
INTRODUCTION
Gliomas are the most common and aggressive primary tumors in adults [1, 2]. Based on the histological appearance, gliomas can be subdivided into astrocytomas, oligodendrogliomas, and mixed oligoastrocytomas [3]. In 2007, World Health Organization (WHO) further subclassified gliomas into grade I (pilocytic astrocytomas), grade II (diffuse infiltrating low-grade gliomas), grade III (anaplastic gliomas, AA), and grade IV (glioblastomas multiforme, GBM) depending on the degree of aggressiveness [4].
In current clinical practice, histological classification is a critical prognostic factor that determines the choice of therapy. The response to therapy and the overall survival (OS) of glioma patients varies in different histological subtypes and grades [4]. Generally, oligodendrogliomas have a better prognosis than mixed oligoastrocytomas, and astrocytomas have the worst prognosis [3]. The median survival time is only 1.6 and 0.4 years for grades III and IV gliomas, respectively [5]. However, histological classification has a limited role in the treatment decision and prediction of individual outcomes due to their subjective criteria and lack of reproducibility [6]. Therefore, recent molecular genetic analyses, such as 1p/19q co-deletion and isocitrate dehydrogenase 1 (IDH1) mutation, have been extensively investigated to develop more objective approaches. Unfortunately, molecular genetic approaches are also limited by the heterogeneity within the tumor groups [7].
Recently, microarray gene analytic tools have been developed for diverse cancers for diagnosis, prognosis or prediction of therapeutic response [8–12]. In various cancers, there are several reports on gene expression profiles for its classification, prognosis and identification of biological processes including cell differentiation and proliferation [13–16]. However, most of the studies on glioma were performed on small number of patients and combined probes of diverse microarray chips. If the patients number is small, gene-expression profiles can vary according to the microarray platform and the analytic strategy, resulting in increased bias [17]. In previously published reports, the gene expression analyses on glioma also have a limitation due to the lack of reflected histology and molecular heterogeneity. In addition, well-defined target genes which predict chemotherapy response in glioma are rare.
In this study, we identified 89 common genes from diverse microarray chips using relatively large number of patients and investigated whether the 89-gene signature could be robustly validated in independent and combined data sets. Moreover, we attempted to establish a prediction model by incorporating the 89 gene set into other clinicohistological factors and molecular markers. Herein, we reported the 89-gene signature that could predict the survival of patients as well as their response to chemotherapy.
RESULTS
Significant association of prognosis with two groups found by hierarchical clustering
We selected four microarray and one RNA-seq data sets, which consisted of GSE16011, TCGA, GSE4412 and GSE4271. GSE16011 was used as the training data set because it had enough number of patients with clinical information such as grade, chemotherapy, radiotherapy, and gene mutation. Detailed informations for these data sets were described in Materials and Methods (Table 1).
Table 1: Clinical and histological characteristics of patients with glioma
Variable |
EUMC GSE16011 |
TCGA |
UCLA GSE4412 |
MDAS GSE4271 |
|---|---|---|---|---|
Patients (n) |
264 |
342 |
85 |
77 |
Male |
177 |
210 |
32 |
52 |
Female |
87 |
132 |
53 |
25 |
Age (years) |
51 (11–82) |
59 (10–89) |
42 (18–82) |
45 (22–82) |
Grade (n) |
||||
I |
6 |
|||
II |
23 |
|||
III |
84 |
26 |
21 |
|
IV |
151 |
342 |
59 |
56 |
Adjuvant chemotherapy (n) |
||||
Yes |
27 |
258 |
||
No |
168 |
61 |
||
N/A |
69 |
23 |
||
Radiotherapy |
||||
Yes |
193 |
280 |
||
No |
51 |
|||
N/A |
71 |
11 |
To generate a potential molecular classifier of glioma, genes with an expression level of at least 2-fold difference relative to median value were selected from the training data set. Then, hierarchical clustering was performed and the results revealed two major groups (n = 154 and n = 110) of glioma that differed in gene expression patterns (Supplementary Figure S1). Next, a stringent threshold cut-off (p < 0.001 and 2.5 fold difference) was applied, and 129 genes whose expression was tightly associated with the two groups were selected (Supplementary Figure S2). Because 89 genes were common among all training and validation data sets, they were used as prognostic signature (Figure 1A). To evaluate groups’ prognosis, Kaplan–Meier survival curves were plotted and the log-rank test showed significant difference in overall survival (OS) (p < 0.001, Figure 1B). Patients were classified into high and low risk groups by risk relied on differences in OS in the training set.
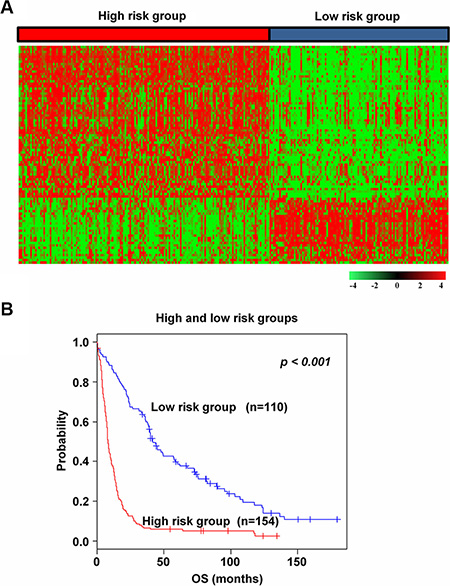
Figure 1: Survival analysis of the training data set. (A) The heatmap of the median centered 89 genes’ expression profiles (red, relative high expression; green, relative low expression) between high and low risk groups in the training data set. (B) Kaplan-Meier plots of overall survival (OS) of the two groups in the training data set. The p values were computed by the log-rank test.
