
The Endoplasmic Reticulum (ER)
The ER is an eukaryotic organelle arranged in a tubular network which is involved in the synthesis, folding, and trafficking of proteins, as well as being a key site for intracellular Ca2+ homeostasis. The highly oxidative, calcium-rich ER lumen facilitates the genesis of proteins destined for secretion or targeted to transmembrane compartments, constituting approximately 30% of the total proteome in eukaryotic cells. In addition, the ER regulates a variety of metabolic processes, such as gluconeogenesis and lipid biosynthesis, as well as biogenesis of autophagosomes and peroxisomes [1].
The ER is not only vital for these key cellular functions, but it also serves as a homeostatic device that monitors the intracellular environment and adjusts metabolic and stress responses accordingly. Stressful conditions such as an accumulation of unfolded proteins (e.g. when the demand for protein secretion is high), imbalance in ER Ca2+ levels, glucose deprivation, or hypoxia can all lead to disruption of ER function termed ER stress [2]. In an attempt to restore ER homeostasis and normal cellular function, several signal transduction pathways are activated. However, if the stress cannot be resolved, the pro-survival signaling switches to a pro-apoptotic one resulting in cell death. Both of these possible outcomes are mediated by the regulation of highly integrated signal transduction pathways, collectively called the unfolded protein response (UPR) [1].
The unfolded protein response
In multicellular eukaryotes the canonical UPR pathways are initiated by three proteins that reside in the ER membrane: inositol requiring-enzyme 1 alpha (IRE1α), activating transcription factor 6 alpha (ATF6α) and protein kinase RNA-like ER kinase (PERK). Upon activation, these ER ‘sensors’ initiate signaling cascades that elicit corrective actions to restore ER homeostasis [1] (Figure 1). Under normal physiological conditions, these transmembrane proteins are held in an inactive configuration by BiP. Upon accumulation of misfolded proteins, or other stimuli that can activate the UPR, BiP dissociates from the ER sensors and binds instead to unfolded proteins in the ER lumen. This results in IRE1α and PERK oligomerization leading to their phosphorylation, as well as translocation of ATF6α to the Golgi where it is cleaved and becomes an active transcription factor. Activation of IRE1α and PERK signaling also activates downstream transcription factors (see below) leading to changes in gene expression and corresponding phenotypic responses in the cell.
UPR signaling aims to restore homeostasis by inducing expression of proteins involved in almost every aspect of the secretory pathway. Gene expression data has demonstrated that the UPR is involved in protein entry into the ER, folding, glycosylation, ER-associated degradation (ERAD), protein quality control, redox metabolism, autophagy, lipid biogenesis and vesicular trafficking [1, 2].
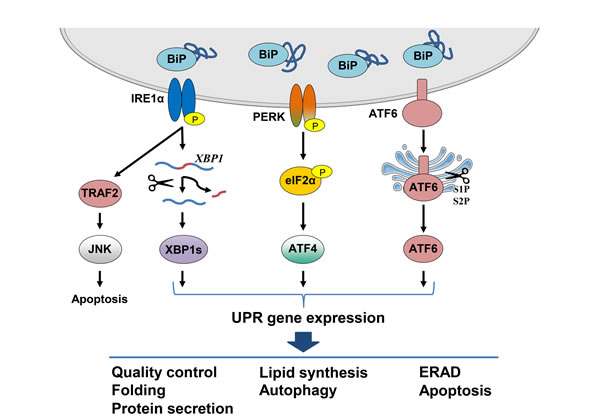
Figure 1: Schematic representation of the canonical UPR signaling pathways. The UPR is activated by the accumulation of unfolded proteins in the ER lumen as BiP dissociates from the three ER stress sensors IRE1α, ATF6α and PERK. Oligomerization of IRE1α leads to its activation and the generation of the transcription factor XBP1s which translocates to the nucleus and induces expression of genes whose products are involved in protein folding and ERAD. Additionally, IRE1α activation leads to degradation of ER-associated mRNAs through RIDD and induces JNK signaling, both of which result in induction of apoptosis. Activated ATF6α is cleaved in the Golgi by the S1P and S2P proteases to produce a transcription factor which translocates to the nucleus and induces chaperone gene expression. UPR signaling leads to a translational block through the PERK mediated phosphorylation of eIF2α. Despite the inhibition of global protein synthesis, ATF4 is translated, leading to the induction of genes involved in autophagy and amino acid metabolism. Signaling through the UPR aims to restore ER homeostasis by blocking further build-up of unfolded proteins, enhancing the folding capacity and initiating degradation of misfolded proteins. Upon persistent ER stress, however, pro-apoptotic signaling is induced and the cell undergoes programmed cell death.
IRE1
IRE1 is both a site-specific endoribonuclease and a Ser/Thr kinase; in mammals, it exists as two isoforms, IRE1α and IRE1β. IRE1α is expressed in all tissues, whereas IRE1β is only found primarily in the gastrointestinal and respiratory tracts, and thus the α isoform has been more extensively studied [3]. In response to unfolded proteins in the ER lumen, IRE1α dimerizes and oligomerizes, resulting in trans-autophosphorylation of the kinase domains. This leads to activation of the cytosolic RNase domain and the highly sequence specific endoribonucleolytic cleavage and subsequent splicing of the mRNA encoding a transcription factor called X-box binding protein 1 (XBP1) [4]. IRE1α excises a 26-nucleotide-long intron in the XBP1 mRNA that results in a frame shift giving rise to an active and stable transcription factor termed spliced XBP1 (XBP1s). When translocated to the nucleus, XBP1s induces UPR target gene expression [4]. The mammalian ligase responsible for joining the two exons of XBP1 mRNA upon removal of the intron, RtcB, was recently identified [5]. In contrast to the spliced form, the unspliced XBP1 (XBP1u) encodes a protein that is more labile, represses UPR target genes, and negatively regulates XBP1s and ATF6α by promoting their degradation [6].
XBP1s alone, or in conjunction with other transcription factors, launches a transcriptional program that activates the production of chaperones, proteins involved in ER biogenesis, phospholipid synthesis that is required for ER expansion under ER stress, ERAD, and secretion (e.g. ER degradation-enhancing alpha-mannosidase-like 1 (EDEM), ER-localized DnaJ 4 (Erdj4), and protein disulfide isomerase (PDI)) [7]. IRE1α-XBP1s signaling is therefore one of the major pathways for enhancing the folding capacity of the ER and coping with ER stress.
XBP1 mRNA was first thought to be the only known substrate for IRE1α; however, IRE1α was later found to also control gene expression via an XBP1-independent post-transcriptional mechanism [8]. Notably, the IRE1α ribonuclease activity can, under certain conditions, target and degrade other ER-associated mRNAs in a process termed regulated IRE1-dependent decay (RIDD) [8-10]. A number of mRNAs as well as miRNAs involved in different cellular processes have been shown to be targeted by the RIDD activity of IRE1α resulting in different cellular outcomes [11, 12]. It has been suggested that the RIDD activity of IRE1α was dependent on the oligomeric state of IRE1α [13]. In contrast, a recent study in yeast proposed the opposite view where RIDD is favored when yeast IRE1 acts as a monomer/dimer, while oligomerization promotes splicing of the mRNA for yeast homologue of XBP1, HAC1 [14].
Phosphorylation and activation of IRE1α also leads to the recruitment of the adaptor protein tumor necrosis factor receptor (TNFR)-associated factor 2 (TRAF2) and apoptosis signal regulating kinase 1 (ASK1) to the cytoplasmic leaflet of the ER membrane. This initiates a cascade of phosphorylation events that result in the activation of c-Jun N-terminal kinase (JNK) [15, 16]. As JNK activity has been closely linked to cell death [17-19], this connects ER stress-induced IRE1α signaling to apoptosis under certain settings.
