
INTRODUCTION
Osteosarcoma (OS) is the most common bone tumor in children, representing 6% of all childhood cancers [1]. Although great efforts have been made to understand the underlying mechanisms of OS carcinogenesis, survival of OS has reached a plateau and the prognosis of advanced OS remains poor [2]. Therefore, novel diagnostic biomarkers and therapeutic targets for OS are needed.
MicroRNAs (miRNAs) are a class of small non-coding RNAs, which regulate gene function by targeting mRNA for translational repression or degradation [3]. miRNAs have been implicated in the pathogenesis of a variety of human diseases, including neoplasms [4]. Over-expression of oncogenic miRNAs or low expression of tumor suppressor miRNAs promote tumorigenesis [5]. A number of studies have reported an association between miRNA dysregulation and OS. In comparisons of miRNA expression profiles, miR-9, miR-18a, miR-145 and miR-451 were consistently decreased in both cell lines and clinical samples compared with normal bone tissues [6–8].
Among these miRNAs, miR-145 is a highly conserved gene among different species and is highly expressed in vascularized tissues such as liver, lung, and heart [9, 10]. Loss of miR-145 results in elevated tumor proliferation, migration, and survival, as well as increased leukocyte adhesion and disorganized tumor vasculature [11]. Recently, a cohort of genes related to cancer pathways have been identified and validated as targeted genes of miR-145, such as P70S6K, C-MYC, PAI-1, FASCIN, and SOX-2 [12–16], suggesting that miR-145 is an oncogene that plays a pivotal role in the initiation and progression of cancer. However, the function of miR-145 in OS is largely unknown.
Friend leukemia virus integration 1 (FLI-1), a member of the ETS transcription factor family, is the target of insertional activation by the Friend murine leukemia virus and is preferentially expressed in vascular endothelial cells, embryonic tissue, and tumors [17, 18]. FLI-1 plays a critical role in normal development, hematopoiesis, and oncogenesis by functioning either as a transcriptional activator or repressor [19–22]. Knocking down FLI-1 expression in cancer cells leads to growth inhibition and cell death, demonstrating a possible therapeutic approach to induce tumor suppression [23, 24]. Anti-FLI-1 compounds have demonstrated strong anti-leukemic activity in a mouse model that over-expresses FLI-1, making it possible to target FLI-1 as an anti-tumor treatment [25]. However the role of the miR-145/FLI-1 pathway has not been elucidated in osteosarcoma.
In this study, we explored the expression of miR-145 in 13 OS tumor tissues using PCR. We then completed a series of cellular functional experiments to investigate the role of miR-145 and its target genes in OS cell growth.
RESULTS
Down-regulation of miR-145 and up-regulation of FLI-1 expression in OS tissues and cell lines
To explore the expression of miR-145 and FLI-1 in OS carcinogenesis, we examined 13 pairs of OS and matched normal tissues using TaqMan RT-PCR analysis. Relative to matched normal tissues, more than half of the OS tissues exhibited under-expression of miR-145 mRNA and all of the OS tissues had high expression of FLI-1 mRNA (Figure 1A & 1C). Expression of miR-145 in OS tissues was lower than in normal tissues (Figure 1C). We next explored the expression of miR-145 and FLI-1 in four OS cell lines (HOS, Saos-2, U2OS, and MG-63 ). Compared with the normal human osteoblast cell line (NHOst), miR-145 expression was reduced in the four OS cell lines (Figure 1B). Interestingly, expression of FLI-1 mRNA in all of the four OS cell lines was higher than in the NHOst cells (Figure 1D). These results suggested that the under-expression of miR-145 and over-expression of FLI-1 mRNA are common features of OS tissues and cell lines.
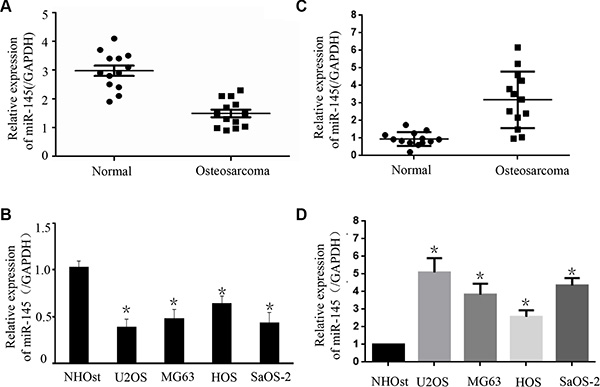
Figure 1: The expression of miR-145 and FLI-1 in osteosarcoma tissues and cell lines. The expression of miR-145 in 13 pairs of OS tissues and adjacent normal bone tissues was detected by TaqMan qRT-PCR. Data are shown as the ratio of OS tissues relative to normal bone tissues. (A) Statistical analysis of relative miR-145 expression in OS tissues and compared normal tissues. Compared with normal bone tissues, the expression of miR-145 in tumor tissues was down-regulated; and (B) Using qRT-PCR, the expression of miR-145 in four OS cell lines (HOS, Saos-2, U2OS and MG-63) was analyzed. The expression of miR-145 in these OS cell lines was down-regulated relative to normal osteoblast cells (NHOst). (C) Expression of FLI-1 mRNA was analyzed in the OS tissues by qRT-PCR. (D) Using qRT-PCR, the expression of FLI-1 mRNA in four OS cell lines (HOS, Saos-2, U2OS and MG-63) and one normal cell line was analyzed. FLI-1 was up-regulated compared with the normal osteoblast cells (NHOst). *p < 0.01 compared with normal tissues or normal cells.