A previous report showed that patients with grade II survived for more than 5 years, whereas the median survival time of patients in grade III was 1.6 years, indicating malignant gliomas [5]. The Kaplan–Meier plots and the log-rank test showed significantly different OS in all of the grades in training data set (p < 0.001, Supplementary Figure S3). Unfortunately, patients in grades II and III could not be stratified in the training set contrary to general findings in histological classification. However, our gene signature demonstrated a prognosticvalue beyond the standard clinical classification of grades.
Prognostic gene signature and clinical relevance
To investigate the association between prognostic gene signature and clinicohistological characteristics, including gender, grade and survival, Chi-square (χ2) test was performed in training data set (Table 2). The grade (p = 0.03) was significantly correlated to our gene signature, while other covariates were not associated. To evaluate prognostic accuracy of the 89-gene signature in combination with covariates, including patient age at diagnosis, gender, grade, and adjuvant chemotherapy, univariate and multivariate Cox proportional hazards regression analyses were performed using the training data set. In both of univariate and multivariate analyses, grade was significantly associated with OS (HR: 2.66, 95% CI 2.11–3.36, p = 1.2e–16 and HR: 1.65, 95% CI 1.24–2.21, p = 0.001, respectively). Notably, the 89-gene signature showed stronger prognostic ability over histological grade (HR: 0.27, 95% CI 0.20–0.36, p = 3.8e–18 and HR: 0.23, 95% CI 0.15–0.34, p = 8.8e–18, respectively) in both of univariate and multivariate analyses (Table 3). No significant difference was obtained in other covariates.
Table 2: Clinical and histological feature of two subgroups of gliomas patients in the EUMC (n = 264)
Variable |
High risk group |
Low risk group |
p-value |
|---|---|---|---|
No. of Patients |
154 |
110 |
0.42 |
Male |
102 |
75 |
|
Female |
52 |
35 |
|
Grade |
0.03 |
||
I |
5 |
1 |
|
II |
22 |
1 |
|
III |
66 |
18 |
|
IV |
61 |
90 |
Table 3: Univariate and multivariate Cox proportional hazard regression analyses of OS in the EUMC (n = 264)
Variable |
Univariate |
Multivariate |
||
|---|---|---|---|---|
HR (95% CI) |
p Value |
HR (95% Cl) |
p Value |
|
Gender (Male or Female) |
0.91 (0.69–1.19) |
0.511 |
1.02 (0.74–1.40) |
0.88 |
Age (< 40, > 40) |
2.77 (2.02–3.78) |
< 0.001 |
1.75 (1.22–2.51) |
0.002 |
Adjuvant chemotherapy |
1.50 (0.96–2.32) |
0.070 |
1.50 (0.96–2.35) |
0.070 |
Grade (I, II, III, IV) |
2.66 (2.11–3.36) |
1.2e–16 |
1.65 (1.24–2.21) |
0.001 |
Gene signature (High/Low risk group) |
0.27 (0.20–0.36) |
3.8e–18 |
0.231 (.15–.34) |
8.8e–18 |
Validation of prognostic gene expression signature in independent validation data sets
To evaluate the robustness of the newly identified 89-gene signature, validation processes were done on three independent microarray and one RNA-seq data sets of glioma. A flow chart of the validation procedure was described in Figure 2A. During leave-one-out cross-validation (LOOCV), the specificity and the sensitivity for predicting groups in all validation data sets were 0.94 and 0.93, respectively. To identify whether the gene signature could be a more accurate prediction model, we validated in the combined three validation data sets. As expected, the gene signature significantly classified patients into high and low risk groups (p = 4.9e–10, Figure 2B). Also, Kaplan-Meier plots predicted significant differences in prognosis in all independent validation data sets: TCGA (p = 0.001, Figure 2C), UCLA (p = 0.0002, Figure 2D) and MDAS (p = 0.005, Figure 2E). We also validated RNA-seq data from TCGA based on the 89-gene signature (p = 0.035, Supplementary Figure S4).
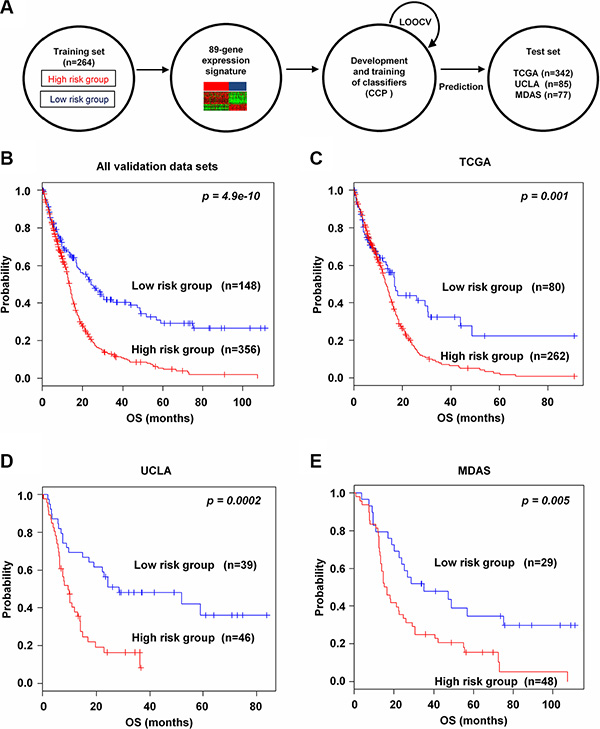
Figure 2: Prognostic significance of the 89-gene signature in independent validation data sets. (A) Schematic overview of the strategy used for the construction of the prediction model and evaluation of predicted outcomes in three independent data sets by the 89-gene signature. (B) All combined validation data sets were stratified by the 89-gene signature into two groups. The p values were computed by the log-rank test. (C–E) Kaplan-Meier survival plots of overall survival (OS) of the two groups in three independent data sets: TCGA, UCLA, and MDAS.
