
INTRODUCTION
Molecular chaperones assist in the folding of nascent polypeptides and the correct assembly or disassembly of protein complexes [1, 2]. A majority of chaperones are the so-called heat-shock proteins (HSPs), which are expressed in response to increased temperature or a variety of other cellular stresses. Among them, heat shock protein 90 (HSP90) is a conserved molecular chaperone and is involved in stabilizing and activating more than 200 proteins[2]. Since many HSP90 ‘clients’ are known oncogenic proteins, such as tyrosine kinases[3-5], steroid hormone receptors[6], AKT[7], HIF1α[8] and MMP2[9], that are known to sustain cancer cell growth, differentiation and survival. HSP90 chaperone machinery enables mutated oncoproteins to escape from misfolding and degradation and allows for malignant transformation [2, 10, 11]. Therefore, HSP90 is considered as a synthetic lethal target [12-14].
After the first HSP90 inhibitor, 17-AAG (tanespimycin), entered clinical trials in 1999, thirteen different HSP90 inhibitors are currently undergoing clinical evaluation in cancer patients in twenty-three active oncology trials [15]. Each of these inhibitors disrupts HSP90 activity by replacing ATP in the N-terminal nucleotide-binding pocket [11, 15]. NVP-AUY922 and NVP-HSP990 are novel, non-geldanamycin-derivative HSP90 inhibitors [16]. Both compounds showed significant antitumor activity in a wide range of mutated and wild-type cancer cell lines, primary tumor cells and animal models of cancer, including melanoma, myeloma, gastric cancer, non-small-cell lung cancer(NSCLC), hepatocellular cancer, sarcoma, and breast cancer [16-19]. Progress has also been made in terms of identifying sensitive cancer indications and effective drug combinations: in HER2+ breast cancer, HSP90 inhibitors block HER2 signaling and suppress tumor growth as the stability of HER2 protein is dependent on HSP90. In a Phase II clinical trial following combination of trastuzumab with 17-AAG treatment, a response rate of 24% was reported and clinical benefit was observed in more than 57% of evaluated patients[2, 20].
Although significant progress has been made and promising results have been seen in breast cancer patients receiving HSP90 inhibitor treatment, HSP90 inhibitor was also shown to lack efficacy in certain cancer types, such as melanoma. In a Phase II trial of 17-AAG in patients with metastatic melanoma, no objective anti-melanoma responses were observed [21]. Therefore, understanding the resistant mechanisms of cancer cells in response to HSP90 inhibition will help us to develop the effective combinational therapy with HSP90 inhibitor.
To identify the genetic modulators of HSP90 inhibition, we performed pooled shRNA screening to search the potential combinational targets of HSP90 inhibitor, and identified HSF1 as a sensitizer of HSP90 inhibitor. HSF1 is a conserved transcription factor and a major regulator of the heat shock response [22, 23]. Beyond heat shock response, HSF1 also regulates a transcriptional program highly specific to malignant cell including cell cycle, cell signaling, metabolism, adhesion and translation [23-25]. Recently, eliminating HSF1 was showed to protect mice from tumors induced by mutation of the RAS oncogene or a hot spot mutation in tumor suppressor p53 and from DEN-induced hepatocellular carcinoma (HCC) formation [24, 26]. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis through dysregulated MAPK signaling [27]. Moreover, HSF1 knock-out or knock-down cells were shown to be more sensitive to HSP90 inhibitor [28-31]. Those studies indicate that HSF1 may play an important role in tumor initiation, development and maintenance, and contribute to cell sensitivity to HSP90 inhibitor. However, the functional role of HSF1 in human cancer cell resistance to HSP90 inhibitors and the mechanisms underlying the combination effect of HSF1 knockdown and HSP90 inhibitors are not fully understood. Moreover, the downstream targets of HSF1 which may play a role in attenuating the effect of HSP90 inhibitor are not fully appreciated.
In this study, we observed that HSF1 knockdown combined with HSP90 inhibitors led to striking inhibitory effects on cancer cell proliferation in vitro and tumor growth in vivo. HSF1 knockdown combined with HSP90 inhibition facilitates the degradation of oncogenic proteins, induces cancer cell apoptosis, and decreases activity of the ERK pathway. HSF1 expression is significantly up-regulated in HCC, suggesting a tumor type that may be targeted by combinational treatment. Finally, we identify DEDD2 as a HSF1 target gene involved in the resistance to HSP90 inhibition.
RESULTS
Pooled shRNA screening reveals that HSF1 as a top sensitizer to HSP90 inhibitor
To identify genes that modulate the efficacy of HSP90 inhibition on tumor cell growth, we performed a large-scale RNA interference (RNAi) genetic screen with a collection of short hairpin RNA (shRNA) vectors targeting 1,000 human genes in A375 (Fig. 1A). A barcoding technique was used to identify genes whose suppression caused resistance or sensitivity to two separate concentrations of NVP-AUY922 (Fig. 1B). 163 and 360 shRNA constructs were significantly depleted form either low- or high-dose NVP-AUY922 treated samples (FDR<=0.15). Among those shRNA hits, 84 hits (including 81 genes) were common shRNA hits as shown in Venn diagram (Fig. 1C) and sensitizing genes or rescuing genes were also shown (Z score≥3, or Z score≤-3, Supplementary Table. S1). Among of these shRNA hits, HSF1 and heat shock protein 90 alpha, class B member 1(HSP90AB1) knockdown scored as the most top sensitizers to HSP90 inhibition in A375 cells, and are known to regulate the cell response to heat shock conditions (Fig. 1D), which may reflect the potential feedback mechanism of HSP90 inhibition. Taking together, HSF1 is identified as a sensitizer of HSP90 inhibitor through pooled shRNA screening.