miR-145 suppresses OS cell growth
The under-expression of miR-145 in both human OS tissues and OS cell lines prompted us to explore its possible biological role in OS carcinogenesis. We sought to compensate for the reduced expression of miR-145 by transfecting MG-63 and U2OS cells with miR-145 mimic. The intracellular level of miR-145 was about 4-fold higher in U2OS and MG-63 cells transfected with the miR-145 mimic relative to the scramble control group (Figure 2A). Cell proliferation was measured using CCK-8 assays. Ectopic expression of miR-145 led to a decrease in cell proliferation in both OS cell lines (Figure 2B). As proliferation directly links to cell apoptosis, we next examined the effect of miR-145 on apoptosis. As expected, the percentage of apoptotic cells was increased in both OS cell lines upon transfection with the miR-145 mimic (Figure 2C). To further understand the molecular mechanism of miR-145-induced apoptosis in OS cell lines we performed western blots. Ectopic expression of miR-145 reduced expression of apoptosis-related proteins, caspase3 and PARP (Figure 2D) in the two OS cell lines. Finally, given that migration promotes tumor metastasis, we investigated the effects of miR-145 on OS cell migration. As shown in Figure 2E, over-expression of miR-145 in OS cells reduced the number of migrating cells, suggesting a suppressive effect of miR-145 on OS cell migration. Taken together, these results indicated that miR-145 inhibits OS cell progression, and may function as a tumor suppressor.
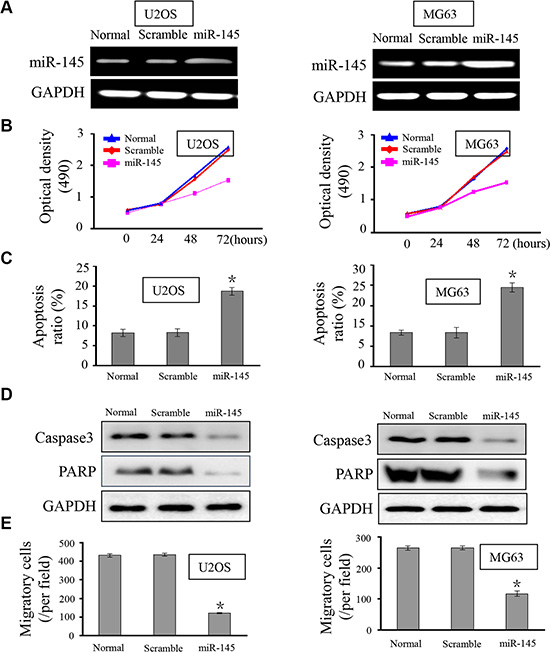
Figure 2: miR-145 suppresses OS cell proliferation, apoptosis and migration. (A) RT-PCR was performed to detect the expression of miR-145 in OS cell lines U2OS and MG-63. Upon transfection with the miR-145 mimic, expression of miR-145 in U2OS and MG-63 cells was restored; (B) CCK-8 assay was performed to analyze the effect of miR-145 on U2OS and MG-63 cell proliferation. Ectopic expression of miR-145 inhibited cell proliferation; (C) The influence of miR-145 on cell apoptosis was analyzed. Transfection with miR-145 revealed more cells undergoing early apoptosis; (D) Caspase 3 and PARP expression were assayed by western blot in the OS cell lines; (E) The effects of miR-145 on cell migration were detected using transwell assays. Over-expression of miR-145 dampened cell migratory capacity; *p < 0.001 compared with scramble group.
miR-145 regulates FLI-1 expression in OS cells
In order to verify FLI-1 as the target of miR-145, we used a luciferase reporter assay with plasmids containing the 3′UTR of human FLI-1 co-transfected with miR-145. According to miRTarBase and targetscan, there is one validated binding site and two predicted binding sites for miR-145 in the FLI-1 3′UTR. In the current study, we only analyzed the validated binding site, as shown in Figure 3A. As expected, there was reduced luciferase activity in U2OS and MG-63 cells co-transfected with pGL3-FLI-1 3′-UTR vector and miR-145 mimic compared to cells transfected with the mutant construct (Mut-1, Figure 3B), suggesting that miR-145 suppresses the transcription of FLI-1 by targeting binding sites in the 3′UTR. At 24 h post-transfection, western blot and RT-PCR analysis were performed. As shown in Figure 3C & 3D, over-expression of miR-145 led to a marked decrease in the expression of endogenous FLI-1 protein compared to OS cells transfected with control (U2OS and MG63 cell groups: both P < 0.001).
