
21. Liu J. Control of protein synthesis and mRNA degradation by microRNAs. Curr Opin Cell Biol. 2008; 20:214–221.
22. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011; 12:99–110.
23. Chang KH, Miller N, Kheirelseid EA, Lemetre C, Ball GR, Smith MJ, Regan M, McAnena OJ, Kerin MJ. MicroRNA signature analysis in colorectal cancer: identification of expression profiles in stage II tumors associated with aggressive disease. Int J Colorectal Dis. 2011; 26:1415–1422.
24. Gauger MA, Sancar A. Cryptochrome, circadian cycle, cell cycle checkpoints, and cancer. Cancer Res. 2005; 65:6828–6834.
25. Mazzoccoli G, Panza A, Valvano MR, Palumbo O, Carella M, Pazienza V, Biscaglia G, Tavano F, Di Sebastiano P, Andriulli A, Piepoli A. Clock gene expression levels and relationship with clinical and pathological features in colorectal cancer patients. Chronobiol Int. 2011; 28:841–851.
26. Zeng ZL, Luo HY, Yang J, Wu WJ, Chen DL, Huang P, Xu RH. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res. 2014; 20:1042–1052.
27. Fang L, Yang Z, Zhou J, Tung JY, Hsiao CD, Wang L, Deng Y, Wang P, Wang J, Lee MH. Circadian Clock Gene CRY2 Degradation Is Involved in Chemoresistance of Colorectal Cancer. Mol Cancer Ther. 2015; 14:1476–1487.
28. Mazza T, Mazzoccoli G, Fusilli C, Capocefalo D, Panza A, Biagini T, Castellana S, Gentile A, De Cata A, Palumbo O, Stallone R, Rubino R, Carella M, et al. Multifaceted enrichment analysis of RNA-RNA crosstalk reveals cooperating micro-societies in human colorectal cancer. Nucleic Acids Res. 2016; 44:4025–4036.
29. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20.
30. Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008; 455:64–71.
31. Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 23:175–205
32. Yang M, Lee JE, Padgett RW, Edery I. 2008. Circadian regulation of a limited set of conserved microRNAs in Drosophila. BMC Genomics. 2007; 9:83.
33. Cheng HY, Papp JW, Varlamova O, Dziema H, Russell B, Curfman JP, Nakazawa T, Shimizu K, Okamura H, Impey S, Obrietan K. microRNA modulation of circadian-clock period and entrainment. Neuron. 2007; 54:813–829
34. Na YJ, Sung JH, Lee SC, Lee YJ, Choi YJ, Park WY, Shin HS, Kim JH. Comprehensive analysis of microRNA-mRNA co-expression in circadian rhythm. Exp Mol Med. 2009; 41:638–647.
35. Gao J, Li N, Dong Y, Li S, Xu L, Li X, Li Y, Li Z, Ng SS, Sung JJ, Shen L, Yu J. miR-34a-5p suppresses colorectal cancer metastasis and predicts recurrence in patients with stage II/III colorectal cancer. Oncogene. 2015; 34:4142–4152.
36. Vishnubalaji R, Hamam R, Abdulla MH, Mohammed MA, Kassem M, Al-Obeed O, Aldahmash A, Alajez NM. Genome-wide mRNA and miRNA expression profiling reveal multiple regulatory networks in colorectal cancer. Cell Death Dis. 2015; 6:e1614.
37. Mogilyansky E, Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013; 20:1603–1614.
38. Li Y, Choi PS, Casey SC, Dill DL, Felsher DW. MYC through miR-17–92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014; 26:262–272
39. Guo J, Miao Y, Xiao B, Huan R, Jiang Z, Meng D, Wang Y. Differential expression of microRNA species in human gastric cancer versus non-tumorous tissues. J Gastroenterol Hepatol. 2009; 24:652–657
40. Hiroki E, Akahira J, Suzuki F, Nagase S, Ito K, Suzuki T, Sasano H, Yaegashi N. Changes in microRNA expression levels correlate with clinicopathological features and prognoses in endometrial serous adenocarcinomas. Cancer Sci. 2010; 101:241–249.