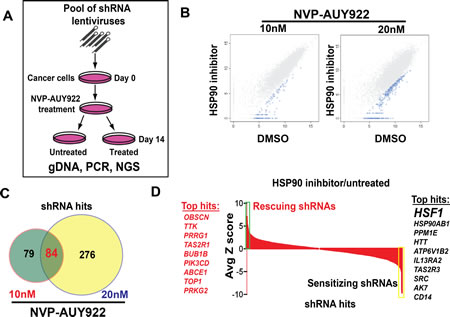
Figure 1: Pooled shRNA screening reveals that HSF1 as a top sensitizer to HSP90 inhibitor. A. The schematic of pooled shRNA screening experiment design. B. Scatter plots of log2 normalized read counts from pooled shRNA screening performed in A375 cells treated with 10nM/20nM NVP-AUY922 or control dimethyl sulfoxide (DMSO) samples. Hairpins that were statistically significantly depleted in NVP-AUY922 treated samples were highlighted in blue color. Each dot in the plot represents one individual shRNA construct. C. Venn diagram showed that 163 shRNAs were identified in lower dose NVP-AUY922 and 360 shRNAs were identified from higher dose NVP-AUY922 from pooled shRNA screening performed in A375 cell. 84 shared shRNAs were found between two experiments. D. Average Z score of shRNA hits from pooled shRNA screening performed in A375 cells was shown in a waterfall plot. Top sensitizing genes including HSF1 were highlighted in yellow color while top rescuing genes were shown in green.
HSF1 knockdown sensitizes cancer cells to HSP90 inhibitor in vitro and in vivo
To validate whether HSF1 was indeed a sensitizer of HSP90 inhibition, two HSF1 inducible shRNA constructs by targeting distinct HSF1 sequence were stably introduced into different cancer cell lines: A375, A2058 and HCT116. When shRNA expression was induced by Doxycycline, robust HSF1 knockdown was achieved in all three cancer cell lines (Fig. 2A). We next tested whether HSF1 knockdown has a combinational effect with NVP-AUY922 or NVP-HSP990. Induction of HSF1 shRNA (but not the NTC shRNA) led to IC50 of NVP-HSP990 shifting from 19nM to 6nM in A375 cell, 12.7nM to 5.2nM in A2058 cell (Fig. 2B). The combination effect was even more dramatically observed in extended colony formation assays (Fig. 2C). In HCT116 cells, HSF1 knockdown led to a significant shift of LD50 of either NVP-HSP990 or NVP-AUY922 (more than 6 fold change) (Fig. 2D). To further validate HSF1 as a sensitizer of HSP90 inhibitor, the combinational effect of HSF1 knockdown with HSP90 inhibitor was tested in A375 xenograft mouse model. HSF1 shRNA alone inhibited tumor growth by 53% T/C, and knockdown was confirmed (Fig. 2E and F). NVP-HSP990 alone at tolerated dosage (10mg/kg PO, qw) inhibited tumor growth by 0.01% T/C (Fig. 2F). More strikingly, HSF1 knockdown & NVP-HSP990 combination led to 76% tumor regression (Fig. 2F). HSP70 level induced by HSP90 inhibition was also significantly reduced upon HSF1 knockdown (Fig. 2E). These results suggest that HSF1 is critical for limiting the efficacy of HSP90 inhibitor in human cancer cells both in vitro and in vivo.
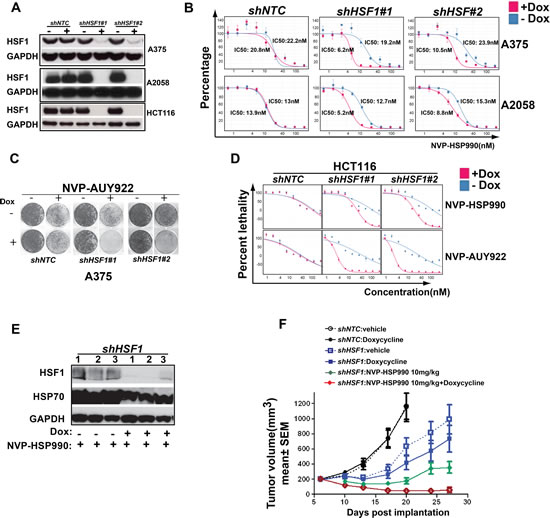
Figure 2: HSF1 knockdown sensitizes cancer cells to HSP90 inhibitor in vitro and in vivo. A. Western blotting analysis of HSF1 knockdown in A375, A2058 and HCT116 cells. shNTC or shHSF1 transduced stable cell lines were treated with Doxycycline for 3 days and cell pellets were collected and western blotting was performed. B. IC50 of HSP90 inhibitor with or without HSF1 knockdown. shNTC or shHSF1 transduced cancer cells were treated with or without Doxycycline for 3 days, then followed by treatment of a serial dilutions of NVP-HSP990 for 5 days. Relative cell growth (average of at least 3 independent experiments) was measured by CellTiter-Glo and normalized to DMSO-treated cells. C. Cell colony formation assay of HSF1 knockdown with HSP90 inhibitor treatment. shNTC or shHSF1 transduced A375 cells were treated or untreated with Doxycycline for 5 days, then followed by compound treatment for 6 days. D. LD50s of HSP90 inhibitors: NVP-AUY922 and NVP-HSP990 with or without HSF1 knockdown in HCT116 cells. shNTC or shHSF1 transduced cancer cells were treated with or without Doxycycline for 3 days, then followed by being treated for 5 days with serial dilutions of NVP-HSP990 or NVP-AUY922. Relative cell growth (average of at least 3 independent experiments) was measured by CellTiter-Glo. Cell relative death rate was calculated. E. Western blotting analysis of tumor samples. Tumor samples were collected at the end of studies and western blotting analysis of HSF1, HSP70 and GAPDH were performed. F. The combinational effect of HSF1 knockdown and HSP90 inhibitor in A375 xenograft mouse model. Tumor growth rate of A375 cells expressing inducible control shRNA or shRNA against HSF1 under Doxycycline and/or NVP-HSP990 were compared at different time points. Tumor inhibition effects were calculated relative to control group at day 20.
HSF1 knockdown sensitizes HCC cells to HSP90 inhibition
To identify the cancer type which may be useful for the stratification of the combinational treatment, we examined HSF1 expression in TCGA database and found that HSF1 is over-expressed in several tumor types (Supplementary Figure S1). Among them, we are particularly interested in HCC since HSF1 has been reported to be a key modulator of HCC development in mouse model [26]. HSF1 mRNA was significantly elevated in HCC tumor in compared to normal control (Fig. 3A). To examine the protein expression of HSF1 in HCC patients, immunohistochemistry (IHC) study for HSF1 was performed on primary human HCC samples and non-neoplastic liver samples and the HSF1 antibody was validated by using Hep3B cell pellets with inducible HSF1 shRNAs. HSF1 staining was significantly decreased in Hep3B cells treated with Doxycycline in comparison with cells without Doxycycline treatment (Supplementary Fig. S2). HSF1 staining was then scored in 50 human HCC tumor samples. Samples that were not immunoreactive (Ki67 negative) or had insufficient tumor were excluded from the analysis. HSF1 expression was not observed in non-neoplastic human hepatocytes of the 45 HCC tumor samples were evaluable, 35 HCC cases showed positive HSF1 staining (Fig. 3B and Table 2). The intensity of staining varied considerably from tumor to tumor. These results suggest that both mRNA and protein level of HSF1 expression are increased in HCC.