Association of the 89-gene signature with molecular pathway and mutation
To investigate whether the 89-gene signature could further stratify glioma patients associated with 1p/19q and IDH1 status, subset analyses were performed only in the training data set, because of the available clinical information. In both 1p/19q co-deletion and wild type groups, the 89-gene signature successfully classified patients into high and low risk groups (p = 2.16e–06 and p = 1.35e–11, Figure 3A–3B, respectively). Similarly, the 89-gene signature significantly classified patients with IDH1 mutation (p = 6.00e–04, Figure 3C) and wild type groups into high and low risk groups (p = 4.57e–09, Figure 3D). Consistent with previous reports demonstrating that the 1p/19q co-deletion and IDH1 mutation generally have favorable prognosis [18–20], our study classified most patients in these groups into low risk, eighty seven (85.3%) and fifty five (70.5%) patients, respectively. On the contrary, the 1p/19q and IDH1 wild type groups were classified into high risk, one hundred sixteen (68.6%) and ninety eight (74.2%) patients, respectively.
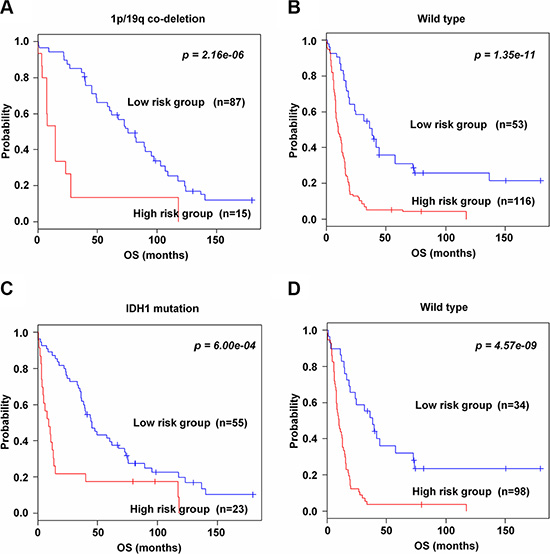
Figure 3: Significant association of the 89-gene signature with molecular pathways and mutation in the training data set. (A–B) Kaplan-Meier curves of patients in 1p/19q co-deletion and wild type groups. (C–D) IDH1 mutation and wild type groups in the training data set. Patients were classified by the 89-gene signature. The p values were computed by the log-rank test.
Subset classification of age groups by the 89-gene signature
To investigate the association of the 89-gene signature with age, patients were classified into under 40 (young patients) and over 40 years of age (old patients) groups. Patients who are diagnosed with gliomas, younger age at diagnosis is a strong predictor of longer patient survival. In both age groups, the 89-gene signature significantly stratified patients in the combined training and validation data sets into high and low risk groups (p = 3.00e–04 and p = 7.7e–16, Figure 4A–4B, respectively). Consistent with recent report showing that patients under 40 years old have more favorable prognosis than patients over 40 years old [21], our study classified most patients under 40 years as low risk and over 40 years of age as high risk. One hundred thirteen (64.6%) patients were classified into low risk in under 40 years of age and 448 (75.5%) patients were classified into high risk in over 40 years of age group. In addition, the gene signature significantly classified patients in over 40 years, even not in under 40 years, in the training set (p < 0.001, Supplementary Figure S5A–S5B). Similar results were shown in patients in both groups in validation data sets (p = 6.00e–04 and p = 0.006, Supplementary Figure S5C-S5D, respectively).
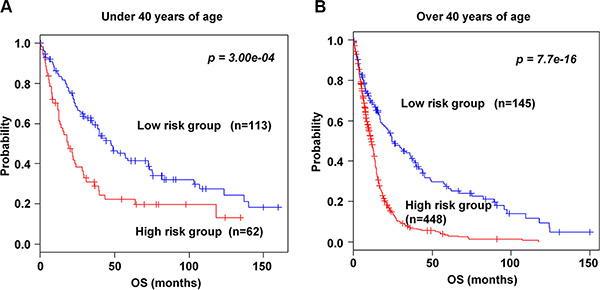
Figure 4: Kaplan-Meier survival analysis of the 89-gene signature in age. (A) Patients under 40 years of age group in the combined training and validation data sets were stratified into high and low risk groups. (B) Patients over 40 years of age group in the combined training and validation data sets were stratified into high and low risk groups. The p values were computed by the log-rank test.
Prognostic subclassification of patients with grades III and IV by the 89-gene signature
To evaluate whether the 89-gene signature could classify patients by grade into high and low risk groups in the training and validation data sets, patients were combined in each grade; I (n = 6), II (n = 23), III (n = 131) and IV (n = 608). The 89-gene signature clearly stratified all combined patients into high and low risk groups (p < 0.001, Figure 5A). It could not significantly classify combined patients in grades I and II into two groups (p = 0.29, Figure 5B). However, the 89-gene signature significantly separated patients in grade III and IV into high and low risk groups (p = 3.18e–12 and p = 2.12e–06, Figure 5C–5D, respectively). Thirty one (23.6%) and 100 (76.3%) patients were classified into high and low risk in grade III, respectively. Four hundreds seventy (77.3%) and 138 (22.7%) patients were classified in grade IV into high and low risk. Similar results were obtained in patients in grade III and IV in training set (Supplementary Figure S6A–S6B, respectively) and all combined validation data sets (Supplementary Figure S6C–S6D, respectively).
