
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common malignancy primary cancer and the third-leading cause of cancer mortality worldwide [1]. The prevalence of this cancer shows remarkable geographic heterogeneity, with the highest rates being observed among East Asian and African populations [1, 2]. Recently, the number of cases increased dramatically in Western countries, and it has been estimated that the annual number of new cases exceeds 700,000 worldwide [3]. Despite the rapid progress in diagnostic and therapeutic modalities, the overall 5-year survival rate for HCC is extremely low (18%) [4]. Many etiological factors for HCC has been reported [5]. Nearly 80% of the HCC are associated with infections with hepatitis B virus or hepatitis C virus. Alcoholic liver diseases and non-alcoholic fatty liver diseases are also major risk factors for HCC [6]. HCC is the combined result of a multi-stage, multi factor of long-term exposure and accumulation [7–10]. Multiple studies have revealed that cancer is a genetic disease, and is thought to develop through the acquisition of genetic alterations [11–13]. Many efforts have been devoted to uncover genetic aberrations in HCC [8, 14–16], such as point mutation in p53 (TP53) and β–catenin (CTNNB1), etc. However, our understanding of genetic landscape in HCC is still far from complete and the key drivers of HCC tumourigenesis remain poorly to be understood.
Up to date, numerous works enrolling tens of thousands of subjects have been performed to examine the role of genetic variations in HCC carcinogenesis in the past two decades. Many sequence polymorphisms have been identified as potential genetic factors associated with HCC susceptibility. Notably, many studies might investigate the association between a specific polymorphism with HCC risk, however, the results of these works are not always consistent. Systematic review covering all tested polymorphisms is necessary. Here, we comprehensively evaluate the candidate-gene association studies of HCC risk, and perform meta-analyses for variants with sufficient data. We provided a systematic synopsis of our current understanding of the genetic basis of HCC susceptibility.
RESULTS
Characteristics of the eligible studies
In this work, we totally identified 505 eligible articles, comprising 282,042 subjects (case: 124,452, 44.1%). A total of 255 polymorphisms in 198 genes were eligible in our analysis (Figure 1). Most of these works (n=496, 98.22%) have been published since 2000. We conducted meta-analyses for 69 polymorphisms in 46 genes that had at least three data sources (301 eligible articles left). For the 69 main meta-analysis works, the mean sample size was 4087 (range from 607 to 14425) with an average of 7.6 independent studies.
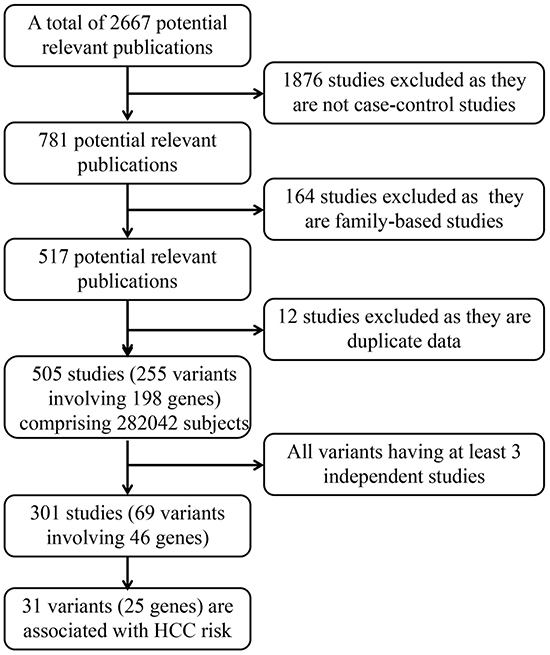
Figure 1: Profiles of literature search, meta-analysis and evaluation of cumulative evidence.