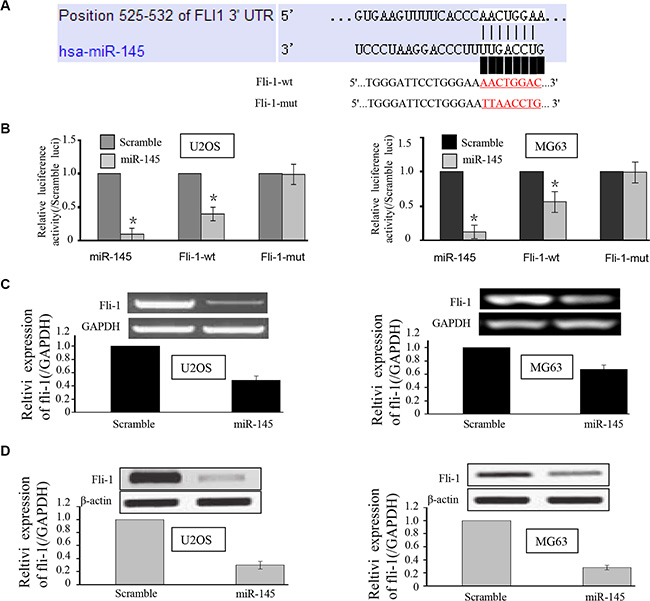
Figure 3: miR-145 targets the FLI-1 gene in U2OS and MG-63 cells. (A) Schematic representation of the FLI-1 3′UTR showing putative miRNA target sites. There was one putative binding site in the 3′UTR of miR-145; (B) Relative luciferase activity of the indicated FLI-1 reporter constructs in U2OS and MG-63 cells, co-transfected with miR-145 mimics or scramble mimics. In cells co-transfected with pGL3-FLI-1 3′-UTR vector and miR-145 mimic, luciferase activity was suppressed relative to mutant construct groups; and (C, D) qRT-PCR and Western blot assays were performed to detect the expression of FLI-1 upon transfection with miR-145 mimics or scramble mimics. Both mRNA and protein expression were suppressed upon transfection with miR-145. *p < 0.01 compared with scramble group.
miR-145 suppresses cell proliferation by targeting FLI-1
To explore whether miR-145-mediated growth inhibition in OS cells via direct targeting of FLI-1, we performed a rescue experiment. We generated a new construct containing the full ORF of the FLI-1 gene (pcDNA-FLI-1). As expected, FLI-1 expression was rescued when pcDNA-FLI-1 was transfected into U2OS and MG-63 cells that had been treated with miR-145 mimic for 24 h (Figure 4A). In agreement with the restored expression of FLI-1, increased cell proliferation (Figure 4B), accompanied by decreased cell apoptosis (Figure 4C) were also observed in OS cells transfected with pcDNA-FLI-1 following treatment with miR-145. Moreover, upon transfection with the FLI-1 construct, the suppression of miR-145-mediated cell migration (Figure 4A & 4B) in U2OS and MG-63 cells was also abolished. We also tested siRNA of FLI-1 in OS cells and performed a proliferation assay. Knock-down of FLI-1 expression by siRNA inhibited OS cell proliferation compared with the control siRNA (Figure 4D). Taken together, these results demonstrate that the repression of OS cell progression by miR-145 is a consequence of decreased FLI-1 expression.
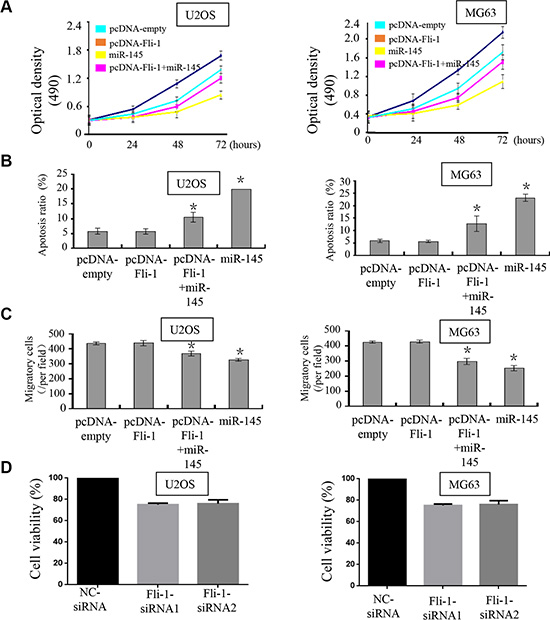
Figure 4: miR-145 suppresses tumor progression through targeting FLI-1. Upon transfection with FLI-1 construct, we rescued the expression of FLI-1 in U2OS and MG-63 cells. The expression of FLI-1 protein was validated by western blot assays; (A) CCK-8 assays were used to detect in U2OS and MG-63 cells co-transfected with miR-145 mimic and pcDNA-FLI-1-plasmid; (B) Cell apoptosis of U2OS and MG-63 cells treated as described was detected by Annexin V-PI staining; and (C) Transwell assays were performed to detect the effects on cell migration of U2OS and MG-63 cells treated as described. Upon transfection with the FLI-1 plasmid, miR-145-mediated suppression of cell proliferation and cell migration was abolished, and promotion of cell apoptosis was inhibited. (×200); (D) We detected the effect of FLI-1 siRNA in MG63 and U2OS cells by CCK8 assay. *p < 0.05 between two groups as indicated with a line.