ATF6
ATF6α and ATF6β are ER stress transducers that belong to the basic leucine zipper (bZIP) family of transcription factors [1]. In response to stress, ATF6 translocates to the Golgi, where it is processed by site-1 proteases (S1P) in its ER luminal domain and by site-2 proteases (S2P) within the region spanning the Golgi bilayer (Figure 1). This releases a cytosolic fragment which then translocates to the nucleus and functions as a transcription factor that binds to ER stress response elements (ERSE) in target genes [20]. The dissociation of BiP from ATF6α in response to stress within the ER lumen is proposed to unmask a Golgi-localization signal in the protein, allowing it to react with COPII, and be transported to the Golgi [21]. There is also evidence for calreticulin involvement in ATF6 transport from the ER to the Golgi, since under-glycosylated ATF6 as a result of ER stress did not interact with calreticulin which led to its transport to the Golgi for processing [22]. Furthermore, a recent study has suggested that PERK signaling is involved in regulating ATF6α trafficking and thus its activation, underlying the crosstalk between the canonical UPR arms [23]. In addition, the protein disulfide isomerase A5 (PDIA5) has been linked to ATF6 activation upon ER stress [24].
Target genes of ATF6α include chaperones BiP and GRP94, ERAD components and the UPR genes XBP1, protein kinase inhibitor of 58 kDa (P58IPK/DNAJC3), and C/EBP homologous protein (CHOP/GADD153) [4, 25-28]. ATF6α can also heterodimerize with XBP1s to regulate transcription from UPR elements (UPRE) in target genes, another example of crosstalk between UPR arms [26]. Similar to ATF6α, ATF6β is cleaved and translocates to the nucleus upon ER stress. However, ATF6β is a poor transcriptional activator and appears to repress ATF6α-mediated induction of UPR targets [29].
PERK
The third canonical UPR sensor is PERK, an ER transmembrane Ser/Thr kinase that attenuates translation in response to ER stress [30]. Upon accumulation of unfolded proteins in the ER lumen, PERK dimerizes, autophosphorylates and subsequently phosphorylates Ser51 in eukaryotic translation initiation factor 2 (eIF2) α-subunit resulting in attenuation of global translation [31] (Figure 1). The decrease in global translation quickly reduces the amount of newly synthesized proteins entering the ER, enabling it to recover. Despite a halt in translation, a few selected mRNAs with short upstream open reading frames (uORF) in the 5’-UTR escape this translational inhibition [31]. The best characterized example of this in mammals is activating transcription factor 4 (ATF4), which regulates the expression of genes involved in redox balance, amino acid metabolism, protein folding, autophagy and cell survival [2, 32]. Among the ATF4 target genes is CHOP, encoding a transcription factor involved in regulation of apoptosis [1]. CHOP has been shown to promote apoptosis demonstrated by Chop-/- MEFs displaying enhanced cell survival when treated with ER stressors compared to wild type cells [33]. CHOP can trigger apoptosis possibly through the transcriptional induction of proapoptotic BIM and the downregulation of antiapoptotic BCL-2 expression leading to apoptosis [34, 35]. PERK signaling is fine-tuned by the CHOP target gene growth arrest and DNA damage-inducible 34 (GADD34) which associates with the phosphatase PP1 and promotes dephosphorylation of eIF2α, thereby alleviating translational inhibition [1, 2]. Thus, PERK signaling is central in the switch between the adaptive response phase and chronic ER stress leading to apoptosis. In addition, CHOP promotes oxidative protein folding in the ER through the induction of ER oxidoreductin-1 alpha (ERO1α) expression. However, the resulting increase in disulphide bond formation generates reactive oxygen species (ROS) [36, 37]. This might suggest that under conditions of chronic ER stress the CHOP-mediated increase in protein flux into the ER through GADD34, and the subsequent increase in ROS formation can in turn lead to an enhanced stress leading to apoptosis [37, 38].
UPR signaling can counter the effect of ROS by launching an anti-oxidant response. PERK activates the transcription factors ATF4 and nuclear factor E2 related factor 2 (NRF2), which induce genes involved in anti-oxidation [1]. NRF2 is held in the cytoplasm through its association with KEAP; phosphorylation of NRF2 by PERK triggers its dissociation and the nuclear import of NRF2 [39]. PERK-eIF2α signaling also activates NF-κB through translational repression of inhibitor of kappa B (IκB), resulting in regulation of apoptosis [40, 41]. The co-chaperone P58IPK, a target of both XBP1s and ATF6α, inhibits PERK signaling by interacting with the kinase domain of PERK and impairing eIF2α phosphorylation [42-44]. This is another example of the self-controlling, auto-corrective nature of the UPR response.
Another aspect adding to the complexity is that UPR signaling through the three canonical branches induce the expression of various miRNAs. Conversely, a number of miRNAs have been shown to target UPR components, [45, 46]. This layer of regulation is another reminder that the UPR needs to be regulated in multiple ways in a manner that allows versatile response to ques from the environment.
Another example of this is that in humans, there are three other eIF2α kinases that can phosphorylate eIF2α and regulate translation: General control non-derepressible-2 (GCN2) which is activated by nutrient deprivation, heme-regulated initiation factor 2 alpha kinase (HRI) activated by heme deficiency and oxidative stress, and protein kinase interferon-inducible double stranded RNA dependent (PKR) which is activated by viral infection. They all converge on the phosphorylation of the same residue in eIF2α and are collectively referred to as the integrated stress response (ISR) [1, 36].
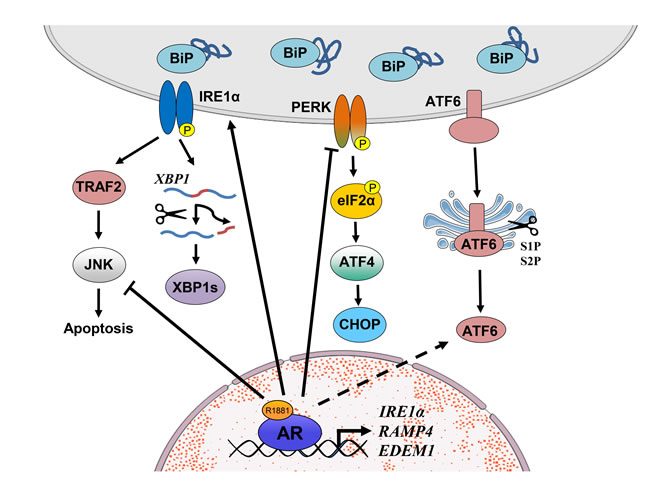
Figure 2: Schematic representation of UPR signaling in prostate cancer. Liganded AR binds in the vicinity of the IRE1α, RAMP4 and EDEM1 genes to increase their expression, thus activating the IRE1α pathway but at the same time inhibits pro-apoptotic JNK signaling which is also activated by IRE1α. In contrast, androgens inhibit PERK and eIF2α activity. However, contrary to expectations (see text), ATF4 and CHOP expression are modestly increased in response to androgen; thus, the exact role of the PERK pathway and its regulation by androgens in prostate cancer requires further investigation. It is currently unknown whether the ATF6 pathway is regulated by androgen signaling in prostate cancer cells. Arrows with solid lines indicate a direct established effect whereas those with dashed lines indicate indirect or currently unexplored interactions.
UPR in cancer
Solid tumors and their metastatic derivatives, often experience hypoxia, nutrient deprivation, lactic acidosis, and oxidative stress which compromise ER function leading to ER stress. In many respects the UPR can be viewed as a cytoprotective mechanism, favoring tumor growth by enabling cancer cells to survive under very unfavorable conditions. However, from another perspective, UPR can serve to protect the host by initiating cell death pathways [2]. UPR signaling has been implicated in various aspects of the tumor cell biology including including angiogenesis, invasion, mitochondrial function, intercellular communication, and tumor-associated inflammation [2]. The connections between ER stress and UPR in oncogenesis and cancer development have been extensively reviewed elsewhere [2, 12, 32].
The majority of the studies in cancer cells so far point to UPR activation as an adaptive survival mechanism. For example, consistent with this notion, increased levels of GRP94 and BiP have been correlated with gastric cancer [47]. The IRE1α arm is also implicated in tumor tolerance to hypoxia and pro-angiogenetic mechanisms, as XBP1s was increased in hypoxic cells and provided a survival advantage [48]. However, despite the large amount of data implicating ER stress and UPR activation in many forms of cancer, it is still not clear whether UPR ultimately promotes or inhibits tumor growth.