Meta-analyses
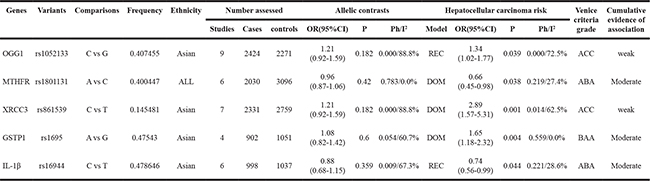
Detailed meta-analysis results were recorded for each of the polymorphisms. At first, we evaluated these polymorphisms using additive model. Among all the allele contrast meta-analysis, 21 (30%) polymorphisms in 19 genes showed nominally significant associations with HCC risk (P-value < 0.05). The number of subjects enrolled in the meta-analyses ranged from 607 to 14425 (mean: 4087, Table 1). The genotype distributions of these polymorphisms in the control group were all in accordance with the Hardy-Weinberg equilibrium (HWE). Strong associations with HCC (ORs > 2) have been detected for only one polymorphism (PNPLA3 rs738409, OR=2.01). Moderate associations with HCC (ORs 1.5-2.0 0.5-0.8) were identified for 13 polymorphisms (Table 1). Five additional variants were significantly associated with HCC risk in meta-analyses stratified by ethnicity (Table 1). Three polymorphisms (IL8 rs4073, OR=1.22; COX-2 rs20417, OR=1.94; MTHFR rs1801133 OR=1.09) had associations with HCC risk only in Asian people. One polymorphism (HFE rs1799945, OR=1.73) only showed significantly association with HCC risk among African people, and one polymorphism (mEPHX rs1051740, OR=1.46) has the association with HCC risk only in Caucasian. Next, we performed ethnicity-specific analyses using either dominant or recessive genetic models to further evaluate the associations of genetic variants with HCC risk. Another 5 significant associated variants in 5 genes were identified using either dominant or recessive models (Table 2). Hepatitis B virus (HBV) infection is particular important risk factor for HCC. Many works have documented the association between genetic polymorphisms and risk of HBV-related HCC. Here, we explored these associations of 8 polymorphisms (Supplementary Table S1). The result showed that two polymorphisms are significantly correlated with HBV-related HCC (TNF-α rs1800630, OR=1.76; IL28B rs12979860 OR=1.70).
Table 1: Genetic variants nominally significantly associated with HCC risk in meta-analyses using additive model
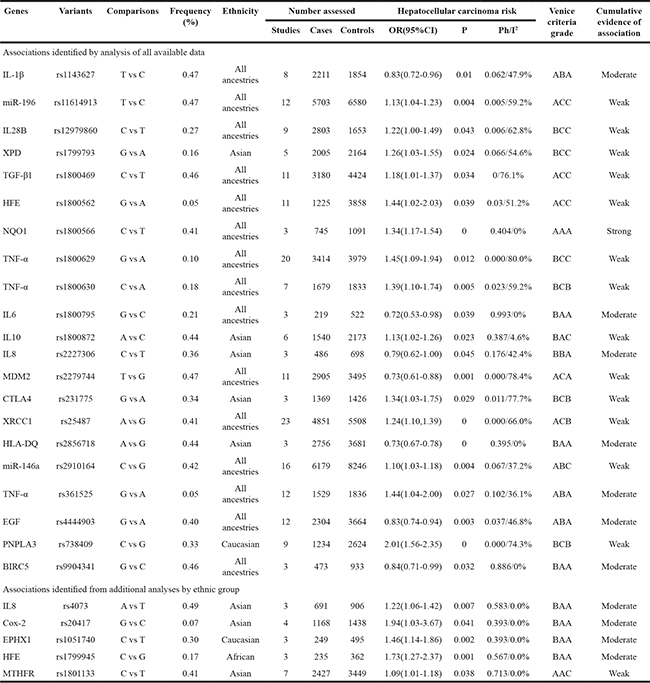
Table 2: Genetic variants nominally significantly associated with HCC risk in meta-analyses using dominant and recessive model
To assess the cumulative epidemiologic evidence for significant meta-analysis, Venice criteria [17] were applied. Epidemiological credibility were scored as ‘strong’, ‘moderate’ or ‘weak’ by a composite assessment, including the amount of evidence, extent of replication, and protection from bias. For the amount of evidences, 14 grades of ‘A’, 16 grades of ‘B’, and 0 grades of ‘C’ were given. For the extent of replication, 10 grades of ‘A’, 7 grades of ‘B’, and 13 grades of ‘C’ were given. For the protection from bias, 16 grades of ‘A’, 4 grades of ‘B’, and 10 grades of ‘C’ were given. One polymorphism (NQO1 rs1800566) was graded strong for evidence of association with HCC risk using Venice criteria result. Moreover, moderate and weak for the evidence of true association with HCC were assigned to 14 and 16 polymorphisms, respectively.