Table 1: HSF1 knockdown sensitizes cancer cells to HSP90 inhibitor. The combinational effect of HSF1 knockdown and HSP90 inhibition was observed among 2 melanoma cell lines, 2 HCC lines and 1 colon cancer cell line |
|||
|
In vitro |
|
In vivo |
Cell line |
IC50 |
LD50 |
|
A375 |
2.9 |
N/A |
Regression |
A2058 |
2 |
N/A |
|
HCT116 |
N/A |
6.5 |
|
Hep3B |
2.1 |
1.9 |
Stasis |
Huh7 |
3.1 |
7 |
|
Average fold change of IC50 or LD50 of HSP90 inhibitor caused by HSF1 knockdown |
|||
Next, we tested whether HSF1 knockdown could inhibit the proliferation of HCC and whether there is a combinational effect of HSF1 knockdown and HSP90 inhibitor in HCC cells. Indeed, both the cell proliferation and colony formation assay showed that HSF1 knockdown inhibited the growth of HCC in vitro (Fig. 3C and D). In addition, HSF1 knockdown sensitized both Hep3B and Huh7 cells to HSP90 inhibitor at different levels in vitro (Table 1). To evaluate whether the phenotype mediated by HSF1 shRNA knockdown and the combination effect mediated by HSF1 shRNA knockdown and HSP90 inhibition was on-target, RNAi-resistant HSF1 (“HSF1R”) cDNA was expressed in Hep3B cells with inducible HSF1 knockdown. HSF1R cDNA restored the expression of HSF1 and HSP70 in Hep3B cell lines when endogenous HSF1 and HSP70 expression was decreased by Doxycycline treatment (Fig. 3E). HSF1R cDNA expression rescued both growth phenotype medicated by HSF1 knockdown (Fig. 3F) and the combinational effects caused by HSF1 knockdown and HSP90 inhibition (Fig. 3G), which indicate that the effects of the HSF1 shRNA constructs are indeed on-target effects. The effect of HSF1 knockdown on the proliferation of tumor cells was also tested in Hep3B xenograft mouse model. HSF1 shRNA alone inhibited tumor growth by 71%, and knockdown was confirmed (Fig. 3H) and NVP-HSP990 alone at tolerated dosage (10mg/kg PO, qw) inhibited tumor growth by 47% (Fig. 3I). However, HSF1 knockdown & NVP-HSP990 combination reduced the HSP90 inhibitor induced cell stress response and led to tumor stasis (Fig. 3H and I).
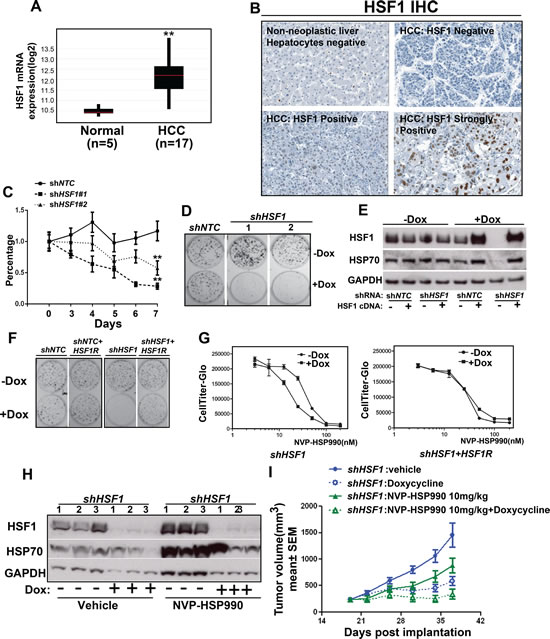
Figure 3: HSF1 knockdown sensitizes hepatocellular cancer cells to HSP90 inhibitor in vitro and in vivo. A. The mRNA level of HSF1 was measured among tumor samples from HCC patients compared to normal control. B. IHC showed the expression of HSF1 in primary human hepatocellular cancer. C. The cell growth curve of Hep3B cell with or without HSF1 knockdown at different time points. shNTC or shHSF1 transduced Hep3B cells were treated with or without Doxycycline for 7 days. Relative cell growth (average of at least 3 independent experiments) was measured by CellTiter-Glo and normalized to cells without Doxycycline treatment. D. Cell colony formation assay of HSF1 knockdown in Hep3B cells. shNTC or shHSF1 transduced Hep3B cells were treated with Doxycycline for 12 days. E. Western blotting analysis of Hep3B cells expressing the indicated shRNA and cDNA contruct. The inducible lenti-virus construct (HSF1R cDNA) was used to transduce Hep3B cells and expression of HSF1R cDNA with or without HSF1 knockdown were tested. HSF1, HSP70 and GAPDH were detected by western blotting. F. Cell colony formation assay of over-expression of HSF1R cDNA in Hep3B cells with or without HSF1 knockdown. G. The comparison of dose response of NVP-HSP990 in Hep3B cells with inducible expression of either HSF1 shRNA alone or both HSF1 shRNA and HSF1R cDNA. HSF1 shRNA alone or both HSF1 shRNA and HSF1R cDNA transduced cancer cells were treated with or without Doxycycline for 3 days, then followed by being treated for 5 days with serial dilutions of NVP-HSP990. H. Western blotting analysis of tumor samples. Tumor samples were collected at the end of studies and western blotting analysis of HSF1, HSP70 and GAPDH were performed. I. The combinational effect of HSF1 knockdown and HSP90 inhibitor in Hep3B xenograft mouse model. Tumor growth rate of Hep3B cells expressing inducible control shRNA or shRNA against HSF1 under Doxycycline and/or NVP-HSP990 were compared at different time points.