UPR activation as a cytoprotective response is supported by the fact that XBP1 overexpression in cancer cells directly promotes tumorigenesis, such as in chronic lymphocytic leukemia [49]. In multiple myeloma (MM), targeting the activated IRE1α and subsequent XBP1 splicing by a number of compounds has been tested with promising results in preclinical models. Furthermore, there were synergistic effects in combination with bortezomib, the FDA approved proteasome inhibitor for MM [50]. Another study demonstrated that XBP1s loss may confer MM cells resistance to bortezomib [51]. XBP1 interacts with hypoxia-inducible factor 1-alpha (HIF1α) in triple negative breast cancer and drives tumor progression by inducing hypoxia signature gene expression [52]. Interestingly, a recent study also showed that XBP1 can blunt the antitumor activity by interfering with the function of tumor-associated dendritic cells [53]. The XBP1s and ATF6α target, P58IPK, has been linked to survival of malignant cells facing ER stress, mediating an adaptive response to chronic UPR signaling [54].
PERK signaling has been reported to both inhibit and promote tumor growth. PERK deficient breast cancer cells display impaired cell growth and increased ROS production [55]. In contrast, inactivation of PERK in a murine model of medulloblastoma promotes tumor growth [56]. Furthermore, the oncoprotein MYC was shown to induce PERK and thereby promote cell survival through cytoprotective autophagy [57]. A recent study also connects ER homeostasis to epithelial-to-mesenchymal transition (EMT), showing that cells undergoing EMT have elevated BiP expression and activated PERK-eIF2α pathway that may be responsible for the increased secretory potential and sensitivity to ER stress inducers [58]. Other studies, however, have shown that CHOP induces apoptosis in cancer cells [54]. A PERK-ATF4 dependent miRNA, miR211, downregulates CHOP expression and promotes tumor cell survival in breast cancer [59]. These examples highlight the diverse effects of PERK signaling on apoptosis.
The outcome of UPR signaling in different cancer types is variable and depends on a multitude of factors such as tissue of origin, tumor stage and the tumor microenvironment. In addition, the heterogeneity of tumor cells represents a challenge when studying the consequence of UPR signaling in cancer.
Genetic manipulation of UPR components in vivo has revealed a spectrum of phenotypes, corroborating the fact that these signaling pathways are essential for the normal functioning of a number of organs. Similarly, naturally occurring mutations in the UPR sensors in humans lead to a range of malignancies such as metabolic diseases and cancer [60]. For example, somatic mutations in IRE1 have been detected in several human cancers, such as hepatocellular carcinoma, glioblastoma, ovarian cancer, lung cancer, renal cancer and gastric cancer [61-63]. Furthermore, XBP mutations have been found in samples from patients with multiple myeloma [64, 65]. Data from the Catalogue of Somatic Mutations in Cancer (COSMIC) database have revealed a range of different mutations in IRE1, PERK and ATF6 which show specific mutation patterns and tissue distribution [12]. The biological effects of the different mutations on tumor progression are at present not clear; however, a number of the IRE1α mutations identified in human cancers appear to provide a survival advantage that can still splice XBP1, but cannot induce RIDD [13].
UPR in prostate cancer
Prostate cancer (PCa) represents a major health issue worldwide as the most commonly diagnosed cancer after skin cancer and the second leading cause of cancer deaths among men in the western world [66]. It is estimated that approximately one of seven men will be diagnosed with PCa in their lifetime. One of the most central signaling pathways in all stages of PCa is that mediated by androgens. If the disease is not organ confined and thus curable by surgery alone, the standard therapeutic option for PCa is androgen ablation leading to an initial regression of the tumor in the vast majority of the cases. However, in most cases the tumors relapse into castration resistant PCa (CRPC) for which there is currently no curative treatment available [67, 68].
Being a major secretory organ, the prostate is particularly reliant on proper functioning of the ER and is vulnerable to agents or conditions that cause ER stress. It has been suggested that ER stress and UPR activation are involved in PCa as earlier studies indicated a negative correlation of UPR marker gene expression and PCa progression in model systems in vitro. For example, XBP1 was found downregulated in PCa compared with normal prostate and displayed an inverse correlation with pathological grade [69]. This study, however, did not differentiate between the spliced and unspliced forms of XBP1. The three UPR branches were also found to be downregulated during prostate tumorigenesis in both the NKX3.1:PTEN mutant and MYC-overexpression PCa mouse models [70].
In contrast, a number of studies have pointed to a positive association between ER/UPR markers and PCa development, mainly driven by androgens and androgen receptor (AR) signaling. Androgens regulated the expression of different ER stress associated genes in PCa, including N-myc downstream-regulated gene 1 protein (NDRG1), protein disulfide isomerase-related protein (PDIR/PDIA5), homocysteine-responsive ER resident ubiquitin-like domain member 1 protein (HERPUD), and oxygen-regulated protein 150 (ORP150) [71]. Furthermore, gene expression profiling of dihydrotestosterone (DHT)-treated ventral prostates from rats revealed that genes whose products are involved in protein synthesis, degradation and processing are differentially expressed. These included calnexin, calreticulin, BiP, GRP94 and PDI [72]. Another study found that expression of genes associated with protein synthesis, folding and secretion, such as ribosomal proteins and seminal vesicle secretion proteins, were significantly induced in mouse prostates treated with DHT further strengthening the connection between androgen signaling and ER function [73].
In spite of the pivotal role of AR signaling throughout all stages of PCa development, its role in regulation of canonical UPR arms was not understood until recently. We found that androgens activated the IRE1α-XBP1 arm and simultaneously inhibited the PERK-eIF2α pathway [74]. Moreover, activated AR directly bound in the vicinity of IRE1, as well as XBP1s targets RAMP4 and EDEM1 genes, in PCa cells. Coupled with the complete loss of androgen regulation upon AR knockdown, these data document that these genes are direct AR targets. Consistent with these findings, AR and UPR gene expression were correlated in human PCa samples whereas XBP1s protein expression is significantly increased in cancer compared to normal prostate [74].
In contrast to the findings on the IRE1α arm, the mechanisms behind androgen-mediated inhibition of PERK-eIF2α signaling is not clear at present. Whereas PERK activation and thus eIF2α phosphorylation is downregulated by androgens, expression of downstream targets ATF4 and CHOP were increased at the protein level [74]. This is despite the fact that ATF4 mRNA expression is not significantly affected whereas CHOP mRNA expression is decreased by androgens. One possible explanation of these observations is that upon dephosphorylation of PERK and eIF2α by androgen treatment, in PCa cells there is a general increase in protein synthesis which compensates for the effects observed at the mRNA level. Alternatively, CHOP may act as a survival factor in PCa as was suggested in some other settings [32]. In addition, in different PCa cell lines regulation of the PERK pathway may be different. For example, in the CRPC model 22Rv1 cells androgens activate eIF2α phosphorylation [75]. Further analyses are thus required to uncover the details in the regulation of the PERK pathway by androgens in PCa cells and the corresponding phenotypic outcomes.
Potential functions of UPR in PROSTATE CANCER
Compared to PERK and ATF6, there is relatively more data on the functional role of IRE1α-XBP1s signaling in PCa. In LNCaP cells, depletion of IRE1α or its downstream target XBP1 leads to inhibition of cell growth in vitro and in vivo, a likely consequence of decreased proliferation and increased apoptosis [74]. Targeting this arm using toyocamycin, a small molecule drug inhibiting IRE1α kinase activity and XBP1 splicing, profoundly inhibits both LNCaP and VCaP cell growth in vitro as well as tumor formation in vivo [74]. Increased XBP1 splicing in the AR negative PC3 cells by treatment of Sphingosine 1-phosphate, a bioactive lysophospholipid, induces autophagy and exerts a cytoprotective effect [76]. Consistently, inhibition of IRE1α kinase activity and subsequent XBP1 splicing impairs proliferation of these cell lines in vitro [77]. However, the exact role of the IRE1α-XBP1s pathway in AR-negative cell lines is not clear at present and further investigation is warranted. In summary, the IRE1α-XBP1s arm plays a pro-survival role in PCa suggesting that targeting IRE1α signaling may be a novel therapeutic strategy for PCa.