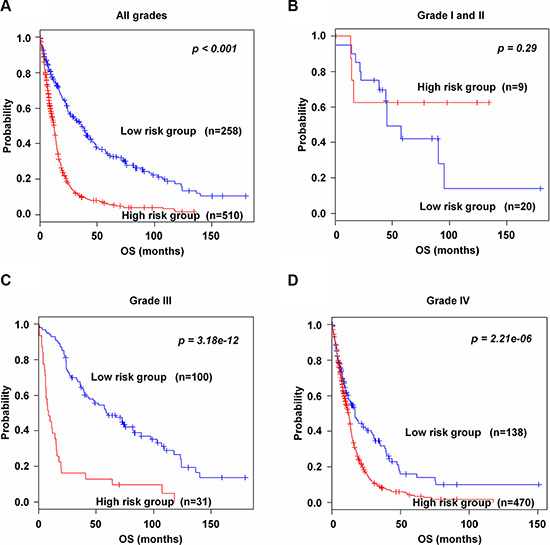
Figure 5: Kaplan-Meier survival analysis of the 89-gene signature in grades. (A) Patients in all grades in the combined training and validation data sets. (B) Patients in grades I and II in the combined training and validation data sets. (C–D) Patients in grades III and IV in the combined training and validation data sets. Each group was classified into high and low risk groups. The p values were computed by the log-rank test.
Association of the 89-gene signature with benefits of adjuvant chemotherapy and radiotherapy
To find the association of the 89-gene signature with response to chemotherapy and radiotherapy, subset analyses were performed in TCGA data set, for which therapeutic information were available. As shown in Figure 6A and 6B, patients in both high and low risk groups benefitted from radiation therapy (p = 1.03e–27 and p = 4.89e–05, respectively). By incorporating the 89-gene signature into chemotherapy information, only high risk group was shown to obtain benefit compared to patients without chemotherapy. In high risk group, over half of patients (80.5 %) benefited from chemotherapy (p = 3.33e–16, Figure 6C). On the contrary, low risk group did not have significant benefit from chemotherapy (p = 0.062, Figure 6D). Interestingly, high risk group had better response to combined therapies (p = 0.02, Figure 6E), while low risk groups did not get benefit from combined therapies (p = 0.74, Figure 6F). Additionally, with the EUMC data set, similar results were observed in chemotherapy and radiotherapy (Supplementary Figure S7A–S7F).
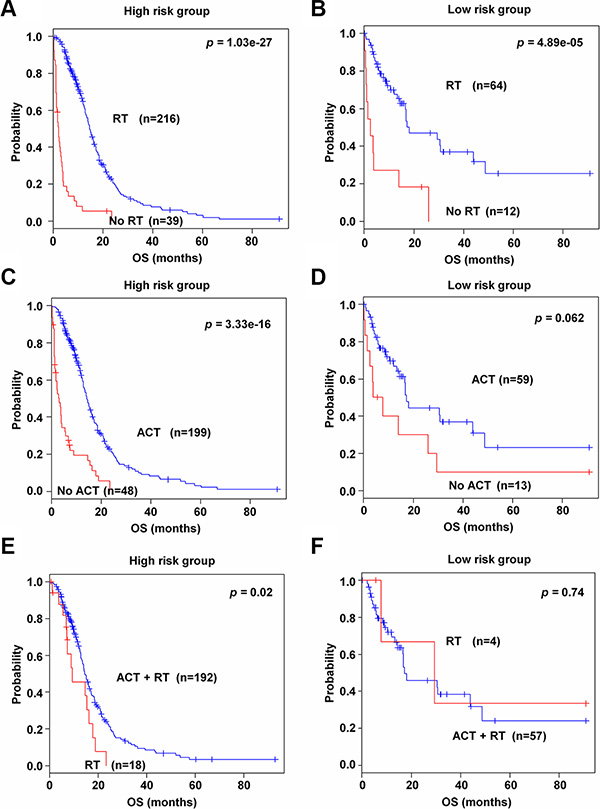
Figure 6: Kaplan-Meier survival analysis of the 89-gene signature with adjuvant chemotherapy and radiation therapy. (A–B) Patients in high and low risk groups with radiotherapy in TCGA data set. (C–D) Patients in high and low risk groups with chemotherapy in TCGA data set. (E–F) Patients in high and low risk groups with combined therapies in TCGA data set. Each group was stratified according to chemotherapy, radiotherapy, and combined therapies. The p values were computed by the log-rank test.
Protein network and gene ontology in the 89-gene signature
The 89 probe sets corresponded to 89 annotated genes (Supplementary Table S1). So, we could clarify protein interactions of 89 genes in the 89-gene signature. To understand how these genes could be involved in networks related to glioma, we performed analysis using the STRING database that was able to elaborate physical and functional associations among proteins. The results showed that 81 out of 89 genes were closely connected in a single network (Supplementary Figure S8). We also investigated interactions using the Ingenuity Pathway Analysis (IPA) software and found putative networks related to NF-KB, STAT3, and AP-1 transcription factors (Supplementary Figure S9). In addition, 23 genes were involved in the activation of these transcription factors pathways. To identify the biological function of genes in the 89-gene signature, we performed GO enrichment analysis in DAVID, and then revealed 89 significant biological pathways involved in glioma. The top 61 important pathways with p < 0.05 were selected. They are involved in multiple cancer-related processes, such as cell-cell signaling, wound healing, inflammatory response (Supplementary Table S2).