Table 2: Immunohistochemistry study showed that the expression of HSF1 is significantly up-regulated in primary human hepatocellular cancer. The expression of HSF1 was measured by immunohistochemistry (IHC) in hepatocellular carcinomas (50 Hep B virus positive cases, 2 cores/case). |
|||
Hepatocellular carcinoma |
|||
Case coordinates |
HSF1 |
Ki67 |
IgG |
A1,2 |
POS-very weak |
POS |
NEG |
A3,4 |
POS |
POS |
NEG |
A5,6 |
POS-strong |
TI |
NEG |
A7,8 |
POS |
TI |
NEG |
B1,2 |
TI |
TI |
TI |
B3,4 |
POS-strong |
POS |
NEG |
B5,6 |
POS |
POS |
NEG |
B7,8 |
POS-weak |
POS |
NEG |
B9,10 |
POS-very weak |
TI |
NEG |
B11,12 |
NEG |
TI |
NEG |
C1,2 |
POS |
POS |
NEG |
C3,4 |
NEG |
POS |
NEG |
C5,6 |
NEG |
POS |
NEG |
C7,8 |
POS |
POS |
NEG |
C9,10 |
POS |
POS |
NEG |
C11,12 |
POS |
POS |
NEG |
D1,2 |
POS |
POS |
NEG |
D3,4 |
NEG |
POS |
Brown pigment c/w lipofuscin |
D5,6 |
POS |
POS |
NEG |
D7,8 |
NEG |
POS |
NEG |
D9,10 |
POS |
POS |
NEG |
D11,12 |
POS-weak |
POS |
NEG |
E1,2 |
NEG |
POS |
NEG |
E3,4 |
POS-very weak |
POS |
NEG |
E5,6 |
NEG |
POS |
Brown pigment c/w bile |
E7,8 |
POS |
POS |
NEG |
E9,10 |
POS-strong |
POS |
NEG |
E11,12 |
POS-weak |
POS |
NEG |
F1,2 |
POS-weak |
POS |
NEG |
F3,4 |
POS-weak |
POS |
NEG |
F5,6 |
NEG |
POS |
NEG |
F7,8 |
POS-very weak |
POS |
NEG |
F9,10 |
NEG |
POS |
NEG |
F11,12 |
POS-weak |
POS |
NEG |
G1,2 |
POS |
POS |
NEG |
G3,4 |
|
NEG |
NEG |
G5,6 |
|
NEG |
NEG |
G7,8 |
POS-weak |
POS |
NEG |
G9,10 |
POS-weak |
POS |
NEG |
G11,12 |
POS-weak |
POS |
NEG |
H1,2 |
POS-very weak |
POS-very weak |
NEG |
H3,4 |
POS |
POS |
Minimal brown pigment |
H5,6 |
POS-very weak |
POS |
NEG |
H7,8 |
POS-very weak |
POS-very weak |
NEG |
H9,10 |
POS-very weak |
POS |
NEG |
H11,12 |
POS |
POS |
NEG |
I1,2 |
|
NEG |
NEG |
I3,4 |
POS-very weak |
POS |
NEG |
I5,6 |
NEG |
POS |
NEG |
I7,8 |
NEG |
POS |
NEG |
A9,10 (non-neoplastic) |
Hepatocytes negative; inflammatory cells positive |
TI |
NEG |
A11,12 (non-neoplastic) |
Hepatocytes negative; inflammatory cells positive |
TI |
Brown pigment c/w lipofuscin |
Combination of HSF1 knockdown and HSP90 inhibition leads to a decreased level of p-ERK and an increase of cell apoptosis
To understand the mechanism of the combination effects of HSF1 knockdown and HSP90 inhibition, we tested: 1) whether HSF1 knockdown may facilitate the degradation of HSP90 client protein by HSP90 inhibition, such as BRAF or HER2 oncogenic proteins; 2) whether HSP90 inhibition may enhance the attenuation of MAPK signaling mediated by HSF1 knockdown as recent finding suggests that HSF1 deficiency attenuates MAPK signaling in mice[27] and 3) HSF1 may regulate other target genes rather than HSP70, which may play a role in attenuating the effect of HSP90 inhibition. Therefore, we examined the status of HSP90 client proteins and the downstream effects in cell treated with either HSF1 shRNA or HSP90 inhibitor or combination of HSF1 shRNA and HSP90 inhibitor. HCT116 cells were treated with different doses of NVP-HSP990 and HSF1 knockdown in combination with NVP-HSP990 (5nM) reduced the HSP70, p-ERK and HER2 levels significantly while NVP-HSP990 or HSF1 knockdown alone did not(Fig. 4A). HSF1 knockdown in combination with NVP-HSP990 (25nM) led increased cleaved PARP (Fig. 4A). The combination also led an enhanced degradation of BRAF in A375 cells (Fig. 4B). A decreased level of HSP70, p-ERK and increased level of cleaved PARP were also observed in melanoma cells (Fig. 4B) and hepatocellular cancer cells (Supplementary Fig. S3). To understand how HSF1 knockdown affects the cell proliferation under HSP90 inhibitor treatment, cell cycle analysis was performed. HSF1 knockdown didn’t affect the percentage of cancer cells in cell cycle while HSP90 inhibitor caused more cancer cells into S+G2M phase (data not shown). In contrast, the percentage of cancer cells in the S+G2M phase was significantly decreased in HSF1 knockdown group than in the control group under HSP90 inhibitor treatment(Fig. 4C), indicating that under HSP90 inhibition the knockdown of HSF1 blocks cancer cells to enter the cell cycle, thereby decrease the proliferation of cancer cells. Next, we examined whether HSF1 knockdown may enhance apoptosis of cancer cells under HSP90 inhibitor treatment by staining the cells with 7AAD and Annexin V. Similarly, HSF1 knockdown didn’t affect the apoptosis of cancer cells while HSP90 inhibitor induced the apoptosis of cancer cells (data not shown). HSF1 knockdown further enhanced the apoptotic proportion of cancer cells under HSP90 inhibitor treatment (Fig. 4D). Thus, the combination treatment of HSF1 knockdown and HSP90 inhibition facilitates the degradation of HSP90 client proteins, such as BRAF and HER2, inhibits MAPK growth signaling and results in cell cycles arrest and cell apoptosis.