miR-145 suppresses OS xenograft tumor growth and FLI-1 expression in vivo
To explore whether ectopic expression of miR-145 affects tumor growth in vivo, we performed a xenograft tumor model assay by subcutaneously injecting U2OS cells stably over-expressing miR-145 or scrambled miRNA in the dorsal flank of nude mice. The tumors in the group injected with U2OS cells stably over-expressing miR-145 grew at a slower rate and had smaller volumes than the scrambled control group (Figure 5A). The average bodyweight was not lower in the miR-145 over-expressing group than the scrambled control group (Figure 5D). To identify whether the expression of miR-145 and FLI-1 were changed in the xenograft tumors we used RT-PCR and IHC, respectively. Compared with the scrambled miRNA group, the expression of miR-145 was increased in the over-expressing group (Figure 5F). In contrast, the expression of FLI-1 was lower in the miR-145 over-expressing tumors (Figure 5E). These results suggested that miR-145 suppresses OS tumor growth and FLI-1 protein expression in vivo.
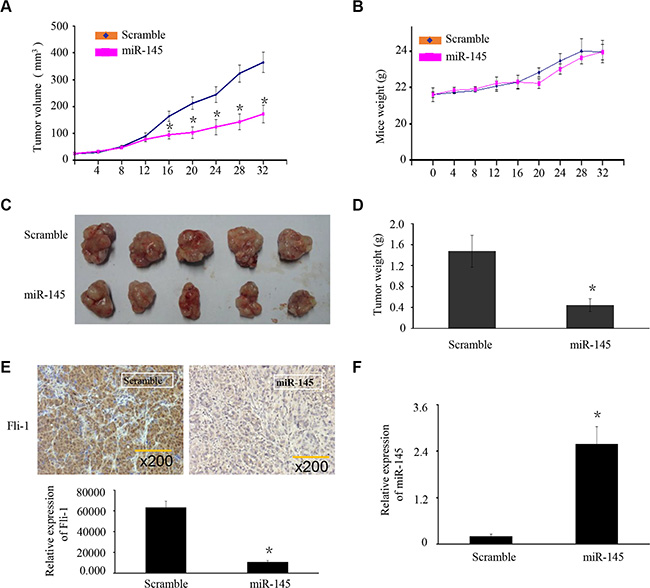
Figure 5: miR-145 suppresses OS xenograft tumor growth and FLI-1 expression in vivo. (A) U2OS cells stably over-expressing miR-145 or scrambled miRNA were subcutaneously injected into nude mice. Four weeks later, U2OS cells stably over-expressing miR-145 had smaller tumors than controls. (A) The growth curves of tumor volumes. (B) Average body weights. (C) Representative image of tumors formed. (D) Tumor weights. (E) Expression of FLI-1 in the zenograft tumor was detected by IHC. (F) Expression of miR-145 in the zenograft tumor was detected by RT-PCR. Data are presented as the mean ± SD. *P < 0.01 compared with control using the student’s t-test.
DISCUSSION
In this study, we provide important evidence in support of miR-145 functioning as a tumor suppressor by targeting the expression of FLI-1 in OS. First, we detected the expression of miR-145 in 13 paired OS tissues and normal bone tissues. Our study validated the under-expression of miR-145 in OS tissues and cell lines. Our results are consistent with the results of Xu et al, which found that miR-145 is under-expressed in bladder cancer cell lines, and that over-expression of miR-145 inhibits cell proliferation by targeting FSCN1 [13]. Along with our findings, these data suggest the tumor suppressor role of miR-145.
As miR-145 expression is decreased in OS tissues and cell lines, we sought to compensate for its loss through exogenous transfection with miR-145 mimic into U2OS and MG-63 cells. Restored expression of miR-145 markedly inhibited cell proliferation and induced cell apoptosis. Moreover, miR-145-transfected cells showed a dramatic decrease in cell migration, accompanied with suppressed expression of FLI-1, which contains a putative binding sites of miR-145 in its 3’UTR identified by the prediction program MicroCosm Targets. Using luciferase assays, we confirmed the function of these binding sites; upon transfection with miR-145, the expression of FLI-1 mRNA and protein were both suppressed. To our knowledge, this is the first observation of miR-145/FLI-1 interaction in OS cells.