In contrast to the IRE1α arm, little is known about the functions of PERK-eIF2α and ATF6 in PCa progression. ATF4 expression has been shown to be induced by leucine or androgen deprivation in PCa cells [78]. ATF4 subsequently activates transcription of L-type amino acid transporter 1 (LAT1) which is important for leucine uptake and cell growth [78]. Furthermore, ATF4 mediates the pro-survival role of six-transmembrane protein of prostate 2 (STAMP2), a protein which impacts both cell growth and cell death pathways in PCa cells [79]. In addition, ATF4 expression is significantly upregulated in CWR22R refractory tumors, implicating it in progression to CRPC [79].
In contrast, there is currently no direct evidence that links the ATF4 target CHOP to survival or apoptosis in PCa cells. CHOP directly induced the expression of death receptor 5 (DR5) in response to either tunicamycin or acetyl-keto-β-boswellic acid treatment, both of which trigger apoptosis in PCa cells [80, 81]. Further work is required to delineate the role of PERK signaling in PCa.
ER chaperones in PROSTATE CANCER
Expression of molecular chaperones with cytoprotective roles is elevated in several types of cancer [82] including in PCa (for a review, see [83]). The most studied ER chaperone in PCa cells is BiP. Increased BiP expression is correlated with greater risk of PCa recurrence and worse survival [84]. Consistently, double knockout of Bip and Pten in prostates of mice completely reverts the invasive adenocarcinoma phenotype normally observed upon Pten deletion, further establishing BiP as a key regulator of PCa progression [85]. Notably, the intracellular localization of BiP seems to be critical for its function; BiP has been detected on the cell surface in several cancers, including PCa, but not on normal cells, suggesting that it is a cancer-specific cell surface marker [86]. Translocation of BiP from the ER lumen to the cell surface is implicated in the hormonal resistance of PCa as well as in breast cancer cells, which may be mediated by enhanced PI3K signaling [87].
A variety of strategies have been employed to specifically target cell surface BiP, such as siRNA, antibodies, peptides, fusion proteins and nanoparticles, all resulting in reduced growth and increased apoptosis both in vitro and in vivo [88-92]. A recent study confirmed that targeting surface BiP with the peptide ligand SNTRVAP suppressed castration-resistant osteoblastic bone metastases in vivo [93]. In addition, prostate apoptosis response 4 (PAR-4), a pro-apoptotic protein secreted by cancer cells, was shown to bind to cell surface BiP and induce apoptosis of PCa cells [94]. Another study has linked androgen-induced impairment of autophagy to an increase in BiP protein levels independent of PERK-eIF2α-ATF4 pathway activation; however, the mechanism behind this effect is currently unclear [95]. These studies suggest that BiP may be a biomarker and therapeutic target for PCa, but further investigations are required to evaluate this possibility.
In addition to BiP, expression of several other molecular chaperones, such as heat shock protein 27 (HSP27) and HSP90, have been associated with aggressive human PCa [96-98]. Overexpression of HSP27 alleviates MG132-induced UPR and inhibits apoptosis of PC3 cells [99]. HSP27 also drives EMT and metastasis in PCa [100]. Furthermore, combining the non-invasive low energy focused ultrasound (LOFU) with a non-toxic dose of the HSP90 inhibitor 17AAG has displayed promising results in PCa xenografts by shifting the pro-survival UPR to a pro-apoptotic response [101]. Chemical inhibition of both HSP90 and HSP27 exerts a synergistic inhibitory effect on LNCaP cell growth in vitro and in vivo [102]. In summary, ER chaperones appear to be key players in tumor survival and treatment resistance, making them potentially valuable therapeutic targets.
Other ER-associated proteins in PROSTATE CANCER
An important biosynthetic function of the ER is glycoprotein production. The glycosylation process involves the addition of an oligosaccharide consisting of N-acetylglucosamine, glucose and mannose moieties to the NH2-side chain of asparagine and is termed N-linked glycosylation. Several proteins involved in ER N-glycosylation have been linked to PCa. For example, the ER-localized protein ectonucleoside triphosphate diphosphohydrolase 5 (ENTPD5), a downstream target of AKT, is important for N-linked glycosylation and increases aerobic glycolysis. ENTPD5 is elevated in PCa and confers a survival advantage to PCa cells, connecting ER function to PCa metabolism [103]. Another example is UDP-N-acetylglucosamine pyrophosphorylase 1 (UAP1), an enzyme of the hexosamine biosynthetic pathway involved in N-glycosylation, that is androgen regulated and highly overexpressed in PCa. Increased levels of UAP1 in PCa cells provides a growth advantage as it confers resistance of PCa cells towards inhibitors of N-linked glycosylation, such as tunicamycin and 2-deoxyglucose [104]. Furthermore, inhibition of N-linked glycosylation in PC3 and DU145 cells, either by tunicamycin or glycosydase, leads to the generation of a lower molecular-weight pattern of death receptor 4 associated with apoptosis [105]; however, how this isoform triggers apoptosis is not known. Recently, tumor suppressor candidate 3 (TUSC3), a member of the oligosaccharyltransferase complex, has also been implicated in N-glycosylation and growth of PCa cells [106]. Loss of TUSC3 leads to increased proliferation, viability, migration, and invasion of DU145 and PC3 cells, which is accompanied by elevated N-glycosylation and AKT phosphorylation, indicating that TUSC3 exerts its tumor suppressor activity by influencing these processes [106]. The involvement of these proteins in the ER glycosylation processes in PCa cells is particularly intriguing given the altered glycosylation patterns found in PCa [107].
Another relevant ER stress associated protein is N-myc downstream regulated gene-1 (NDRG1) which is a potent metastasis suppressor that plays a key role in regulating signaling pathways involved in PCa [108]. NDRG1 interactome map in LNCaP cells in response to androgens identified a group of ER chaperones and proteins involved in ER stress. Interestingly, BiP levels were unaffected upon NDRG1 knockdown, whereas GRP94 expression was decreased [109]. Speckle-type POZ protein (SPOP), a component of the CUL3-RBX1 E3 ubiquitin ligase complex, has been shown to interact with CHOP and induce its degradation. PCa cells expressing SPOP mutants showed a defect in inducing CHOP ubiquitination and underwent CHOP-mediated apoptosis [110]. These findings highlight the importance of ER-associated proteins in PCa, even if they may not be directly involved in ER stress. However, the majority of the findings to date on these proteins are mainly correlative and their possible functional impact in PCa is currently not known. Another ER-associated protein is PACE-4, a proprotein convertase that is shown to be upregulated in PCa tissues. A recent in vitro study using multiple PCa cell lines showed that siRNA knockdown of PACE4, led to apoptosis accompanied with increased PERK and eIF2α phosphorylation [111]. However, how PACE4 regulates these UPR arms and the molecular mechanisms of apoptosis in this context is currently not known.
Skp2, a critical component of the Skp2-SCF complex E3 ligase, is found highly expressed in numerous cancers including PCa [112]. Induction of eIF2α phosphorylation and ATF4 expression were credited to be partially responsible for the cellular senescence triggered by loss of Skp2 in mouse embryonic fibroblasts [113]. Interestingly, BiP and phospho-PERK level remained unchanged upon Skp2 loss, suggesting that other eIF2α kinases and compensatory mechanisms may be involved. Another study using a panel of human PCa cell lines revealed an inverse relationship between Skp2 and ATF4 expression upon caffeic acid phenethyl ester treatment, the chief extract from honeybee hive propolis [114]. However, whether Skp2 plays a role in the context of ER stress and UPR in PCa is largely unknown.