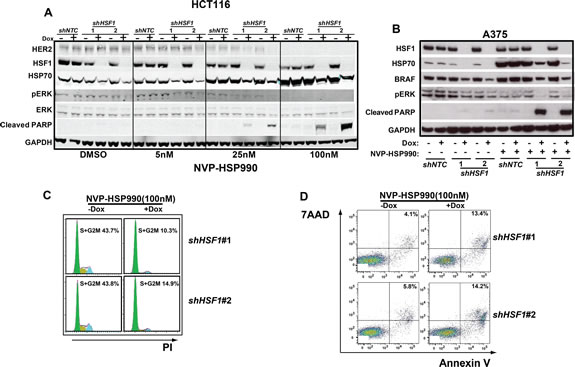
Figure 4: Combination of HSF1 knockdown and HSP90 inhibitor leads to a decreased level of p-ERK and an increased cell apoptosis rate. A. Western blotting analysis of HCT116 cells expressing the inducible shHSF1 treated with different doses of NVP-HSP990. shNTC or shHSF1 transduced HCT116 cells were treated with or without Doxycycline for 3 days and were further treated with different doses of NVP-HSP990 for 48h. B. Western blotting analysis of A375 cells expressing the inducible shRNA treated with different doses of NVP-HSP990. shNTC or shHSF1 transduced A375 cells were treated with or without Doxycycline for 3 days and were further treated with NVP-HSP990 100nM for 48h. C. Cell cycle analysis of A375 cells treated with NVP-HSP990 with or without HSF1 knockdown. shHSF1 transduced A375 cells were treated with or without Doxycycline for 3 days and were further treated with NVP-HSP990 100nM for 48h. The percentage of S+G2M cells were determined by PI staining. D. Cell apoptosis analysis of A375 cells with HSF1 knockdown and NVP-HSP990 treatment. shHSF1 transduced A375 cells were treated with or without Doxycycline for 3 days and were further treated with NVP-HSP990 100nM for 48h. The apoptotic cells represented by 7AAD+AnnexinV+ were determined by FACS.
DEDD2 is a HSF1-target gene involved in attenuation of the effect of HSP90 inhibitor
Previous studies suggest that HSP70/HSC70 knockdown has a combinational effect with HSP90 inhibitor, which might partially account for certain extend of the combinational effect of HSF1 knockdown with HSP90 inhibitor [32]. To identify additional HSF1-target genes, we compared the gene profiles in A375 cells with or without HSF1 knockdown treated or untreated with HSP90 inhibitor. Several genes are up-regulated by HSP90 inhibitor treatment and this upregulation are diminished by HSF1 knockdown (Fig. 5A). Most of those genes belong to well-known HSF1-regulated cell stress pathway, such as: HSPA1L, HSPA1A, HSPA6, HSPB1, HSPA4C and DNAJR1 (Fig. 5A). Among these top hits, apart from heat-shock pathway genes regulated by HSF1, BAG3 and DEDD2 are involved in cell death [33, 34]. Previously, BAG3 was identified as a HSF1 target gene and DEDD2 was defined as one of genes included in the molecular signature in response to the HSP90 inhibitor [35-37]. BAG3, a member of the Bcl-2-associated athanogen family, was reported as a mediator of a novel macroautophagy pathway that uses the specificity of HSP70 to misfolded proteins [33]. DEDD2 associates with DEDD and is involved in the regulation of nuclear events mediated by the extrinsic apoptosis pathway [37]. DEDD2 might be an important mediator for death receptors and target caspases to the nucleus [34]. However, the functional roles of BAG3 and DEDD2 in cancer cell response to HSP90 inhibition are not explored. Therefore, we decided to further validate whether BAG3 and DEDD2 may play a role in the combinational effect mediated by HSF1 knockdown and HSP90 inhibition. Like HSP70, both BAG3 and DEDD2 mRNA expression were increased upon HSP90 inhibition, which were abolished upon HSF1 knockdown (Fig. 5B and Supplement Fig. S4). DEDD2 protein expression was not significantly regulated by HSP90 inhibition, but its basal level was decreased upon HSF1 knockdown (Fig.5C). In comparison, HSP70 protein level was significantly increased by HSP90 inhibition, which was abolished by HSF1 knockdown (Fig.5C). Furthermore, DEDD2 expression was elevated by over-expression of HSF1 (Fig. 5D). ChIP experiment also showed that HSF1 is bound to DEDD2 promoter in response to HSP90 inhibition (Fig. 5E). These results suggest that DEDD2 is a direct target gene of HSF1. DEDD2 knockdown alone mildly enhances effect of HSP90 inhibitor (Fig. 5F). In comparison, BAG3 knockdown did not affect the activity of HSP90 inhibitor (Supplement Fig. S5). Taking consideration of the possible redundancy function of DEDD and DEDD2, knockdown of both DEDD and DEDD2 was performed by using siRNA in the combinational experiment and a significant combinational effect with HSP90 inhibitor was observed (Fig. 5F). Thus, these results suggest that DEDD2 is involved in compensation mechanism of HSP90 inhibitor.
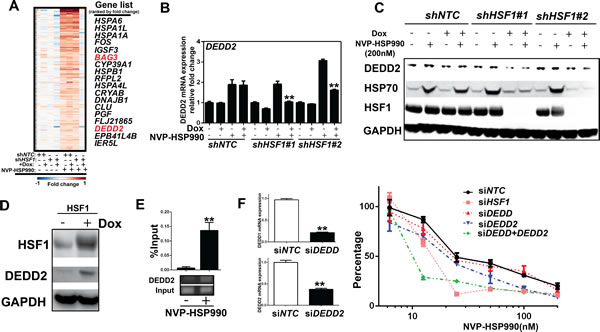
Figure 5: DEDD2 is a HSF1-target gene involved in attenuation of the effect of HSP90 inhibitor. A. Heat map showing that genes were up-regulated by HSP90 inhibitor, but the upregulation was abolished by HSF1 knockdown. shNTC or shHSF1 transduced A375 cells were treated with or without Doxycycline for 3 days and were further treated with NVP-HSP990 100nM for 3h. Total RNA were collected and microarray was performed. B. Real-time PCR analysis of the expression of DEDD2 gene in cells under HSP90 inhibitor treatment with or without HSF1 knockdown. C. Western blotting analysis of protein expression in cells treated with HSP990 and HSF1 shRNA. shHSF1 transduced A375 cells were treated with or without Doxycycline for 3 days and were further treated with NVP-HSP990 100nM for 24h. D. Western blotting analysis of DEDD2 expression in cells with over-expression of HSF1. Inducible HSF1 over-expressed cells were treated by Doxycycline for 3 day. The expression of HSF1 and DEDD2 were measured by western blotting. E. ChIP with HSF1 antibody in cells treated with NVP-HSP990. A375 cells were treated with HSP90 inhibitor for 1 hour. Chromatin was immunoprecipotated with anti-HSF1 antibody and amplified by quantitative real-time PCR using primers around HSE element of DEDD2 gene promoter. F. Dose response of NVP-HSP990 in cancer cells with knockdown of either or both DEDD and DEDD2. A375 cells were treated with or without siRNA for 2 days, then followed by treatment of a serial dilutions of NVP-HSP990 for 3 days. Total RNA were also collected and real-time PCR was performed. Relative cell growth was measured by CellTiter-Glo and normalized to DMSO-treated cells.