FLI-1 acts as an oncogene [26, 27], and the etiology of a number of virally induced leukemias, as well as human Ewing′s sarcoma, has been associated with FLI-1 over-expression [28]. Aberrant expression of FLI-1 has been recognized as a seminal event in the initiation of certain types of malignant transformation. FLI-1 is also expressed at high levels in F-MuLV-induced erythroleukemias, malignant melanoma, small cell carcinomas of the lung and adenocarcinomas [29, 30]. Suppression of FLI-1 inhibits cell proliferation and induces cell apoptosis of human cancer cell by targeting Rb, GATA-1, and BCL-2 [31–33]. Anti-FLI-1 compounds demonstrate strong anti-leukemic activity in a mouse erythroleukemia model that over-expresses FLI-1, making it possible to target FLI-1 as a treatment strategy [34]. In this paper, we found that the activation of FLI-1 might be a consequence of constitutive suppression of miR-145. Moreover, we restored the expression of FLI-1 in miR-145-transfected cells to explore the role of FLI-1 in miR-145-mediated tumor suppression. As expected, restoring the expression of FLI-1 in OS cells abolished miR-145-mediated suppression, suggesting that FLI-1 might have a key role in miR-145 mediated OS tumor suppression.
In summary (Figure 6), this study identified FLI-1 as a novel gene target of miR-145 in OS. These results demonstrate that miR-145 acts as a tumor suppressor, targeting the 3′-UTR of FLI-1 mRNA. Down-regulation of FLI-1 has a profound effect on the growth and migration of OS cells. Its inhibition of growth in OS cells is compatible with the well-known ability of FLI-1 to stimulate cell proliferation. miR-145 regulates FLI-1 at the both the levels of protein and mRNA, although the specific mechanism requires further investigation. These studies extend our knowledge concerning FLI-1 as an oncogene involved in OS tumorigenesis.
Figure 6: Summary of the signaling pathways regulated by miR-145 and directly influencing OS cell proliferation, apoptosis, and migration.
MATERIALS AND METHODS
Specimens
Thirteen fresh specimens of osteosarcoma and matched normal tissues were collected from the Department of Orthopedics, Xiang Ya Hospital of the Central South University. The matched normal tissues were obtained 5 cm distant from the tumor margin, which were confirmed by pathologists. No patients underwent any therapy before study recruitment. Use of the tissue samples and all experiments were approved by the Institutional Ethics Committee.
Cell lines and cell culture
Human osteosarcoma cell lines (HOS, Saos-2, U2OS and MG-63) or normal osteoblast cells (NHOst) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were maintained in Dulbecco’s modified Eagle medium (DMEM, Gibco, Life Technologies, Darmstadt, Germany), supplemented with 10% fetal bovine serum (FBS; PAA, Pasching, Austria), streptomycin (100 μg/mL), and penicillin (100 U/mL). Cultures were incubated in a humidified atmosphere of 5% CO2 at 37°C.
Reverse transcription and quantitative real-time PCR
Total RNA was extracted from cultured cells, fresh OS tissues, and xenograft mouse tumors with Trizol reagent (Ambion) according to the manufacturer’s protocol. miRNA cDNA was synthesized using a One-Step PrimeScript miRNA cDNA Synthesis Kit (Takara). Quantitative real-time PCR (qRT-PCR) was performed with SYBR green Premix Ex Taq II (Takara) with a StepOne Plus Real-Time PCR System (Applied Biosystems). Expression of U6 was used as an endogenous control. Specific primers qualified with a probe for reverse transcription were used as follows: MIR-145: 5′-GTCCAGTTTTCCCAGGAATCCCT-3′; and U6: 5′-AACGCTTCACGAATTTGCGT-3′. The expression of GAPDH was used as an internal control and Oligo(dT) was used as the primer for FLI-1 and GAPDH reverse transcription. Then, qRT-PCR was performed to quantify relative expression of FLI-1 with the following primers:
FLI-1: sense, 5′-CAGTCGCCTAGCCAACCCTG and antisense, 5′-GCAATGCCG TGGAAGTCAAAT. GAPDH: sense, 5′-TCAACGACCACTTTGTCAAGC TCA-3′ and anti-sense, 5′-GCTGGTG GTCCAGGGGTC TTACT-3′. PCR efficiencies were calculated with a relative standard curve derived from a complementary DNA mixture that gave regression coefficients > 0.95. The thermal profile for the qPCR was 95°C for 1 min, followed by 35 cycles of 93°C for 15 sec, 62°C for 30 sec and 72°C for 10 sec on a Bio-Rad CFX96 qRT-PCR system (Bio-Rad, Hercules, CA, USA). All qPCR experiments were performed in triplicate.