An active field of investigation in PCa in recent years has been the interplay between ER stress and autophagy. Autophagy is another typical adaptive mechanism by which the cells react to metabolic, toxic, and even infectious stressors [115]. It is dynamically regulated by a number of factors, for instance the PI3K-AKT-mTOR signaling pathway which is frequently found activated in human PCa due to the loss of PTEN [68, 115-117]. Various agents and chemicals that induce ER stress have also been shown to induce autophagy in PCa cells [118, 119]. The ER stress-triggered autophagy appears to play a protective role under these conditions by clearing polyubiquitinated protein aggregates and reducing cellular vacuolization [118]. On the other hand, acutely-stimulated ER stress has been shown to induce expression of the proapoptotic protein, PAR-4, which switches protective autophagy to apoptosis in androgen-independent PCa cells by inhibiting autophagy-related proteins BCL2 and BECLIN-1 [120]. In turn, disruption of autophagy may give rise to ER stress through feedback mechanisms. For instance, in a prostate-specific Pten-deficient mouse model, additional knockout of autophagy-related-7 (Atg7) markedly delayed tumor development under both castration-naïve and castration-resistant conditions [121]. The double knockout phenotype displayed impaired autophagy and increased ER stress, underscoring the importance of protein homeostasis in PCa progression [121]. There are a number of detailed recent reviews on the possible roles of PI3K-mTOR-Akt pathway as well as autophagy in PCa [115, 117, 122].
Targeting the ER stress response for PROSTATE CANCER therapy
Given the significance of maintaining proteostasis in cancer cells, targeting the ER homeostasis is emerging as a new therapeutic strategy [123], also in PCa. In addition to the compounds exemplified above targeting the IRE1α arm, various small molecule drugs and chemical extracts that disrupt ER homeostasis in vitro in PCa cells have been identified. For example, selenium and its metabolites had anti-cancer activity on PCa cells through, at least in part, activation of ER stress and subsequent induction of apoptosis, an effect which was attenuated by BiP overexpression [124]. In line with this, BiP knockdown greatly enhanced the growth inhibition of PC3 cells by selenium [125]. Metformin, a first-line anti-diabetic drug, has been shown to decrease the risk of PCa in people who use it for metabolic disturbances [126]. Research has shown that this may be mediated by the activation of the miR-708-5p/neuronatin pathway, which subsequently leads to ER stress-induced apoptosis [127]. However, combined application of ER stress inducers should be carefully examined, as the outcome may not always be detrimental. This is exemplified by a recent study in PC3 cells, where epigallocatechin gallate (EGCG), a major bioactive green tea polyphenol known to induce ER stress, failed to further promote cell death triggered by either bortezomib or MG132. Instead, EGCG activated cytoprotective autophagy, reduced CHOP levels and thereby protected from bortezomib-caused ER stress and apoptosis [128].
A number of natural compounds that have anti-cancer activities interfere with ER function and induce PCa cell death. In addition, several enzyme inhibitors have been shown to trigger ER stress and induce apoptosis in PCa. Molecules that have been identified to date that target ER homeostasis are summarized in Table 1. However, it should be noted that the data are mainly from either in vitro studies or limited preclinical models; thus, additional translational studies investigating these compounds in an in vivo setting are needed.
Table 1: Molecules targeting ER homeostasis in prostate cancer.
Molecule |
Origin |
Phase |
Readout |
Refs |
Toyocamycin |
Actinomycete |
Preclinical, LNCaP and VCaP |
Inhibition of XBP1 splicing |
[74] |
N-butylidenephthalide |
Angelica sinensis |
Preclinical, LNCaP |
Induction of CHOP, IRE1 and BiP |
[129] |
Tanshinone IIA |
Salviae Miltiorrhizae Radix |
Preclinical, LNCaP |
Induction of CHOP, IRE1 and BiP |
[130] |
SMI-4a |
Pim kinase inhibitor |
Preclinical, LNCaP |
Activation of eIF2α, ATF4, CHOP and induction of XBP1 splicing |
[131] |
Curcumin |
Turmeric |
Preclinical, PC-3M |
Induction of IRE1, eIF2α, CHOP and BiP |
[132] |
Clofoctol |
Antibiotic for upper respiratory tract infections |
Preclinical, PC3 |
Activation of IRE1, PERK and ATF6 pathways |
[133] |
Monascuspiloin |
Monascus pilosus M93-fermented rice |
Preclinical, PC3 |
Activation of IRE1 and eIF2a |
[134] |
Marchantin M |
Bryophytes |
Preclinical, PC3 |
Induction of CHOP and BiP |
|
Nelfinavir |
HIV protease inhibitor |
In vitro, PC3 and DU145 |
Activation of ATF6, BiP and S2P target gene expression |
[137] |
Shikonin |
Lithospermum erythrorhizon |
In vitro, PC3 and DU145 |
Activation of PERK, eIF2α, CHOP and BiP |
[138] |
Methylseleninic acid |
Selenium |
In vitro, PC3 |
Activation of PERK, eIF2α, CHOP and BiP |
[124] |
Celastrol |
Autoimmune diseases, chronic inflammation, asthma and neurodegenerative disease |
In vitro, PC3 |
Activation of IRE1, PERK and BiP |
[139] |
Polyphenon E |
Green tea |
In vitro, PC3 |
Activation of CHOP |
[140] |
Diindolylmethane |
Indole-3-carbinol |
In vitro, DU145 |
Activation of IRE1 |
[141] |
Capsaicin |
Hot chilli peppers |
In vitro, PC3 |
Activation of eIF2α, ATF4 and CHOP |
[142] |
4,5,6,7-tetrabromobenzotriazole |
Protein kinase CK2 inhibitor |
In vitro, PC3 |
Activation of CHOP |
[143] |
VN/124-1 |
17A-hydroxylase/17,20 lyase inhibitor |
In vitro, PC3 |
Activation of eIF2α and BiP |
[144] |
Proteasome inhibitor-I |
In vitro, PC3 |
Inhibition of IRE1 phosphorylation and induction of CHOP |
[145] |
|
Triptolide |
Tripterygium wilfordii Hook F. |
In vitro, PC3 |
Inhibition of BiP and activation of IRE1α, PERK and eIF2α |
[146] |
Metformin |
First-line antidiabetes drug |
In vitro, C4-2B |
Induction of miR-708-5p and inhibition of neuronatin |
[127] |
Conclusions
The data that have accumulated in the last few years indicate that ER stress and the UPR play an important role in PCa. Among these intriguing findings are the androgen regulation of UPR activation, the functional roles of different UPR components, as well as the differential regulation of ER chaperone expression. These data suggest that targeting the adaptive survival aspects of the UPR and interfering with ER homeostasis is potentially a novel and promising approach for PCa therapy.
Acknowledgments
We would like to thank members of the FS laboratory for helpful discussions and critically reading the manuscript. This work was supported by grants to FS from the Norwegian Research Council, Norwegian Cancer Society, and Health South East Norway.
Conflicts of Interest
No potential conflicts of interest were disclosed by the authors.
References
1. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012; 13:89-102.
2. Clarke HJ, Chambers JE, Liniker E and Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer cell. 2014; 25:563-573.
3. Tabas I and Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011; 13:184-190.
4. Yoshida H, Matsui T, Yamamoto A, Okada T and Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001; 107:881-891.
5. Lu Y, Liang FX and Wang X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol Cell. 2014; 55:758-770.
6. Yoshida H, Oku M, Suzuki M and Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol. 2006; 172:565-575.
7. Lee AH, Chu GC, Iwakoshi NN and Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 2005; 24:4368-4380.
8. Hollien J and Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006; 313:104-107.
9. Hollien J, Lin JH, Li H, Stevens N, Walter P and Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol. 2009; 186:323-331.
10. Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA and Papa FR. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell. 2009; 138:562-575.
11. Wang M and Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016; 529:326-335.
12. Chevet E, Hetz C and Samali A. Endoplasmic Reticulum Stress-Activated Cell Reprogramming in Oncogenesis. Cancer discovery. 2015; 5:586-597.
13. Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell. 2014; 158:534-548.
14. Tam AB, Koong AC and Niwa M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell reports. 2014; 9:850-858.
15. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP and Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000; 287:664-666.
16. Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002; 16:1345-1355.
17. Dhanasekaran DN and Reddy EP. JNK signaling in apoptosis. Oncogene. 2008; 27:6245-6251.
18. Lorenzo PI and Saatcioglu F. Inhibition of apoptosis in prostate cancer cells by androgens is mediated through downregulation of c-Jun N-terminal kinase activation. Neoplasia. 2008; 10:418-428.
19. Hubner A, Mulholland DJ, Standen CL, Karasarides M, Cavanagh-Kyros J, Barrett T, Chi H, Greiner DL, Tournier C, Sawyers CL, Flavell RA, Wu H and Davis RJ. JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:12046-12051.