DISCUSSION
Under heat shock condition, HSF1 monomers dissociate from HSP90, undergo trimerization, nuclear translocation and subsequently upregulate expression of heat shock proteins, including HSP70 and HSP27 [32]. The inhibition of HSP90 also increases HSF1 trimer stability and prolongs the heat shock response [38]. Previously, studies suggest that HSF1 transcriptional activity is upregulated by HSP90 inhibition, which may limit the efficacy of HSP90 inhibitors through the activation of heat-shock responsive genes including HSP27 and HSP70 [32]. However, the functional role of HSF1 itself in response to HSP90 inhibition is not fully appreciated. We found that knockdown of HSF1 sensitizes cancer cells to HSP90 inhibitors in many different cancer lineages, including melanoma (A375, A2058), hepatocellular carcinoma (Hep3B, Huh7) and colon cancer (HCT116). More importantly, the combination of HSF1 knockdown with HSP90 inhibition prevents tumor growth significantly in xenograft mouse model (stasis or regression). We also observed that the combined treatment with HSF1 shRNA and HSP90 inhibitor leads to reduction of HSP90 inhibitor-induced HSP70 expression and p-ERK signal and induction of cleaved PARP.
Targeting HSF1 in combination with HSP90 inhibitor in HCC is particularly interesting because HSF1 expression level, both in mRNA and protein, is remarkable higher in primary HCC sample than normal hepatocytes. HSF1 knockout mice are viable and healthy, suggesting that inhibition of HSF1 might not be toxic to normal tissues [24]. Apart from combination with potential HSF1 inhibition, HSP90 inhibitor was also proposed to combine with mTOR inhibitor since blocking HSP90 may disrupt rapamycin-induced activation of alternative signaling pathways in HCCs and substantially improve the growth-inhibitory effects of mTOR inhibition in vivo [39]. HCC is an aggressive human cancer and current therapies are not very effective. HSP90 inhibitor in combination with HSF1 inhibitor or mTOR inhibitor may provide an additional therapeutic strategy for HCC.
The question is how to drug HSF1 as a transcription factor. A promising approach to target HSF1 is to use therapeutic siRNA to knockdown HSF1. Although a significant progress has been made in developing siRNA therapy, there are still many hurdles to overcome [31]. Many efforts have also been put into drugging the HSF1 pathway. A couple of small compounds have been identified to have the ability to inhibit heat shock-induced upregulation of HSP and other HSF1 targets in cells, such as quercetin and KNK437[40]. In particular, the anti-malaria drug quinacrine (QC) was showed to prevent heat shock response in cancer cells and suppresses HSF1 induced HSP70 expression in a relatively selective manner [41]. In future, developing the potent and selective HSF1 pathway inhibitors might eventually be useful for treating human cancer in combination with HSP90 inhibitor or other agents.
We also compared the gene profile between melanoma cells treated with HSP90 inhibitor with or without HSF1 knockdown. As expected, most genes regulated by HSF1 in response to HSP90 inhibitors are related to heat shock response. Some of them, such as HSP70, have been shown to have a combinational effect with HSP90 inhibitor previously [32]. DEDD2 is up-regulated by HSP90 inhibition and this up-regulation is reversed by HSF1 knockdown. Interestingly, only DEDD2 is regulated by HSF1 while DEDD is not regulated by HSF1 under HSP90 inhibitor treatment (data not shown). Knockdown of both DEDD2 and DEDD, but not either alone, enhances HSP90 inhibitor efficacy, suggesting the redundancy function of these proteins.
In this study, we identified that HSF1 transcriptional activities are induced by HSP90 inhibitors, which may provide a resistance mechanism through up-regulating a protective “heat shock” response and other transcriptional targets, such as DEDD2. However, a couple of questions still remain to be answered: 1) Are there any other transcriptional targets regulated by HSF1 required for HSF1-mediated resistance to HSP90 inhibitor in different settings? 2) Are there any co-regulators whose activities are required for HSF1-mediated resistance to HSP90 inhibitor? These questions will further prompt us to initiate large-scale screens to directly identify the HSF1-key-targets in different settings and HSF1-cofactors that are important for HSF1 transcriptional activities induced by HSP90 inhibitors in future. While a subset of HSF1-dependent-targets may play important roles in attenuating the efficacy of HSP90 inhibitors, the newly identified HSF1-target genes and/or HSF1-cofactors will not only help us to understand how HSF1 transcriptional function is regulated but may also reveal novel therapeutic targets in combination with HSP90 inhibitors.
METHOD AND MATERIALS
Cell Culture
A375, A2058, HCT116, Hep3B and Huh7 cells were obtained from American Type culture Collection. All cell lines were maintained in Dulbecco’s Modification of Eagle’s Medium, McCoy’s 5a medium or advanced RPMI medium 1640 (Invitrogen) with 10% FBS (Invitrogen). Infected cell lines were maintained under 1 μg/mL of puromycin (MP Biomedicals) for selection.
Cell Viability Assay
Cell viability at starting and ending day of compound treatment was determined by measuring cellular ATP content using the CellTiter-Glo luminescence assay (Promega). CellTiter-Glo reagent was added to each well and luminescence recorded on an Envision plate reader (Perkin Elmer). Luminescence values were used to calculate the inhibition of cell viability relative to DMSO-treated cells (0% inhibition) to calculate. Half maximal inhibitory concentration (IC50) and median lethal dosage (LD50) were further calculated.