Cell transfection and reporter gene analysis
miR-145 mimic and scramble mimic were purchased from Dharmacon (Austin, TX, USA). All oligonucleotides were transiently transfected into U2OS and MG-63 cells lines with a final concentration of 50 nM using Lipofectamine 3000 Reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Briefly, the cells were seeded in 6-well, 24-well, or 96 well plates at 50% confluence the day before transient transfection into the OS cell lines. miR-145 precursor, miR-145 inhibitor, and microRNA control (50 nM each), were used for each transfection.
Plasmid construction and primer sequences
The full-length FLI-1 gene open reading frame (ORF) was amplified by PCR and cloned into a pCDNA3.1 construct to generate the pCDNA3.1-FLI-1 construct. The empty pCDNA3.1 construct was used as a control. Primer sequences used for PCR were, sense: 5′-AAACGAGGGAAATGGGAG-3′ and anti-sense: 5′-TACCAACGGTGTCAACCTG-3′. Following bioinformatic analysis, the putative microRNA regulatory element (MRE, Figure 3A) of miR-145 was chemically synthesized and cloned into a pGL3 control vector (Promega) at the Xba1 restriction site. To construct the Luc-FLI-1-3′UTR-Full vector, the full length 3′UTR of FLI-1 was amplified from the cDNA of U2OS cells using FLI-1 PCR primers; sense: 5′CTAGAGAAGCCCATCCTGCACACT 3′ and antisense: 5′CTAGACGTTGTTTTTCCCAGAGCT 3′, and then cloned into the pGL3 control vector at the Xba1 site. To create the mutated FLI-1-3′UTR vector, eight nucleotides (TGGGATTCCTGGGAATTAACCTG) were changed for the reporter construct. All of the constructs were designed and synthesized by Cyagen Biosciences, Inc.
Proliferation assay
Before analysis of cell proliferation, U2OS and MG-63 cells were seeded into 96-well plates at a concentration of 8 × 103 cells/well. The Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan) was added to the wells at 0, 24, 48, and 72 hours post-transfection, and cells were diluted in normal culture medium at 37°C. The absorbance values of optical density (OD) in each well were measured with a microplate reader set at 490 nM (OD490). All experiments were performed three times and the average percentages of cells shown.
Apoptosis assay
Cells were transfected with miR-145 mimics or control mimics for 24 h. Forty-eight hours after transient transfection, according to the Annexin V-PI Apoptosis Detection Kit I (BD Pharmingen, CA, USA) protocol, cells were harvested and re-suspended with 500 μL of binding buffer. The cell suspension was incubated with 5 μL annexin-V-FITC and propidium iodide buffer at room temperature for 20 min. The stained cells were analyzed on a Flow Cytometer (BD Biosciences, NJ) and apoptotic cells were quantified by apoptosis ratio. The experiment was repeated three times.
Migration assays
Migration assays were carried out in modified Boyden chambers (BD Biosciences, San Jose, CA, USA) with 8 μm pore filter inserts in 24-well plates. Twenty-four hours after transfection, 3 × 105 cells suspended in serum-free DMEM were added to the upper chamber. DMEM containing 20% FBS was added to the lower chambers as a chemoattractant. Twenty-four hours later, the non-filtered cells were gently removed with a cotton swab. Filtered cells located on the lower side of the chamber were stained with crystal violet, air-dried, and photographed. The transmembrane cells in each treatment group were counted. Three independent experiments were performed.
Luciferase activity assays
The entire 3′-UTR of the FLI-1 gene was cloned into the pGL-3-vector (Promega, WI, USA) immediately downstream of the Renilla luciferase gene. Mutations in the 3′-UTR of the FLI-1 gene with miR-145 target sites deleted (Mut-1) were generated with the QuickChange Site-Directed Mutagenesis kit (Stratagene, CA, USA). Approximately 1 × 105 MG-63 cells per well were seeded into 24-well plates for 24 h before transfection. Cells were co-transfected with 50 ng pGL-3 firefly luciferase reporter, 10 ng pRL-TK Renilla luciferase reporter, and 50 nM miR-145 mimics or scramble mimics using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). A luciferase reporter construct containing the miR-145 consensus target sequence served as a positive control and the pRL-TK vector served as an internal control. Cell lysates were prepared using Passive Lysis Buffer (Promega, Madison, WI, USA) 48 h after transfection, and luciferase activity was measured using the Dual-Luciferase Reporter Assay (Promega, Madison, WI, USA). Results were normalized to the Renilla luciferase, and the scramble groups as the standard, which was divided by the treatment groups. Experiments were independently repeated three times.
Western blot analysis
Cells were lysed in RIPA buffer. Cellular proteins were collected and subjected to 10% SDS-PAGE, and transferred onto PVDF membranes (Amersham Pharmacia Biotech, Piscataway, NJ). The membranes were then incubated with specific primary antibodies. Anti-FLI-1 was purchased from Santa Cruz Biotechnology, Inc. Anti-β-ACTIN was purchased from Epitomics, Inc. This was followed by incubation with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibodies from Santa Cruz Biotechnology, and antigen-antibody complexes were visualized using the Western Bright ECL detection system (Advansta, CA).