DISCUSSION
Several reports have been published to predict prognosis in gliomas [3, 13–15]. Studies based on gene expression profiles have been reported to classify patients according to known prognostic factors; however, no report has yet predicted chemotherapy response in gliomas. An unsupervised clustering approach was integrated to construct the 89-gene signature from the training set. The 89-gene signature was validated for its prognostic significance in three microarray independent data sets (TCGA, UCLA and MDAS) and one RNA-seq data (TCGA). Univariate and multivariate analyses showed significant association of the prognostic gene signature with survival after adjusting clinical covariates. In addition, the 89-gene signature has the ability to identify patients benefiting from chemo-and radiotherapy. Therefore, the established gene signature might be helpful in clinical management.
Tumor grade is the major clinical variable used to make glioma treatment decisions [3]. However, heterogeneity observed in therapeutic response among patients within the same histological grade indicates that histological classification is not an adequate predictor of the clinical behavior of a tumor [22]. Moreover, histological classification is based on subjective criteria, and lacks reproducibility [6]. In the present study, although patients in low risk group more often were presented with grade IV glioma than those in high risk group (Table 2), patients in high risk group showed poorer OS than those in low risk group. Considering the above results, we concluded that the 89-gene signature was more correlated with survival than histological classification, a finding which is also supported by previous reports [6, 14]. Additionally, the 89-gene signature stratified patients with grades III and IV glioma into high and low risk groups. Interestingly, patients with grades I and II glioma could not be stratified into two risk groups using this signature. Although we cannot definitively assert why the 89-gene signature could not stratify patients with grades I and II glioma, we hypothesize that this is due to the small number of patients analyzed in this study. Considering that grades III and IV gliomas are rapidly progressive malignant tumors [4], it is noteworthy that the 89-gene signature could stratify these patients even harboring tumor heterogeneity.
Recently described molecular markers, such as IDH1 mutation and 1p/19q co-deletion, are considered predictive of clinical outcomes for glioma patients [18, 23]. The IDH1 mutation is a strong predictor of outcome irrespective of histological type and grade [18, 19]. However, several studies have shown a higher rate of malignant transformation in IDH-mutated low grade glioma than in wild-type IDH1 tumors, showing that a subset of patients with the IDH1 mutation are characterized as having secondary glioblastoma [24, 25]. Additionally, oligodendrogliomas with 1p/19q co-deletion have been shown to progress more slowly and respond better to treatment [20, 26, 27]. However, the prognostic value of 1p/19q co-deletion in glioblastoma remains unknown [28]. Considering of diverse studies, molecular markers should be further defined according to molecular heterogeneity. In the present study, the 89-gene signature could stratify patients with IDH1 mutation and 1p/19q co-deletion status into high and low risk groups. In agreement with previous studies, most patients with the IDH1 mutation and 1p/19q co-deletion were classified as low risk, whereas most patients who had wild type for these markers were classified as high risk. However, it is meaningful that we could precisely predict the clinical behavior of tumors in individual patients within same molecular state. Additionally, these results suggest that the 89-gene signature has overcome the limitations of genetic molecular approaches in assessing tumor heterogeneity. By incorporating our gene signature into clinical information, patients could get more benefits in clinical practice.
Recent reports have shown that glioblastoma patients under the age of 40 years survive longer than those patients over 40 years [21]. In the present study, young age (< 40) was an independent significant prognostic factor. However, it remains unclear whether young age confers a favorable prognosis for children with glioblastoma [29]. One previous study reported that the prognosis was unfavorable for pediatric patients with glioblastoma. Another study reported favorable prognosis for glioblastoma patients over 70 years old, with a 2-year overall survival rate of 20% [30]. Considering the heterogeneity in prognosis within a single age group, more defined prognostic factors might be required to stratify such patients. Our study showed that the 89-gene signature could further stratify both young and old patients into high and low risk groups. As expected based on previous reports, most young patients (64.6%) were classified as low risk, whereas most old patients (75.5%) were classified as high risk, suggesting that the 89-gene signature could be of clinical value by subclassifying patients within same age group and so helping to make treatment plan decisions for individual patients.
Generally, adjuvant chemotherapy (ACT) and radiotherapy (RT) after surgery constitute the standard treatment in glioblastoma grade IV [31, 32]. However, addition of ACT to RT remains controversial in grade III anaplastic gliomas [3]. Unfortunately, no gene signatures related to ACT sensitivity in glioma have been discovered yet, although one prior study involving the EUMC data set with small number of patients who had ACT demonstrated that some genes were implicated in chemoradiation sensitivity [13]. Our subset analysis of patients with available chemotherapy information suggested that the 89-gene signature could predict patients who would benefit from ACT. Our study showed that patients in high risk group had significantly improved outcome with ACT, whereas patients in low risk group did not get significant benefit from ACT in all patients with available treatment data sets. Considering that high risk group carried a poorer prognosis than those in low risk group, our 89-gene signature has the potential to facilitate clinical decisions on using ACT for grade III glioma because of a poorer prognosis. The utility of the gene signature for treatment management in glioma still needs to be further evaluated in a prospective ACT clinical trial.