Previous meta-analyses works have independently analyzed the association between genetic variants and risk of HCC [18–22]. Here, we comprehensively compiled these works, and compared the differences with our results (Supplementary Table S2). The result showed that a total of 42 polymorphisms have been reported by previous meta-analyses, and the results of seven polymorphisms (rs11614913, rs1143627, rs25487, rs2910164, rs1801131, rs17401966 and rs861539) are inconsistent with our work because of their limited data resources. A total of 27 polymorphisms have been comprehensively evaluated for the first time.
HCCdb: a database of HCC-related polymorphisms
To facilitate HCC related polymorphisms integration and online query, we subsequently constructed a database of HCC-related polymorphisms (HCCdb). Currently, HCCdb contains 69 polymorphisms in 46 genes. The current version of HCCdb provides a user friendly search engine, which allows to search the basic content of gene or literature information. Furthermore, HCCdb provided a module to carry out a direct meta-analysis on the polymorphisms. Users can select the genetic model and effect models when performing online meta-analysis. In HCCdb, the OR and 95% CI can be measured to evaluate the strength of associations between polymorphisms and HCC risk. The database is freely available at http://donglab.ecnu.edu.cn/databases/HCCdb/.
DISCUSSION
Sequence polymorphisms in genes have been considered as underlying candidates in hepatocellular carcinogenesis. In this work, we described the results of the first systematic synopsis and meta-analysis in the field of genetic predisposition to HCC. We found that 31 polymorphisms in 25 genes showed significant associations with HCC risk. Our work provided a comprehensive research synopsis of candidate-gene association study of HCC risk. Using Venice criteria results, we graded one polymorphism strong for cumulative epidemiological evidence of association with HCC risk (NQO1 rs1800566). NQO1 is a cytosolic enzyme and plays an important role in protecting cells against oxidative stress by catalyzing two-electron reduction of numerous quinoid compounds into their less toxic form. Many epidemiological studies have investigated the effect of NQO1 rs1800566 polymorphism on carcinogenesis [23, 24], and the effect seems diverse in different malignant tumors. This work showed that the NQO1 rs1800566 polymorphism is a critical risk factor for HCC risk (OR=1.34). However, we failed to evaluate the susceptibility of NQO1 rs1800566 polymorphism to HCC in specific populations because of the limited eligible published case–control studies.
There are still several deficiencies in this work. First, although we have thoroughly searched the literature in PubMed database to identify eligible studies, it is possible that some studies might have been missed. To extend our search, we also checked the related meta-analysis in Google Scholar linking multiple databases. Second, we did not evaluate gene-gene interactions or gene-environment interactions. More additional studies specifically designed to detect these interactions are needed. Third, although Venice criteria offer the advantage for assessing various sources of potential bias, some of the indicators are difficult to measure, such as genotyping error, population stratification and phenotype misclassification.
To the best of our knowledge, this work is the largest and most comprehensive assessment of the literature on the genetic association with HCC susceptibility conducted to date. This work not only summarizes the current literature linking to genetic epidemiology of HCC, but also gives comprehensive data and helpful clues for designing future studies to further investigate genetic risk factors for HCC. In the future, the updating of web-based data collection for disease related studies would help to improve the cumulative evidence for genetic associations in HCC.