Immunohistochemistry
Immunohistochemistry was used to detect FLI-1 expression in xenograft mouse model tumors. Sections (4 μm) were cut from formalin-fixed paraffin-embedded tissue blocks and then deparaffinized in xylene and rehydrated in successive washes of ethanol. The sections were then heated in a microwave oven at medium power for 2 minutes in citrate buffer (pH 6.0) for heat-induced epitope retrieval. The sections were subjected to blockade of endogenous peroxidase activity and nonspecific binding of the primary antibody, and then target protein localization with the first antibody and visualization with the secondary antibody and color reaction as described above. Subsequently, the slides were incubated with rabbit monoclonal antibody anti-FLI-1 (1:100 dilution), for overnight at 4°C in a moist chamber. The slides were sequentially incubated with a secondary antibody for 1 hour at room temperature, and stained with 3,3-diaminobenzidine. Finally, the sections were counterstained with Mayer’s hematoxylin, dehydrated, and mounted. A negative control was obtained by replacing the primary antibody with PBS.
Animal experiments
To determine the effect of miR-145 on OS tumor growth in a xenograft model, six-week old athymic Nu/nu nude mice were maintained in a pathogen-free environment. Five NC and five miR-145 mice groups were used (for a total of 10 mice). In brief, U2OS cells (2 × 106) were inoculated subcutaneously into the flank of the nude mice. Once palpable tumors developed (average volume 50 mm3), mice were treated with 100 nM synthetic miR-145 complexed with 100 μl transfection reagent in 50 μl PBS delivered nine times intratumorally every three days. Tumor growth was followed for 32 days from first injection until tumors reached about 400 mm3 total volume, at which time mice were euthanized. Tumor volume was calculated using the following formula: V = (width2 x length)/2. Body weights were also recorded. Primary analyses involved the comparison (for each time point separately) between the NC group and the miR-145 group. Animal experiments were approved by the Animal Research Committee of Central South University and were performed in accordance with established guidelines.
Statistical analysis
All data are expressed as the mean ± standard deviation of at least three independent experiments. Statistical analysis was carried out using SPSS 18.0 software (SPSS Inc.; Chicago, IL, USA). p-values < 0.05 were considered significant.
ACKNOWLEDGMENTS
This work was supported by the fund from the Department of Education Foundation in Hunan Province (12C0356).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
1. Pierz KA, Womer RB, Dormans JP. Pediatric bone tumors: osteosarcoma ewing’s sarcoma, and chondrosarcoma associated with multiple hereditary osteochondromatosis. J Pediatr Orthop. 2001; 21:412–418.
2. Li XP, Cao GW, Sun Q, Yang C, Yan B, Zhang MY, Fu YF, Yang LM. Cancer incidence and patient survival rates among the residents in the Pudong New Area of Shanghai between 2002 and 2006. Chin J Cancer. 2013; 32:512–519.
3. Ambros V. microRNAs: tiny regulators with great potential. CELL. 2001; 107:823–826.
4. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857–866.
5. Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006; 25:6188–6196.
6. Miao J, Wu S, Peng Z, Tania M, Zhang C. MicroRNAs in osteosarcoma: diagnostic and therapeutic aspects. Tumour Biol. 2013; 34:2093–2098.
7. Namlos HM, Meza-Zepeda LA, Baroy T, Ostensen IH, Kresse SH, Kuijjer ML, Serra M, Burger H, Cleton-Jansen AM, Myklebost O. Modulation of the osteosarcoma expression phenotype by microRNAs. Plos One. 2012; 7:e48086.
8. Wu PF, Liang JY, Yu F, Zhou ZB, Tang JY, Li KH. MiR-125b inhibits stromal cell proliferation in giant cell tumor of bone by targeting parathyroid hormone 1 receptor. Iran J Basic Med Sci. 2015; 18:705–709.
9. Meister J, Schmidt MH. miR-126 and miR-126*: new players in cancer. ScientificWorldJournal. 2010; 10:2090–2100.
10. Nikolic I, Plate KH, Schmidt MH. EGFL7 meets miRNA-126: an angiogenesis alliance. J Angiogenes Res. 2010; 2:9.
11. Liu B, Peng XC, Zheng XL, Wang J, Qin YW. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer. 2009; 66:169–175.
12. Villadsen SB, Bramsen JB, Ostenfeld MS, Wiklund ED, Fristrup N, Gao S, Hansen TB, Jensen TI, Borre M, Orntoft TF, Dyrskjot L, Kjems J. The miR-143/-145 cluster regulates plasminogen activator inhibitor-1 in bladder cancer. Br J Cancer. 2012; 106:366–374.