Most of the identified 89 genes play a critical role in aggressiveness, angiogenesis, local invasion, migration, and proliferation. These genes included COL1A2, COL3A1, CLO6A3 [33], FABP7 [16, 34-37], GDF-15, SH3GL2 [38, 39], ADM, VEGFA, and PTX3 [40, 41]. Our gene signature also contained proneural genes, BMP2, DCX, IGFBP2, PDPN, and PLAT, which are associated with anaplastic oligodendroglioma harboring 1p/19q co-deletion [42]. The proneural genes indicate a better prognosis of malignant glioma [43]. The 89-gene signature consists of a number of hypoxia and inflammation-related genes such as AKR1C3 [44], PTX3, PLAT [45] and IGFBP2 [46], showing that these two inseparable hallmarks are involved in tumor progression [47, 48] and play significant roles in glioma pathogenesis. Purinergic signaling related genes such as GPR17 [49], VEGFA [50] and CCL2 [51] are involved in inflammation leading to glioma growth [52, 53]. In addition, BCAT1 was reported to promote cell proliferation in gliomas carrying wild-type IDH1 [54]. Our gene signature also possessed several genes related to clinical characteristics of recurrent glioblastoma. Furthermore, genes such as AKR1C3, ETNPPL, FXYD1, SH3GL2, SH3GL3, SNAP91, and SYT1 have important role in reprograming and are also involved in drug resistance [55]. Additionally, IPA revealed that most of the genes were controlled by NF-KB, STAT3, and AP-1 transcription factors. These transcription factors play a vital role in cancer and are important regulators of immune and inflammatory functions [56–58]. Finally, many novel genes including CHRNA9, CNGA3, FERMT1, FZD7, GABRB3, METTL7B, USD5, and SYT4 were also identified, suggesting that our 89-gene signature contains novel information which may provide new biomarkers to assist in clinical decision making concerning new opportunities for targeted treatment of individual patients.
In conclusion, we identified the 89-gene signature as a highly discriminative predictor of prognosis. The prognostic value of the 89-gene signature was statistically significant in a reliable and reproducible manner across independent and combined data sets. Furthermore, our study revealed that patients could be stratified into high and low risk groups with different OS regardless of histology classification and molecular markers. In addition, the 89-gene signature might suggest which patients would benefit from ACT. Therefore, we propose that the 89-gene signature has an independent and accurate prognostic value for clinical use. Also, this study offers new opportunities for novel targeted treatment of individual patients.
MATERIALS AND METHODS
Patients and gene expression data
All gene expression data sets were obtained from the National Center for Biotechnology Information Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) and The Cancer Genome Atlas database (http://cancergenome.nih.gov/). Data were selected based on following criteria: raw CEL files and clinical information of patients with survival event and time. The raw data were preprocessed using robust multiarray averaging (RMA) method for normalization. Gene expression data from the GSE16011 (n = 264, Erasmus University Medical Center (EUMC)) was used as the training data set. The Cancer Genome Atlas data (TCGA, n = 342), GSE4412 (n = 85, University of California Los Angeles (UCLA)) and GSE4271 (n = 77, MD Anderson (MDAS)) were used as validation data sets (Table 1). To test the prognostic significance of gene expression signature, we used only gene expression data with available survival data. The information of adjuvant chemotherapy and radiotherapy were available for only 285 and 473 patients respectively from EUMC and TCGA data sets. In addition, RNA-seq data from TCGA (n = 165) was also used as validation data set (https://genome-cancer.ucsc.edu).
Development of the prognostic gene expression signature
A gene expression signature was developed from the EUMC data set. Gene expression was generated by using the Affymetrix GeneChip Human Genome U133 Plus 2.0 chip set. Differentially expressed probe sets were identified among two classes using a random-variancet test. Differences of probe sets’ expression between two classes were considered statistically significant if their p value was less than 0.001. A global and permutation tests were performed to investigate whether the expression profiles differed between the classes. The cluster analysis was performed with Cluster 3.0 (http://bonsai.hgc.jp/~mdehoon/software/cluster) and Tree View (http://www.eisenlab.org/eisen/). Although initially 129 probe sets were identified for constructing prediction models in t test analysis, only 89 probe sets, which were shared in both U133 Plus 2.0 and U133A, were used for all validation data sets (Supplementary Table S1).
Validation of the prognostic gene expression signature
The validation of the gene signature was accomplished on independent data sets. Gene expression data from validation data sets were adjusted individually by subtracting the median expression value across the samples. To integrate each validation data set for constructing prediction models, 89 probe sets were aligned in each data set. To further refine this model and to sub-stratify the predicted outcomes, Compound Covariate Predictor (CCP) was utilized as a class prediction algorithm [59]. Gene expression data in the training set were combined to form a classifier according to CCP. The robustness of the classifier was estimated by the misclassification rate determined during the leave-one-out cross-validation (LOOCV) in the training set. During prediction, the cross-validation process omitted one sample at a time. For each sample omitted, the entire analysis was repeated from scratch, including the determination of genes which were univariately significant on the reduced training sample. From that gene list, a multivariate predictor was constructed and applied to the sample that was omitted. The program recorded whether that prediction was correct or not. This was repeated, omitting all of the samples one at a time.
Kaplan–Meier survival analyses were performed after the patient classification into two predicted subgroups, and Chi-square (χ2) and log-rank tests were used to evaluate the survival risk between two predicted subgroups of patients. The uni-and multi-variate Cox proportional hazards model were used to evaluate independent prognostic factors associated with survival, gene signature, tumor grade, age, and adjuvant chemotherapy as covariates.
Pathway analysis
To perform pathway analysis on these expressed genes between subtypes, we used GO term enrichment analysis, using the Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resource (http://david.abcc.ncifcrf.gov/home.jsp). Pathway analysis was performed to map genes to the Biological Process (BP) categories of GO and then calculate the significance of overrepresented categories in the selected gene list. The p value less than 0.05 was used to define significant pathways.
Gene network analysis
Protein-protein interactions were predicted using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database v10.0 (http://www.string-db.org/). Proteins were linked based on the following six criteria; neighborhood, gene fusion, co-occurrence, co-expression, experimental evidence and existing databases [60]. Ingenuity Pathways Analysis (IPA) (http://www.ingenuity.com) was also used for gene network analysis, using a global molecular network developed from information contained in the Ingenuity Knowledge Database. Identified gene networks were ranked to score (z-score = 02).