MATERIALS AND METHODS
Literature search and selection criteria
To identify the genetic variants associated with HCC risk, we searched the PubMed database by using the following keywords: “(liver cancer OR hepatocellular carcinoma) AND (polymorphism OR polymorphisms)” in title field without language restrictions for studies published up to 18 June 2015. Furthermore, Google scholar was also used to search eligible publications using the same keywords. This search produced 2667 potentially relevant publications. After further evaluation, 505 eligible publications were retained (Figure 1). All the eligible studies should met the following criteria: 1) Publications must be published in a peer-reviewed journal; 2) the study used a case-control and other appropriate cohort design in human beings were included; 3) Family-based studies were excluded; 4) HCC cases were diagnosed by pathological and/or histological examination, excluding liver cirrhosis, chronic hepatitis B, acute liver failure, asymptomatic HBV carriers and so on; 5) sufficient genotype data were presented to calculate the odd ratios (ORs) and 95% confidence intervals (CIs).
Data extraction
Data were independently extracted by two reviewer (PZ and YZ) and then checked by another reviewer (DD). The results of a total of 16 publications have some inconsistent and disagreements. All of these disagreements were encountered because of careless. We collected the disagreements and discuss with DD, and made the final decision after re-performing the work. The study provided enough information for the genotypic or allelic distribution of individual variants for both HCC cases and controls was the one we needed. Following characteristics were collected: first author’s surname, publication year, ethnicity (categorized as Caucasian, Asian, African, or mixed, including more than one ethnic category [25]), dbSNP ID, gene symbol, variants, source of control (hospital based, population based, family based), numbers of cases and controls of different genotypes, genotyping method, PubMed ID, and the type of the infected virus, respectively.
Statistical analysis
For the stabilization of heterogeneity test statistic (I2) and the operation of sensitivity analyses, we performed meta-analyses for the 69 genetic variants with case-control data available containing at least three independent sources. All statistical tests were conducted by STATA, version 12.0. All tests were two-tailed, and only P-value<0.05 was considered significant. To comprehensively analyze the relationship between genetic variants and risk of HCC, we selected three genetic models: additive model, dominant model and recessive model. To illustrate the models, we assume a polymorphism genome locus having two alleles, labeled A and a. A is the high-risk candidate allele and a is the lower-risk allele. Additive model is the same as allele model, represent the effect of the A allele vs. the a allele; dominant model represent the effect of the a/a+A/a vs. the A/A genotypes only when present in two copies of A allele, recessive mode represent the effect of the A/A+A/a vs. the a/a genotypes when present in either one or two copies of A allele. Summary odds ratios (ORs) with 95% confidence intervals (CIs) for alleles or genotypes were used to assess strength of associations between genetic variants and HCC risk by the random-effects method [26]. In the primary analyses, pooled ORs were acquired for allele contrast. In addition, dominant and recessive models were also assessed on all eligible polymorphisms. For some specific variants, like GSTM1 and GSTT1 ‘Present/Null’, conventional comparisons were used in original studies. If data permitted (at least 3 data source), we also performed subgroup analyses by ethnicities. For common variants (MAF≥5%), the minor allele and major allele sometimes were reversed in different ethnicities, it may lead to deviation. To minimize false-negative errors, for variants that showed no evidence of association with HCC risk in the meta-analyses, only those with admission of six independent datasets were selected for presentation.