13. Chiyomaru T, Enokida H, Tatarano S, Kawahara K, Uchida Y, Nishiyama K, Fujimura L, Kikkawa N, Seki N, Nakagawa M. miR-145 and miR-133a function as tumour suppressors and directly regulate FSCN1 expression in bladder cancer. Br J Cancer. 2010; 102:883–891.
14. Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009; 137:647–658.
15. Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo YY. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009; 106:3207–3212.
16. Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, Li D, Lai L, Jiang BH. MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2012; 40:761–774.
17. Ben-David Y, Bernstein A. Friend virus-induced erythroleukemia and the multistage nature of cancer. Cell. 1991; 66:831–834.
18. Klemsz MJ, Maki RA, Papayannopoulou T, Moore J, Hromas R. Characterization of the ets oncogene family member, FLI-1. J Biol Chem. 1993; 268:5769–5773.
19. Truong AH, Ben-David Y. The role of FLI-1 in normal cell function and malignant transformation. Oncogene. 2000; 19:6482–6489.
20. Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000; 20:5643–5652.
21. Liu F, Walmsley M, Rodaway A, Patient R. Fli1 acts at the top of the transcriptional network driving blood and endothelial development. Curr Biol. 2008; 18:1234–1240.
22. Lakhanpal GK, Vecchiarelli-Federico LM, Li YJ, Cui JW, Bailey ML, Spaner DE, Dumont DJ, Barber DL, Ben-David Y. The inositol phosphatase SHIP-1 is negatively regulated by FLI-1 and its loss accelerates leukemogenesis. Blood. 2010; 116:428–436.
23. Cui JW, Vecchiarelli-Federico LM, Li YJ, Wang GJ, Ben-David Y. Continuous FLI-1 expression plays an essential role in the proliferation and survival of F-MuLV-induced erythroleukemia and human erythroleukemia. Leukemia. 2009; 23:1311–1319.
24. Takigami I, Ohno T, Kitade Y, Hara A, Nagano A, Kawai G, Saitou M, Matsuhashi A, Yamada K, Shimizu K. Synthetic siRNA targeting the breakpoint of EWS/FLI-1 inhibits growth of Ewing sarcoma xenografts in a mouse model. Int J Cancer. 2011; 128:216–226.
25. Li YJ, Zhao X, Vecchiarelli-Federico LM, Li Y, Datti A, Cheng Y, Ben-David Y. Drug-mediated inhibition of FLI-1 for the treatment of leukemia. Blood Cancer J. 2012; 2:e54.
26. Riggi N, Suva ML, Suva D, Cironi L, Provero P, Tercier S, Joseph JM, Stehle JC, Baumer K, Kindler V, Stamenkovic I. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008; 68:2176–2185.
27. Peter M, Magdelenat H, Michon J, Melot T, Oberlin O, Zucker JM, Thomas G, Delattre O. Sensitive detection of occult Ewing’s cells by the reverse transcriptase-polymerase chain reaction. Br J Cancer. 1995; 72:96–100.
28. Pereira R, Quang CT, Lesault I, Dolznig H, Beug H, Ghysdael J. FLI-1 inhibits differentiation and induces proliferation of primary erythroblasts. Oncogene. 1999; 18:1597–1608.
29. Athanasiou M, Mavrothalassitis G, Sun-Hoffman L, Blair DG. FLI-1 is a suppressor of erythroid differentiation in human hematopoietic cells. Leukemia. 2000; 14:439–445.
30. Torlakovic EE, Slipicevic A, Florenes VA, Chibbar R, DeCoteau JF, Bilalovic N. FLI-1 expression in malignant melanoma. Histol Histopathol. 2008; 23:1309–1314.
31. Tamir A, Howard J, Higgins RR, Li YJ, Berger L, Zacksenhaus E, Reis M, Ben-David Y. FLI-1, an Ets-related transcription factor, regulates erythropoietin-induced erythroid proliferation and differentiation: evidence for direct transcriptional repression of the Rb gene during differentiation. Mol Cell Biol. 1999; 19:4452–4464.
32. Yi H, Fujimura Y, Ouchida M, Prasad DD, Rao VN, Reddy ES. Inhibition of apoptosis by normal and aberrant FLI-1 and erg proteins involved in human solid tumors and leukemias. Oncogene. 1997; 14:1259–1268.
33. Seth A, Robinson L, Thompson DM, Watson DK, Papas TS. Transactivation of GATA-1 promoter with ETS1, ETS2 and ERGB/Hu-FLI-1 proteins: stabilization of the ETS1 protein binding on GATA-1 promoter sequences by monoclonal antibody. Oncogene. 1993; 8:1783–1790.
34. Mhawech-Fauceglia P, Herrmann FR, Bshara W, Odunsi K, Terracciano L, Sauter G, Cheney RT, Groth J, Penetrante R, Mhawech-Fauceglia P. Friend leukaemia integration-1 expression in malignant and benign tumours: a multiple tumour tissue microarray analysis using polyclonal antibody. J Clin Pathol. 2007; 60:694–700.