Pooled shRNA screening
pLKO.1 lentiviral plasmids encoding shRNAs targeting the kinases and apoptosis related genes were obtained and combined to generate a plasmid pool as well as control shRNAs designed not to target any gene. These plasmid pools were used to generate lentivirus-containing supernatants as described23. For screening, A375 cells were infected with the pooled virus so as to ensure that each cell contained only one viral integrant. Cells were selected for 3 days with 1μg/ml puromycin. After selection, 6x106 cells were collected as Day0 sample. 6x106 cells were further cultured and treated with or without NVP-AUY922 (10 or 20nM) for 14 days. Genomic DNA was isolated from cells by DNA extraction in Qiagen DNA blood and Tissue kit. To amplify the shRNAs encoded in the genomic DNA, PCR was performed for 33 cycles at an annealing temperature of 66 °C using 2-6 μg of genomic DNA, the primer pair indicated below, and DNA polymerase. Forward primer: CGGCGACCACCGAGATCttgggtagtttgcagttttaaaattatgt; reverseprimer1: CATACGAGATCTAGCAttctttcccctgcactgtaccccccaatcc; reverseprimer2: GCATACGAGATCGCATGttctttcccctgcactgtaccccccaatcc. After purification, the PCR products from each tumour were quantified by ethidium bromide staining after gel electrophoresis, pooled at equal proportions, and analysed by high-throughput sequencing (Illumina).
Short Hairpin RNA Constructs
Control short hairpin RNA (shRNA), GGATAATGGTGATTGAGATGG, HSF1 shRNA#1, GCAGGTTGTTCATAGTCAGAA, and HSF1 shRNA#2, GCCCAAGTACTTCAAGCACAA, were cloned into the inducible pLKO-Tet-On puromycin vector as previously described.
Lentivirus and Infection
Lentiviral supernatants were generated according to our previously established protocol. A total of 100 μL of lentivirus was used to infect 300,000 cancer cells in a six-well plate, in 8 μg/mL polybrene (Chemicon). Medium was replaced and after 24 h, cells were selected by puromycin (MP Biomedicals) and expanded. Induction of shRNA was obtained by addition of 100ng/mL Doxycycline (Clontech) to the medium.
RNA Extraction and Quantitative Reverse Transcription-PCR
Total RNA was isolated using the RNeasyMini kit (Qiagen). ABI taqman gene expression assays include HSP70 and DEDD2. VICMGB primers/probe sets (Applied Biosystems) were used in each reaction to coamplify the B2M transcripts. All experiments were performed in triplicate and normalize to B2M levels as indicated.
Immunohistochemistry
A tissue microarray containing hepatocellular carcinoma samples with Hepatitis B-positive patients (50 cases, 2 cores per case) and two non-neoplastic liver samples (2 cores per sample) was purchased from AccuMax (A217). Primary antibodies were Ki67 antibody, Ki67_2 (Vector Laboratories Rabbit, VP-RM04), rabbit IgG isotype control (Southern Biotech, 0111-01) and HSF1 antibody (Cell Signaling Technology, Rabbit, 4356) were used. Secondary antibody incubation was done with either Ventana OmniMap or Ventana UltraMap prediluted HRP-conjugated multimer anti-rabbit secondary antibodies (Cat # 760-4315). Immunohistochemical staining was performed on the Ventana Discovery System. Images were captured using Aperio Scanscope.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was carried out according to the manufacturer’s protocol (chromatin immunoprecipitation assay kit, catalog no. 17-295, Upstate Biotechnology Inc, Lake Placid, NY). Immune complexes were prepared using anti-HSF1 (Cell Signaling, 4356) antibody. The supernatant of immunoprecipitation reaction carried out in the absence of antibody served as the total input DNA control. PCR was carried out with 10 μl of each sample using the following primers: DEDD2 promoter, 5′-GAGTCACGGGCAGGAAGTAG-3′ and 5′-ATTATTACGCCTGCGTCACC-3 ′. This was followed by analysis on 2% agarose gels.
Gene Profiling
RNA was isolated using the Qiagen RNeasy mini kit. Generation of labeled cDNA and hybridization to HG-U133 Plus2 arrays (Affymetrix) were performed as previously described [42]. DNA microarray results have been deposited at the Gene Expression Omnibus (GEO) under accession GSE44867.
Western blotting
Western blotting was performed as follows: total tumor lysates were separated by SDS/PAGE and electrotransferredto nitrocellulose membrane (Invitrogen). Membraneswere blocked in PBS and 0.1% (vol/vol) Tween-20 (PBS-T) and 4% (wt/vol) nonfat dry milk (Bio-Rad) for 1 h on a shaker at room temperature. Primary antibodies were added to the blocking solution at 1:1,000 (HSF1; Cell signaling, 4356), 1:1,000 (HSP70; Cell signaling, 4876), 1:1,000(DEDD2 ; Abcam, ab104350), 1:1,000(p-ERK; Cell signaling, 4370), 1:1,000(ERK; Cell signaling, 4695), 1:1,000(HER2; Cell signaling, 4290), 1:1,000(BRAF; Cell signaling, 9433), 1:1,000(cleaved PARP; Cell signaling, 5625), and 1:10,000 (GAPDH; Cell Signaling Technology, 2118S) dilutions and incubated overnight and a rocker at 4 °C. Immunoblottings were washed three times, 5 min each with PBS-T, and secondary antibody was added at 1:10,000 dilution into PBS-T milk for 1 h on a shaker at room temperature. After several washes, enhanced chemiluminescence (ECL) reactions were performed according to manufacturer’s recommendations (SuperSignal West Dura Extended Duration Substrate; Thermo Scientific).