20. Haze K, Yoshida H, Yanagi H, Yura T and Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999; 10:3787-3799.
21. Shen J, Chen X, Hendershot L and Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002; 3:99-111.
22. Hong M, Luo S, Baumeister P, Huang JM, Gogia RK, Li M and Lee AS. Underglycosylation of ATF6 as a novel sensing mechanism for activation of the unfolded protein response. The Journal of biological chemistry. 2004; 279:11354-11363.
23. Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG and Wek RC. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell. 2011; 22:4390-4405.
24. Higa A, Taouji S, Lhomond S, Jensen D, Fernandez-Zapico ME, Simpson JC, Pasquet JM, Schekman R and Chevet E. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Molecular and cellular biology. 2014; 34:1839-1849.
25. Yoshida H, Haze K, Yanagi H, Yura T and Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. The Journal of biological chemistry. 1998; 273:33741-33749.
26. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A and Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007; 13:365-376.
27. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD and Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007; 13:351-364.
28. Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A and Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008; 33:75-89.
29. Thuerauf DJ, Marcinko M, Belmont PJ and Glembotski CC. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. The Journal of biological chemistry. 2007; 282:22865-22878.
30. Harding HP, Zhang Y and Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999; 397:271-274.
31. Holcik M and Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005; 6:318-327.
32. Wang M and Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature reviews Cancer. 2014; 14:581-597.
33. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL and Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998; 12:982-995.
34. Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P and Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007; 129:1337-1349.
35. McCullough KD, Martindale JL, Klotz LO, Aw TY and Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Molecular and cellular biology. 2001; 21:1249-1259.
36. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM and Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003; 11:619-633.
37. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004; 18:3066-3077.
38. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA and Kaufman RJ. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013; 15:481-490.
39. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ and Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and cellular biology. 2003; 23:7198-7209.
40. Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR and Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Molecular and cellular biology. 2003; 23:5651-5663.
41. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP and Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Molecular and cellular biology. 2004; 24:10161-10168.
42. Lee AH, Iwakoshi NN and Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and cellular biology. 2003; 23:7448-7459.
43. Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D and Katze MG. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:15920-15925.
44. van Huizen R, Martindale JL, Gorospe M and Holbrook NJ. P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2alpha signaling. The Journal of biological chemistry. 2003; 278:15558-15564.
45. Maurel M and Chevet E. Endoplasmic reticulum stress signaling: the microRNA connection. Am J Physiol Cell Physiol. 2013; 304:C1117-1126.
46. Chitnis N, Pytel D and Diehl JA. UPR-inducible miRNAs contribute to stressful situations. Trends Biochem Sci. 2013; 38:447-452.
47. Zheng HC, Takahashi H, Li XH, Hara T, Masuda S, Guan YF and Takano Y. Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Human pathology. 2008; 39:1042-1049.
48. Auf G, Jabouille A, Guerit S, Pineau R, Delugin M, Bouchecareilh M, Magnin N, Favereaux A, Maitre M, Gaiser T, von Deimling A, Czabanka M, Vajkoczy P, Chevet E, Bikfalvi A and Moenner M. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:15553-15558.
49. Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del Valle JR and Hu CC. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. The Journal of clinical investigation. 2014; 124:2585-2598.
50. Hetz C, Chevet E and Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013; 12:703-719.
51. Leung-Hagesteijn C, Erdmann N, Cheung G, Keats JJ, Stewart AK, Reece DE, Chung KC and Tiedemann RE. Xbp1s-negative tumor B cells and pre-plasmablasts mediate therapeutic proteasome inhibitor resistance in multiple myeloma. Cancer cell. 2013; 24:289-304.
52. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014; 508:103-107.
53. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR and Glimcher LH. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell. 2015; 161:1527-1538.
54. Huber AL, Lebeau J, Guillaumot P, Petrilli V, Malek M, Chilloux J, Fauvet F, Payen L, Kfoury A, Renno T, Chevet E and Manie SN. p58(IPK)-mediated attenuation of the proapoptotic PERK-CHOP pathway allows malignant progression upon low glucose. Mol Cell. 2013; 49:1049-1059.
55. Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D and Diehl JA. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene. 2010; 29:3881-3895.
56. Lin W, Lin Y, Li J, Harding HP, Ron D and Jamison S. A deregulated integrated stress response promotes interferon-gamma-induced medulloblastoma. J Neurosci Res. 2011; 89:1586-1595.
57. Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, Li Y, Gao Y, Liu H, Li C, Maity A, Thomas-Tikhonenko A, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. The Journal of clinical investigation. 2012; 122:4621-4634.
58. Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, Jin DX, Reinhardt F, Ploegh HL, Wang Q and Gupta PB. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer discovery. 2014; 4:702-715.
59. Chitnis NS, Pytel D, Bobrovnikova-Marjon E, Pant D, Zheng H, Maas NL, Frederick B, Kushner JA, Chodosh LA, Koumenis C, Fuchs SY and Diehl JA. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol Cell. 2012; 48:353-364.
60. Wang S and Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012; 197:857-867.
61. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153-158.
62. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature genetics. 2012; 44:694-698.
63. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807-1812.
64. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, Baker A, Bergsagel PL, Bernstein BE, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011; 471:467-472.
65. Kortum KM, Langer C, Monge J, Bruins L, Zhu YX, Shi CX, Jedlowski P, Egan JB, Ojha J, Bullinger L, Kull M, Ahmann G, Rasche L, Knop S, Fonseca R, Einsele H, et al. Longitudinal analysis of 25 sequential sample-pairs using a custom multiple myeloma mutation sequencing panel (M(3)P). Ann Hematol. 2015; 94:1205-1211.
66. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
67. Wyatt AW and Gleave ME. Targeting the adaptive molecular landscape of castration-resistant prostate cancer. EMBO molecular medicine. 2015; 7:878-894.
68. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, Vinson J, Cao X, Vats P, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215-1228.
69. Takahashi S, Suzuki S, Inaguma S, Ikeda Y, Cho YM, Nishiyama N, Fujita T, Inoue T, Hioki T, Sugimura Y, Ushijima T and Shirai T. Down-regulation of human X-box binding protein 1 (hXBP-1) expression correlates with tumor progression in human prostate cancers. The Prostate. 2002; 50:154-161.
70. So AY, de la Fuente E, Walter P, Shuman M and Bernales S. The unfolded protein response during prostate cancer development. Cancer Metastasis Rev. 2009; 28:219-223.
71. Segawa T, Nau ME, Xu LL, Chilukuri RN, Makarem M, Zhang W, Petrovics G, Sesterhenn IA, McLeod DG, Moul JW, Vahey M and Srivastava S. Androgen-induced expression of endoplasmic reticulum (ER) stress response genes in prostate cancer cells. Oncogene. 2002; 21:8749-8758.
72. Nantermet PV, Xu J, Yu Y, Hodor P, Holder D, Adamski S, Gentile MA, Kimmel DB, Harada S, Gerhold D, Freedman LP and Ray WJ. Identification of genetic pathways activated by the androgen receptor during the induction of proliferation in the ventral prostate gland. The Journal of biological chemistry. 2004; 279:1310-1322.
73. Ma C, Yoshioka M, Boivin A, Gan L, Takase Y, Labrie F and St-Amand J. Atlas of dihydrotestosterone actions on the transcriptome of prostate in vivo. The Prostate. 2009; 69:293-316.
74. Sheng X, Arnoldussen YJ, Storm M, Tesikova M, Nenseth HZ, Zhao S, Fazli L, Rennie P, Risberg B, Waehre H, Danielsen H, Mills IG, Jin Y, Hotamisligil G and Saatcioglu F. Divergent androgen regulation of unfolded protein response pathways drives prostate cancer. EMBO molecular medicine. 2015; 7:788-801.
75. Overcash RF, Chappell VA, Green T, Geyer CB, Asch AS and Ruiz-Echevarria MJ. Androgen signaling promotes translation of TMEFF2 in prostate cancer cells via phosphorylation of the alpha subunit of the translation initiation factor 2. PloS one. 2013; 8:e55257.