Statistical methods of microarray data
Microarray data and heatmap were analyzed using BRB-Array Tools Version 3.0 (http://linus.nci.nih.gov/BRB-ArrayTools.html). All other statistical analyses were accomplished in the R language environment (http://www.r-project.org) and Statistical Package for Social Sciences (SPSS) software (version 20, SPSS Inc, Chicago, IL, USA). In all statistical analyses, p value of less than 0.05 was considered significant.
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2011-0030072).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest in the research.
REFERENCES
1. Nakazato Y. [The 4th Edition of WHO Classification of Tumours of the Central Nervous System published in 2007]. [Article in Japanese]. No Shinkei Geka. 2008; 36:473–491.
2. Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004; 64:6892–6899.
3. Ricard D, Idbaih A, Ducray F, Lahutte M, Hoang-Xuan K, Delattre JY. Primary brain tumours in adults. Lancet. 2012; 379:1984–1996.
4. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
5. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005; 64:479–489.
6. van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol. 2010; 120:297–304.
7. Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004; 64:6503–6510.
8. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004; 351:2817–2826.
9. Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, Durnez A, Demetris AJ, Thorgeirsson SS. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004; 40:667–676.
10. Tomida S, Koshikawa K, Yatabe Y, Harano T, Ogura N, Mitsudomi T, Some M, Yanagisawa K, Takahashi T, Osada H, Takahashi T. Gene expression-based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene. 2004; 23:5360–5370.
11. Akter S, Choi TG, Nguyen MN, Matondo A, Kim JH, Jo YH, Jo A, Shahid M, Jun DY, Yoo JY, Nguyen NN, Seo SW, Ali L, et al. Prognostic value of a 92-probe signature in breast cancer. Oncotarget. 2015; 6:15662–15680. doi: 10.18632/oncotarget.3525.
12. Nguyen MN, Choi TG, Nguyen DT, Kim JH, Jo YH, Shahid M, Akter S, Aryal SN, Yoo JY, Ahn YJ, Cho KM, Lee JS, Choe W, et al. CRC-113 gene expression signature for predicting prognosis in patients with colorectal cancer. Oncotarget. 2015; 6:31674–31692. doi: 10.18632/oncotarget.5183.
13. Gravendeel LA, Kouwenhoven MC, Gevaert O, de Rooi JJ, Stubbs AP, Duijm JE, Daemen A, Bleeker FE, Bralten LB, Kloosterhof NK, De Moor B, Eilers PH, van der Spek PJ, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009; 69:9065–9072.
14. Nutt CL, Mani DR, Betensky RA, Tamayo P, Cairncross JG, Ladd C, Pohl U, Hartmann C, McLaughlin ME, Batchelor TT, Black PM, von Deimling A, Pomeroy SL, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003; 63:1602–1607.
15. Shirahata M, Oba S, Iwao-Koizumi K, Saito S, Ueno N, Oda M, Hashimoto N, Ishii S, Takahashi JA, Kato K. Using gene expression profiling to identify a prognostic molecular spectrum in gliomas. Cancer Sci. 2009; 100:165–172.
16. Liang Y, Diehn M, Watson N, Bollen AW, Aldape KD, Nicholas MK, Lamborn KR, Berger MS, Botstein D, Brown PO, Israel MA. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci U S A. 2005; 102:5814–5819.
17. Zhang L, Yoder SJ, Enkemann SA. Identical probes on different high-density oligonucleotide microarrays can produce different measurements of gene expression. BMC Genomics. 2006; 7:153.
18. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–773.
19. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, El Hallani S, Boisselier B, Mokhtari K, Hoang-Xuan K, Delattre JY. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol. 2009; 27:4150–4154.
20. Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, Fink K, Souhami L, Laperriere N, Curran W, Mehta M. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013; 31:337–343.
21. Lee Y, Scheck AC, Cloughesy TF, Lai A, Dong J, Farooqi HK, Liau LM, Horvath S, Mischel PS, Nelson SF. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med Genomics. 2008; 1:52.
22. Kros JM, Gorlia T, Kouwenhoven MC, Zheng PP, Collins VP, Figarella-Branger D, Giangaspero F, Giannini C, Mokhtari K, Mork SJ, Paetau A, Reifenberger G, van den Bent MJ. Panel review of anaplastic oligodendroglioma from European Organization For Research and Treatment of Cancer Trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss, and correlations with outcome. J Neuropathol Exp Neurol. 2007; 66:545–551.
23. Idbaih A, Marie Y, Pierron G, Brennetot C, Hoang-Xuan K, Kujas M, Mokhtari K, Sanson M, Lejeune J, Aurias A, Delattre O, Delattre JY. Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol. 2005; 58:483–487.
24. Kanamori M, Kumabe T, Shibahara I, Saito R, Yamashita Y, Sonoda Y, Suzuki H, Watanabe M, Tominaga T. Clinical and histological characteristics of recurrent oligodendroglial tumors: comparison between primary and recurrent tumors in 18 cases. Brain Tumor Pathol. 2013; 30:151–159.
25. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A, Felsberg J, Wolter M, Mawrin C, Wick W, Weller M, Herold-Mende C, Unterberg A, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009; 118:469–474.
26. Lassman AB, Iwamoto FM, Cloughesy TF, Aldape KD, Rivera AL, Eichler AF, Louis DN, Paleologos NA, Fisher BJ, Ashby LS, Cairncross JG, Roldan GB, Wen PY, et al. International retrospective study of over 1000 adults with anaplastic oligodendroglial tumors. Neuro Oncol. 2011; 13:649–659.
27. van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Haaxma-Reiche H, Kros JM, van Kouwenhoven MC, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol. 2006; 24:2715–2722.