41. Fridman E, Dotan Z, Barshack I, David MB, Dov A, Tabak S, Zion O, Benjamin S, Benjamin H, Kuker H, Avivi C, Rosenblatt K, Polak-Charcon S, et al. Accurate molecular classification of renal tumors using microRNA expression. J Mol Diagn. 2010; 12:687–696
42. Krishnan K, Steptoe AL, Martin HC, Pattabiraman DR, Nones K, Waddell N, Mariasegaram M, Simpson PT, Lakhani SR, Vlassov A, Grimmond SM, Cloonan N. miR-139-5p is a regulator of metastatic pathways in breast cancer. RNA. 2013; 19:1767–1780.
43. Dettmer MS, Perren A, Moch H, Komminoth P, Nikiforov Y, Nikiforova M. MicroRNA profile of poorly differentiated thyroid carcinomas. J Mol Endocrinol. 2014; 52:181–189.
44. Gotter AL, Suppa C, Emanuel BS. Mammalian TIMELESS, Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol. 2007; 366:36–52.
45. Leman AR, Noguchi C, Lee CY, Noguchi E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J Cell Sci. 2010; 123:660–670.
46. Leman AR, Noguchi E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle. 2012; 11:3945–3955.
47. Mazzoccoli G, Laukkanen MO, Vinciguerra M, Colangelo T, Colantuoni V. A Timeless Link Between Circadian Patterns and Disease. Trends Mol Med. 2016; 22:68–81.
48. Piepoli A, Tavano F, Copetti M, Mazza T, Palumbo O, Panza A, di Mola FF, Pazienza V, Mazzoccoli G, Biscaglia G, Gentile A, Mastrodonato N, Carella M, et al. Mirna Expression Profiles Identify Drivers In Colon And Pancreatic Cancers. PLoSOne. 2012; 7:e33663.
49. Piepoli A, Palmieri O, Maglietta R, Panza A, Cattaneo E, Latiano A, Laczko E, Gentile A, Carella M, Mazzoccoli G, Ancona N, Marra G, Andriulli A. The expression of leucine-rich repeat gene family members in colorectal cancer. Exp Biol Med (Maywood). 2012; 237:1123–1128.
50. Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions, Nature Methods. 2015; 12: 697.
51. Borgatti SP, Mehra A, Brass DJ, Labianca G. Network analysis in the social sciences. Science. 2009; 323:892–895.
52. Mazzoccoli G, Tomanin R, Mazza T, D’Avanzo F, Salvalaio M, Rigon L, Zanetti A, Pazienza V, Francavilla M, Giuliani F, Vinciguerra M, Scarpa M. Circadian transcriptome analysis in human fibroblasts from Hunter syndrome and impact of iduronate-2-sulfatase treatment. BMC Med Genomics. 2013; 6:3.
53. Dahlén A, Mertens F, Rydholm A, Brosjö O, Wejde J, Mandahl N, PanagopoulosI: Fusion, disruption, and expression of HMGA2 in bone and soft tissue chondromas. Mod Pathol. 2003; 16:1132–1140.
54. Surace C, Pedeutour F, Trombetta D, Burel-Vandenbos F, Rocchi M, Storlazzi CT Episomal amplification of MYCN in a case of medulloblastoma. Virchows Archive. 2008; 452:491–497.
55. Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517.
56. Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006; 126:1203–1217.
57. Mazzoccoli G, Colangelo T, Panza A, Rubino R, De Cata A, Tiberio C, Valvano MR, Pazienza V, Merla G, Augello B, Trombetta D, Storlazzi CT, Macchia G, et al. Deregulated expression of cryptochrome genes in human colorectal cancer. Mol Cancer. 2016; 15:6.
58. Colangelo T, Polcaro G, Ziccardi P, Muccillo L, Galgani M, Pucci B, Rita Milone M, Budillon A, Santopaolo M, Mazzoccoli G, Matarese G, Sabatino L, Colantuoni V. The miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis. 2016; 7:e2108.
59. Colangelo T, Fucci A, Votino C, Sabatino L, Pancione M, Laudanna C, Binaschi M, Bigioni M, Maggi CA, Parente D, Forte N, Colantuoni V. MicroRNA-130b promotes tumor development and is associated with poor prognosis in colorectal cancer. Neoplasia. 2013; 15:1218–1231.