Heterogeneity assumption was estimated by Chi-square based on Q-test [27]. I2 statistic was also used to assess heterogeneity [28]. Generally, I2 values less than 25% correspond to mild heterogeneity, values between 25% and 50% correspond to moderate heterogeneity, and values greater than 50% correspond to large heterogeneity between studies. Sensitivity analyses was performed excluding studies whose allele frequencies in controls exhibited significant deviation from the Hardy-Weinberg Equilibrium (HWE), given that the deviation may denote bias. Moreover, the extent to which the combined risk estimate might be influenced by individual studies was assessed consecutively omitting every study from the meta-analyses (leave-one-out sensitivity analyses). Begg’s funnel plots [29] and Egger’s linear regression test [30] were used to investigate the publication bias.
Evaluation of cumulative evidence
In order to assess statistically significant associations identified by meta-analyses, Venice criteria was employed in this work. It grades the cumulative evidence at three major criteria: (i) amount of evidence; (ii) replication of results; (iii) publication from bias. Amount of evidence was graded by the sum of test alleles or genotypes among both cases and controls in the meta-analysis; ‘A’ for over 1,000, ‘B’ for 100 to 1,000, and ‘C’ for less than 100. Caution was taken when applying this criterion to rare variants with frequency <1%, as an A grade is unobtainable. Replication was graded by the heterogeneity statistic; ‘A’ for I2 < 25%, ‘B’ for I2 between 25% and 50%, and ‘C’ for I2 >50%. Protection from bias may be caused by factors that lead to systematic deviations from the true effect of a genetic association. Biases may operate at the level of a single study, a collection of studies (e.g. meta-analysis), or a research field at large. It can be graded as ‘A’ if there was no observable bias and bias was unlikely to explain the presence of the association, ‘B’ if bias could be present or could explain the presence of the association, or ‘C’ if bias was evident or was likely to explain the presence of the association. Assessment of protection from bias also included consideration of the magnitude of the association; a score of ‘C’ was assigned to an association with a summary OR < 1.15 unless the association had been replicated prospectively by multiple studies with no evidence of publication bias. Cumulative epidemiological evidence was defined as strong, moderate, or weak. If all three grades were A, we considered it was strong. While if one or more grades were C, it was weak. All other combinations was moderate.
Database construction
The HCCdb system is based on a three-tier architecture: client, server and database. It includes a user-friendly web interface, PHP’s DBI module and MySQL database. HCCdb was developed on MySQL v4.1 with the MyISAM storage engine.
ACKNOWLEDGMENTS
This project is supported by Key Construction Program of the National ‘985’ project of East China Normal University to Dong Dong (79633006)
COMPETING INTERESTS
The authors declare that they have no competing interests.
Authors’ contributions
YZ, PZ, ZW and DD carried out the data analysis and drafted the manuscript. DD designed the study and revised the manuscript. All authors read and approved the final manuscript.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer. 2015; 136:E359-386.
2. Bosetti C, Levi F, Boffetta P, Lucchini F, Negri E and La Vecchia C. Trends in mortality from hepatocellular carcinoma in Europe, 1980-2004. Hepatology. 2008; 48:137-145.
3. Bosch FX, Ribes J, Cleries R and Diaz M. Epidemiology of hepatocellular carcinoma. Clinics in liver disease. 2005; 9:191-211, v.
4. Altekruse SF, McGlynn KA, Dickie LA and Kleiner DE. Hepatocellular carcinoma confirmation, treatment, and survival in surveillance, epidemiology, and end results registries, 1992-2008. Hepatology. 2012; 55:476-482.
5. Altekruse SF, McGlynn KA and Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009; 27:1485-1491.
6. Sherman M. Hepatocellular carcinoma: epidemiology, surveillance, and diagnosis. Seminars in liver disease. 2010; 30:3-16.
7. Fattovich G, Stroffolini T, Zagni I and Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004; 127:S35-50.
8. El-Serag HB. Hepatocellular carcinoma. The New England journal of medicine. 2011; 365:1118-1127.
9. Marrero JA, Fontana RJ, Fu S, Conjeevaram HS, Su GL and Lok AS. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. Journal of hepatology. 2005; 42:218-224.