76. Huang YL, Chang CL, Tang CH, Lin YC, Ju TK, Huang WP and Lee H. Extrinsic sphingosine 1-phosphate activates S1P5 and induces autophagy through generating endoplasmic reticulum stress in human prostate cancer PC-3 cells. Cellular signalling. 2014; 26:611-618.
77. Thorpe JA and Schwarze SR. IRE1alpha controls cyclin A1 expression and promotes cell proliferation through XBP-1. Cell stress & chaperones. 2010; 15:497-508.
78. Wang Q, Bailey CG, Ng C, Tiffen J, Thoeng A, Minhas V, Lehman ML, Hendy SC, Buchanan G, Nelson CC, Rasko JE and Holst J. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer research. 2011; 71:7525-7536.
79. Jin Y, Wang L, Qu S, Sheng X, Kristian A, Maelandsmo GM, Pallmann N, Yuca E, Tekedereli I, Gorgulu K, Alpay N, Sood A, Lopez-Berestein G, Fazli L, Rennie P, Risberg B, et al. STAMP2 increases oxidative stress and is critical for prostate cancer. EMBO molecular medicine. 2015; 7:315-331.
80. Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T and Sakai T. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer research. 2005; 65:6364-6370.
81. Lu M, Xia L, Hua H and Jing Y. Acetyl-keto-beta-boswellic acid induces apoptosis through a death receptor 5-mediated pathway in prostate cancer cells. Cancer research. 2008; 68:1180-1186.
82. Trepel J, Mollapour M, Giaccone G and Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature reviews Cancer. 2010; 10:537-549.
83. Azad AA, Zoubeidi A, Gleave ME and Chi KN. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nat Rev Urol. 2015; 12:26-36.
84. Pootrakul L, Datar RH, Shi SR, Cai J, Hawes D, Groshen SG, Lee AS and Cote RJ. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006; 12:5987-5993.
85. Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P and Lee AS. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:19444-19449.
86. Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W and Pasqualini R. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer cell. 2004; 6:275-284.
87. Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R and Lee AS. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PloS one. 2013; 8:e80071.
88. Liu R, Li X, Gao W, Zhou Y, Wey S, Mitra SK, Krasnoperov V, Dong D, Liu S, Li D, Zhu G, Louie S, Conti PS, Li Z, Lee AS and Gill PS. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clinical cancer research. 2013; 19:6802-6811.
89. Delie F, Petignat P and Cohen M. GRP78-targeted nanotherapy against castrate-resistant prostate cancer cells expressing membrane GRP78. Target Oncol. 2013; 8:225-230.
90. Maddalo D, Neeb A, Jehle K, Schmitz K, Muhle-Goll C, Shatkina L, Walther TV, Bruchmann A, Gopal SM, Wenzel W, Ulrich AS and Cato AC. A peptidic unconjugated GRP78/BiP ligand modulates the unfolded protein response and induces prostate cancer cell death. PloS one. 2012; 7:e45690.
91. Backer JM, Krivoshein AV, Hamby CV, Pizzonia J, Gilbert KS, Ray YS, Brand H, Paton AW, Paton JC and Backer MV. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia. 2009; 11:1165-1173.
92. Lu T, Yang W, Wang Z, Hu Z, Zeng X, Yang C, Wang Y, Zhang Y, Li F, Liu Z, Wang D and Ye Z. Knockdown of glucose-regulated protein 78/binding immunoglobulin heavy chain protein expression by asymmetric small interfering RNA induces apoptosis in prostate cancer cells and attenuates migratory capability. Molecular medicine reports. 2015; 11:249-256.
93. Mandelin J, Cardo-Vila M, Driessen WH, Mathew P, Navone NM, Lin SH, Logothetis CJ, Rietz AC, Dobroff AS, Proneth B, Sidman RL, Pasqualini R and Arap W. Selection and identification of ligand peptides targeting a model of castrate-resistant osteogenic prostate cancer and their receptors. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112:3776-3781.
94. Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR and Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell. 2009; 138:377-388.
95. Bennett HL, Fleming JT, O’Prey J, Ryan KM and Leung HY. Androgens modulate autophagy and cell death via regulation of the endoplasmic reticulum chaperone glucose-regulated protein 78/BiP in prostate cancer cells. Cell Death Dis. 2010; 1:e72.
96. Thomas SA, Brown IL, Hollins GW, Hocken A, Kirk D, King RJ and Leake RE. Detection and distribution of heat shock proteins 27 and 90 in human benign and malignant prostatic tissue. Br J Urol. 1996; 77:367-372.
97. Rocchi P, So A, Kojima S, Signaevsky M, Beraldi E, Fazli L, Hurtado-Coll A, Yamanaka K and Gleave M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer research. 2004; 64:6595-6602.
98. Rocchi P, Beraldi E, Ettinger S, Fazli L, Vessella RL, Nelson C and Gleave M. Increased Hsp27 after androgen ablation facilitates androgen-independent progression in prostate cancer via signal transducers and activators of transcription 3-mediated suppression of apoptosis. Cancer research. 2005; 65:11083-11093.
99. Kumano M, Furukawa J, Shiota M, Zardan A, Zhang F, Beraldi E, Wiedmann RM, Fazli L, Zoubeidi A and Gleave ME. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Molecular cancer therapeutics. 2012; 11:1661-1671.
100. Shiota M, Bishop JL, Nip KM, Zardan A, Takeuchi A, Cordonnier T, Beraldi E, Bazov J, Fazli L, Chi K, Gleave M and Zoubeidi A. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer research. 2013; 73:3109-3119.
101. Saha S, Bhanja P, Partanen A, Zhang W, Liu L, Tome W and Guha C. Low intensity focused ultrasound (LOFU) modulates unfolded protein response and sensitizes prostate cancer to 17AAG. Oncoscience. 2014; 1:434-445. doi: 10.18632/oncoscience.48.
102. Lamoureux F, Thomas C, Yin MJ, Fazli L, Zoubeidi A and Gleave ME. Suppression of heat shock protein 27 using OGX-427 induces endoplasmic reticulum stress and potentiates heat shock protein 90 inhibitors to delay castrate-resistant prostate cancer. European urology. 2014; 66:145-155.
103. Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW and Wang X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell. 2010; 143:711-724.
104. Itkonen HM, Engedal N, Babaie E, Luhr M, Guldvik IJ, Minner S, Hohloch J, Tsourlakis MC, Schlomm T and Mills IG. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene. 2015; 34:3744-3750.
105. Yoshida T, Shiraishi T, Horinaka M, Wakada M and Sakai T. Glycosylation modulates TRAIL-R1/death receptor 4 protein: different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncology reports. 2007; 18:1239-1242.
106. Horak P, Tomasich E, Vanhara P, Kratochvilova K, Anees M, Marhold M, Lemberger CE, Gerschpacher M, Horvat R, Sibilia M, Pils D and Krainer M. TUSC3 loss alters the ER stress response and accelerates prostate cancer growth in vivo. Scientific reports. 2014; 4:3739.
107. Drake RR, Jones EE, Powers TW and Nyalwidhe JO. Altered glycosylation in prostate cancer. Adv Cancer Res. 2015; 126:345-382.
108. Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo YY, Fukuda K, Li Y and Watabe K. N-myc downstream regulated gene 1 modulates Wnt-beta-catenin signalling and pleiotropically suppresses metastasis. EMBO molecular medicine. 2012; 4:93-108.
109. Tu LC, Yan X, Hood L and Lin B. Proteomics analysis of the interactome of N-myc downstream regulated gene 1 and its interactions with the androgen response program in prostate cancer cells. Molecular & cellular proteomics : MCP. 2007; 6:575-588.
110. Zhang P, Gao K, Tang Y, Jin X, An J, Yu H, Wang H, Zhang Y, Wang D, Huang H, Yu L and Wang C. Destruction of DDIT3/CHOP protein by wild-type SPOP but not prostate cancer-associated mutants. Hum Mutat. 2014; 35:1142-1151.
111. Yao Z, Sun B, Hong Q, Yan J, Mu D, Li J, Sheng H and Guo H. PACE4 regulates apoptosis in human prostate cancer cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Drug Des Devel Ther. 2015; 9:5911-5923.