28. Kaneshiro D, Kobayashi T, Chao ST, Suh J, Prayson RA. Chromosome 1p and 19q deletions in glioblastoma multiforme. Appl Immunohistochem Mol Morphol. 2009; 17:512–516.
29. Yazici G, Zorlu F, Cengiz M, Ozyigit G, Eren G, Yuce D, Varan A, Akyuz C, Akalan N, Gurkaynak M. High-grade glioma in children and adolescents: a single-center experience. Childs Nerv Syst. 2016; 32:291–297.
30. Minniti G, Scaringi C, Baldoni A, Lanzetta G, De Sanctis V, Esposito V, Enrici RM. Health-related quality of life in elderly patients with newly diagnosed glioblastoma treated with short-course radiation therapy plus concomitant and adjuvant temozolomide. Int J Radiat Oncol Biol Phys. 2013; 86:285–291.
31. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009; 10:459–466.
32. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352:987–996.
33. St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, Kinzler KW. Genes expressed in human tumor endothelium. Science. 2000; 289:1197–1202.
34. Nutt CL, Matthews RT, Hockfield S. Glial tumor invasion: a role for the upregulation and cleavage of BEHAB/brevican. Neuroscientist. 2001; 7:113–122.
35. Wang LH, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci. 1996; 16:6197–6207.
36. Zheng C, Heintz N, Hatten ME. CNS gene encoding astrotactin, which supports neuronal migration along glial fibers. Science. 1996; 272:417–419.
37. Nutt CL, Zerillo CA, Kelly GM, Hockfield S. Brain enriched hyaluronan binding (BEHAB)/brevican increases aggressiveness of CNS-1 gliomas in Lewis rats. Cancer Res. 2001; 61:7056–7059.
38. Roth P, Junker M, Tritschler I, Mittelbronn M, Dombrowski Y, Breit SN, Tabatabai G, Wick W, Weller M, Wischhusen J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin Cancer Res. 2010; 16:3851–3859.
39. Qu S, Yao Y, Shang C, Xue Y, Ma J, Li Z, Liu Y. MicroRNA-330 is an oncogenic factor in glioblastoma cells by regulating SH3GL2 gene. PLoS One. 2012; 7:e46010.
40. Desbaillets I, Diserens AC, de Tribolet N, Hamou MF, Van Meir EG. Regulation of interleukin-8 expression by reduced oxygen pressure in human glioblastoma. Oncogene. 1999; 18:1447–1456.
41. Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, Morgelin M, Bourseau-Guilmain E, Bengzon J, Belting M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A. 2013; 110:7312–7317.
42. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006; 9:157–173.
43. Ducray F, Idbaih A, de Reynies A, Bieche I, Thillet J, Mokhtari K, Lair S, Marie Y, Paris S, Vidaud M, Hoang-Xuan K, Delattre O, Delattre JY, et al. Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer. 2008; 7:41.
44. Ragel BT, Couldwell WT, Gillespie DL, Jensen RL. Identification of hypoxia-induced genes in a malignant glioma cell line (U-251) by cDNA microarray analysis. Neurosurg Rev. 2007; 30:181–187; discussion 187.
45. Locatelli M, Ferrero S, Martinelli Boneschi F, Boiocchi L, Zavanone M, Maria Gaini S, Bello L, Valentino S, Barbati E, Nebuloni M, Mantovani A, Garlanda C. The long pentraxin PTX3 as a correlate of cancer-related inflammation and prognosis of malignancy in gliomas. J Neuroimmunol. 2013; 260:99–106.
46. Minchenko DO, Kharkova AP, Karbovskyi LL, Minchenko OH. Expression of insulin-like growth factor binding protein genes and its hypoxic regulation in U87 glioma cells depends on ERN1 mediated signaling pathway of endoplasmic reticulum stress. Endocr Regul. 2015; 49:73–83.
47. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011; 364:656–665.
48. Eltzschig HK, Bratton DL, Colgan SP. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat Rev Drug Discov. 2014; 13:852–869.
49. Hennen S, Wang H, Peters L, Merten N, Simon K, Spinrath A, Blattermann S, Akkari R, Schrage R, Schroder R, Schulz D, Vermeiren C, Zimmermann K, et al. Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci Signal. 2013; 6:ra93.
50. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA. Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem Pharmacol. 2006; 72:19–31.
51. Braganhol E, Kukulski F, Levesque SA, Fausther M, Lavoie EG, Zanotto-Filho A, Bergamin LS, Pelletier J, Bahrami F, Ben Yebdri F, Fonseca Moreira JC, Battastini AM, Sevigny J. Nucleotide receptors control IL-8/CXCL8 and MCP-1/CCL2 secretions as well as proliferation in human glioma cells. Biochim Biophys Acta. 2015; 1852:120–130.
52. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014; 509:310–317.
53. Idzko M, Ferrari D, Riegel AK, Eltzschig HK. Extracellular nucleotide and nucleoside signaling in vascular and blood disease. Blood. 2014; 124:1029–1037.
54. Tonjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AH, Karra D, Piro RM, Felsberg J, Addington A, Lemke D, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013; 19:901–908.
55. Kwon SM, Kang SH, Park CK, Jung S, Park ES, Lee JS, Kim SH, Woo HG. Recurrent Glioblastomas Reveal Molecular Subtypes Associated with Mechanistic Implications of Drug-Resistance. PLoS One. 2015; 10:e0140528.
56. Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006; 124:823–835.
57. Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994; 264: 1415–1421.
58. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003; 3:859–868.
59. Radmacher MD, McShane LM, Simon R. A paradigm for class prediction using gene expression profiles. J Comput Biol. 2002; 9:505–511.
60. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015; 43:D447–452.