10. Bravi F, Bosetti C, Tavani A and La Vecchia C. Coffee drinking and hepatocellular carcinoma: an update. Hepatology. 2009; 50:1317-1318.
11. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
12. Vogelstein B and Kinzler KW. The multistep nature of cancer. Trends Genet. 1993; 9:138-141.
13. Su JC, Tseng PH, Hsu CY, Tai WT, Huang JW, Ko CH, Lin MW, Liu CY, Chen KF and Shiau CW. RFX1-dependent activation of SHP-1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget. 2014; 5:4909-4919. doi: 10.18632/oncotarget.2054.
14. Laurent-Puig P and Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006; 25:3778-3786.
15. Tinkle CL and Haas-Kogan D. Hepatocellular carcinoma: natural history, current management, and emerging tools. Biologics. 2012; 6:207-219.
16. Tateishi R and Omata M. Hepatocellular carcinoma in 2011: Genomics in hepatocellular carcinoma--a big step forward. Nature reviews Gastroenterology & hepatology. 2012; 9:69-70.
17. Ioannidis JP, Boffetta P, Little J, O’Brien TR, Uitterlinden AG, Vineis P, Balding DJ, Chokkalingam A, Dolan SM, Flanders WD, Higgins JP, McCarthy MI, McDermott DH, Page GP, Rebbeck TR, Seminara D, et al. Assessment of cumulative evidence on genetic associations: interim guidelines. International journal of epidemiology. 2008; 37:120-132.
18. Liu GY, Jiang DK, Shen SQ and Yu L. MDM2 SNP309T>G polymorphism with hepatocellular carcinoma risk: a meta-analysis. Archives of medical research. 2011; 42:149-155.
19. Zhang H, Su YL, Yu H and Qian BY. Meta-Analysis of the Association between Mir-196a-2 Polymorphism and Cancer Susceptibility. Cancer biology & medicine. 2012; 9:63-72.
20. Liu F, Li B, Wei YG, Chen X, Ma Y, Yan LN, Wen TF, Xu MQ, Wang WT and Yang JY. IFN-gamma+874 A/T polymorphism and cancer risk: an updated analysis based on 32 case-control studies. Cytokine. 2011; 56:200-207.
21. Jin F, Qu LS and Shen XZ. Association between the methylenetetrahydrofolate reductase C677T polymorphism and hepatocellular carcinoma risk: a meta-analysis. Diagnostic pathology. 2009; 4:39.
22. Wei Y, Liu F, Li B, Chen X, Ma Y, Yan L, Wen T, Xu M, Wang W and Yang J. Polymorphisms of tumor necrosis factor-alpha and hepatocellular carcinoma risk: a HuGE systematic review and meta-analysis. Digestive diseases and sciences. 2011; 56:2227-2236.
23. Fan Y, Hu D, Feng B and Wang W. The NQO1 C609T polymorphism and hepatocellular carcinoma risk. Tumour Biol. 2014; 35:7343-7350.
24. Lajin B and Alachkar A. The NQO1 polymorphism C609T (Pro187Ser) and cancer susceptibility: a comprehensive meta-analysis. British journal of cancer. 2013; 109:1325-1337.
25. Ioannidis JP, Ntzani EE and Trikalinos TA. ‘Racial’ differences in genetic effects for complex diseases. Nature genetics. 2004; 36:1312-1318.
26. DerSimonian R and Laird N. Meta-analysis in clinical trials. Controlled clinical trials. 1986; 7:177-188.
27. Cochran WG. The combination of estimates from different experiments. Biometrics. 1954; 10:101-129.
28. Higgins JP and Thompson SG. Quantifying heterogeneity in a meta-analysis. Statistics in medicine. 2002; 21:1539-1558.
29. Begg CB and Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994:1088-1101.
30. Egger M, Davey Smith G, Schneider M and Minder C. Bias in meta-analysis detected by a simple, graphical test. Bmj. 1997; 315:629-634.