112. Wang Z, Gao D, Fukushima H, Inuzuka H, Liu P, Wan L, Sarkar FH and Wei W. Skp2: a novel potential therapeutic target for prostate cancer. Biochimica et biophysica acta. 2012; 1825:11-17.
113. Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, Cordon-Cardo C, Teruya-Feldstein J and Pandolfi PP. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010; 464:374-379.
114. Lin HP, Lin CY, Huo C, Hsiao PH, Su LC, Jiang SS, Chan TM, Chang CH, Chen LT, Kung HJ, Wang HD and Chuu CP. Caffeic acid phenethyl ester induced cell cycle arrest and growth inhibition in androgen-independent prostate cancer cells via regulation of Skp2, p53, p21Cip1 and p27Kip1. Oncotarget. 2015; 6:6684-6707. doi: 10.18632/oncotarget.3246.
115. Farrow JM, Yang JC and Evans CP. Autophagy as a modulator and target in prostate cancer. Nat Rev Urol. 2014; 11:508-516.
116. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010; 18:11-22.
117. Bitting RL and Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocrine-related cancer. 2013; 20:R83-99.
118. Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM and Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. The Journal of biological chemistry. 2007; 282:4702-4710.
119. Bhutia SK, Dash R, Das SK, Azab B, Su ZZ, Lee SG, Grant S, Yacoub A, Dent P, Curiel DT, Sarkar D and Fisher PB. Mechanism of autophagy to apoptosis switch triggered in prostate cancer cells by antitumor cytokine melanoma differentiation-associated gene 7/interleukin-24. Cancer research. 2010; 70:3667-3676.
120. Rah B, ur Rasool R, Nayak D, Yousuf SK, Mukherjee D, Kumar LD and Goswami A. PAWR-mediated suppression of BCL2 promotes switching of 3-azido withaferin A (3-AWA)-induced autophagy to apoptosis in prostate cancer cells. Autophagy. 2015; 11:314-331.
121. Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E and DiPaola RS. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016; 30:399-407.
122. Morgan TM, Koreckij TD and Corey E. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 2009; 9:237-249.
123. Maly DJ and Papa FR. Druggable sensors of the unfolded protein response. Nat Chem Biol. 2014; 10:892-901.
124. Wu Y, Zhang H, Dong Y, Park YM and Ip C. Endoplasmic reticulum stress signal mediators are targets of selenium action. Cancer research. 2005; 65:9073-9079.
125. Zu K, Bihani T, Lin A, Park YM, Mori K and Ip C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene. 2006; 25:546-554.
126. Preston MA, Riis AH, Ehrenstein V, Breau RH, Batista JL, Olumi AF, Mucci LA, Adami HO and Sorensen HT. Metformin use and prostate cancer risk. European urology. 2014; 66:1012-1020.
127. Yang J, Wei J, Wu Y, Wang Z, Guo Y, Lee P and Li X. Metformin induces ER stress-dependent apoptosis through miR-708-5p/NNAT pathway in prostate cancer. Oncogenesis. 2015; 4:e158.
128. Modernelli A, Naponelli V, Giovanna Troglio M, Bonacini M, Ramazzina I, Bettuzzi S and Rizzi F. EGCG antagonizes Bortezomib cytotoxicity in prostate cancer cells by an autophagic mechanism. Scientific reports. 2015; 5:15270.
129. Chiu SC, Chen SP, Huang SY, Wang MJ, Lin SZ, Harn HJ and Pang CY. Induction of apoptosis coupled to endoplasmic reticulum stress in human prostate cancer cells by n-butylidenephthalide. PloS one. 2012; 7:e33742.
130. Chiu SC, Huang SY, Chen SP, Su CC, Chiu TL and Pang CY. Tanshinone IIA inhibits human prostate cancer cells growth by induction of endoplasmic reticulum stress in vitro and in vivo. Prostate cancer and prostatic diseases. 2013; 16:315-322.
131. Song JH and Kraft AS. Pim kinase inhibitors sensitize prostate cancer cells to apoptosis triggered by Bcl-2 family inhibitor ABT-737. Cancer research. 2012; 72:294-303.
132. Lee WJ, Chien MH, Chow JM, Chang JL, Wen YC, Lin YW, Cheng CW, Lai GM, Hsiao M and Lee LM. Nonautophagic cytoplasmic vacuolation death induction in human PC-3M prostate cancer by curcumin through reactive oxygen species -mediated endoplasmic reticulum stress. Scientific reports. 2015; 5:10420.
133. Wang M, Shim JS, Li RJ, Dang Y, He Q, Das M and Liu JO. Identification of an old antibiotic clofoctol as a novel activator of unfolded protein response pathways and an inhibitor of prostate cancer. British journal of pharmacology. 2014; 171:4478-4489.
134. Chiu HW, Fang WH, Chen YL, Wu MD, Yuan GF, Ho SY and Wang YJ. Monascuspiloin enhances the radiation sensitivity of human prostate cancer cells by stimulating endoplasmic reticulum stress and inducing autophagy. PloS one. 2012; 7:e40462.
135. Zhang TW, Xing L, Tang JL, Lu JX and Liu CX. Marchantin M Induces Apoptosis of Prostate Cancer Cells Through Endoplasmic Reticulum Stress. Med Sci Monit. 2015; 21:3570-3576.
136. Jiang H, Sun J, Xu Q, Liu Y, Wei J, Young CY, Yuan H and Lou H. Marchantin M: a novel inhibitor of proteasome induces autophagic cell death in prostate cancer cells. Cell Death Dis. 2013; 4:e761.
137. Guan M, Su L, Yuan YC, Li H and Chow WA. Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Scientific reports. 2015; 5:9698.
138. Gara RK, Srivastava VK, Duggal S, Bagga JK, Bhatt M, Sanyal S and Mishra DP. Shikonin selectively induces apoptosis in human prostate cancer cells through the endoplasmic reticulum stress and mitochondrial apoptotic pathway. Journal of biomedical science. 2015; 22:26.
139. Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH, Yang M, Liu X and Guo DA. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. Journal of cellular physiology. 2012; 227:2196-2206.
140. Rizzi F, Naponelli V, Silva A, Modernelli A, Ramazzina I, Bonacini M, Tardito S, Gatti R, Uggeri J and Bettuzzi S. Polyphenon E(R), a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized PNT1a cells by anoikis and tumorigenic PC3 by necroptosis. Carcinogenesis. 2014; 35:828-839.
141. Sun S, Han J, Ralph WM, Jr., Chandrasekaran A, Liu K, Auborn KJ and Carter TH. Endoplasmic reticulum stress as a correlate of cytotoxicity in human tumor cells exposed to diindolylmethane in vitro. Cell stress & chaperones. 2004; 9:76-87.
142. Sanchez AM, Martinez-Botas J, Malagarie-Cazenave S, Olea N, Vara D, Lasuncion MA and Diaz-Laviada I. Induction of the endoplasmic reticulum stress protein GADD153/CHOP by capsaicin in prostate PC-3 cells: a microarray study. Biochemical and biophysical research communications. 2008; 372:785-791.
143. Hessenauer A, Schneider CC, Gotz C and Montenarh M. CK2 inhibition induces apoptosis via the ER stress response. Cellular signalling. 2011; 23:145-151.
144. Bruno RD, Gover TD, Burger AM, Brodie AM and Njar VC. 17alpha-Hydroxylase/17,20 lyase inhibitor VN/124-1 inhibits growth of androgen-independent prostate cancer cells via induction of the endoplasmic reticulum stress response. Molecular cancer therapeutics. 2008; 7:2828-2836.
145. Huong PT, Moon DO, Kim SO, Kim KE, Jeong SJ, Lee KW, Lee KS, Jang JH, Erikson RL, Ahn JS and Kim BY. Proteasome inhibitor-I enhances tunicamycin-induced chemosensitization of prostate cancer cells through regulation of NF-kappaB and CHOP expression. Cellular signalling. 2011; 23:857-865.
146. Zhao F, Huang W, Zhang Z, Mao L, Han Y, Yan J and Lei M. Triptolide induces protective autophagy through activation of the CaMKKbeta-AMPK signaling pathway in prostate cancer cells. Oncotarget. 2016; 7:5366-5382. doi: 10.18632/oncotarget.6783.