Tumor xenografts
Mice were maintained and handled in accordance with Novartis Biomedical Research Animal Care and Use Committee protocols and regulations. A375 and Hep3B cells engineered with Tet-inducible shRNA against HSF1 were cultured in DMEM and EMEM supplemented with 10% Tet-approved FBS. Mice (6–8 weeks old, n=8) were inoculated s.c. with 5 × 106 A375 cells or 7 × 106 Hep3B cells in the right dorsal axillary region. Tumor volume was measured by calipering in two dimensions and calculated as (length × width) / 2. Drug treatment started 6 days (A375) or 19 days (Hep3B) after implant when average tumor volume was around 200 mm3. Animals received vehicle (5% dextrose, 10 ml/kg, orally, qw), Doxycycline (25mg/kg, orally, qd) or NVP-HSP990 (10 mg/kg, orally, qw) for the duration of the study. At termination of the study, tumor tissues were excised and snap frozen in liquid nitrogen for immunoblotting analyses of biomarkers. Data were expressed as mean ± SEM, and differences were considered statistically significant at P < 0.05 by Student t test.
Patient tumor sample analysis
RNAseq data for breast cancer (BRCA) and liver (LIHC) was retrieved from the TCGA data portal for both tumor and matched normal samples. The normalized RSEM values for each transcript were log2 transformed, and then Z score normalized for performing survival analysis.
Statistics
Statistical analyses were performed by using Student t Test (*: p<0.05, **: p<0.01) (GraphPad Prism v5.01 software for Windows, GraphPad Software, San Diego, CA USA).
ACKNOWLEDGEMENTS
We thank William Sellers, Nicholas Keen, Heather Keane, Markus Warmuth, Li Li, Junxia Min, Alan Huang, Qing Sheng, Michael Morrisey, Kornelia Polyak and the Novartis post-doctoral fellows for helpful discussions of this work.
AUTHORS’ CONTRIBUTIONS
YC, JC and WZ designed the experiments. YC, JC, AL, LB, DR, RG and MM performed the experiments. SJ, JY and JK analyzed the data. FC, FF, PZ, FS, RP and DP helped with the experiments. YC and WZ wrote the paper.
Disclosure of Potential Conflicts of Interest
Y.C is a presidential postdoctoral fellow from Novartis Institutes for Biomedical Research. JC, AL, LB, DR, RG, MM, SJ, JY, JK, FC, FF, PZ, FS, RP, DP and WZ are all employees of Novartis Institutes for Biomedical Research.
GRANT SUPPORT
This work was supported by presidential postdoctoral fellowship from Novartis Institutes for Biomedical Research.
References
1. Wandinger SK, Richter K and Buchner J. The Hsp90 chaperone machinery. The Journal of biological chemistry. 2008; 283(27):18473-18477.
2. Trepel J, Mollapour M, Giaccone G and Neckers L. Targeting the dynamic HSP90 complex in cancer. Nature reviews. 2010; 10(8):537-549.
3. Munster PN, Marchion DC, Basso AD and Rosen N. Degradation of HER2 by ansamycins induces growth arrest and apoptosis in cells with HER2 overexpression via a HER3, phosphatidylinositol 3’-kinase-AKT-dependent pathway. Cancer research. 2002; 62(11):3132-3137.
4. Schulte TW, Blagosklonny MV, Romanova L, Mushinski JF, Monia BP, Johnston JF, Nguyen P, Trepel J and Neckers LM. Destabilization of Raf-1 by geldanamycin leads to disruption of the Raf-1-MEK-mitogen-activated protein kinase signalling pathway. Molecular and cellular biology. 1996; 16(10):5839-5845.
5. Stepanova L, Leng X, Parker SB and Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes & development. 1996; 10(12):1491-1502.
6. Beliakoff J, Bagatell R, Paine-Murrieta G, Taylor CW, Lykkesfeldt AE and Whitesell L. Hormone-refractory breast cancer remains sensitive to the antitumor activity of heat shock protein 90 inhibitors. Clin Cancer Res. 2003; 9(13):4961-4971.
7. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P and Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. The Journal of biological chemistry. 2002; 277(42):39858-39866.
8. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F and Neckers LM. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway. The Journal of biological chemistry. 2002; 277(33):29936-29944.
9. Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, Scroggins BT, Neckers L, Ilag LL and Jay DG. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nature cell biology. 2004; 6(6):507-514.
10. Solimini NL, Luo J and Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007; 130(6):986-988.
11. Whitesell L and Lindquist SL. HSP90 and the chaperoning of cancer. Nature reviews. 2005; 5(10):761-772.
12. Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P and Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget. 2011; 2(3):209-221.
13. Martins AS, Davies FE and Workman P. Inhibiting the molecular evolution of cancer through HSP90. Oncotarget. 2012; 3(10):1054-1056.
14. Subjeck JR and Repasky EA. Heat shock proteins and cancer therapy: the trail grows hotter! Oncotarget. 2011; 2(6):433-434.
15. Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L and Trepel JB. Update on Hsp90 inhibitors in clinical trial. Current topics in medicinal chemistry. 2009; 9(15):1479-1492.
16. Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, Cheung KM, Collins I, Davies NG, Drysdale MJ, Dymock B, Eccles SA, Finch H, Fink A, Hayes A, Howes R, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. Journal of medicinal chemistry. 2008; 51(2):196-218.
17. Menezes DL, Taverna P, Jensen MR, Abrams T, Stuart D, Yu GK, Duhl D, Machajewski T, Sellers WR, Pryer NK and Gao Z. The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol Cancer Ther. 2012; 11(3):730-739.
18. Walsby E, Pearce L, Burnett AK, Fegan C and Pepper C. The Hsp90 inhibitor NVP-AUY922-AG inhibits NF-kappaB signaling, overcomes microenvironmental cytoprotection and is highly synergistic with fludarabine in primary CLL cells. Oncotarget. 2012; 3(5):525-534.
19. Flandrin-Gresta P, Solly F, Aanei CM, Cornillon J, Tavernier E, Nadal N, Morteux F, Guyotat D, Wattel E and Campos L. Heat Shock Protein 90 is overexpressed in high-risk myelodysplastic syndromes and associated with higher expression and activation of Focal Adhesion Kinase. Oncotarget. 2012; 3(10):1158-1168.
20. Chandarlapaty S, Scaltriti M, Angelini P, Ye Q, Guzman M, Hudis CA, Norton L, Solit DB, Arribas J, Baselga J and Rosen N. Inhibitors of HSP90 block p95-HER2 signaling in Trastuzumab-resistant tumors and suppress their growth. Oncogene. 2009; 29(3):325-334.
21. Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, Teitcher J, Wolchok JD, Germino FJ, Krown SE, Coit D, Rosen N and Chapman PB. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res. 2008; 14(24):8302-8307.
22. de Billy E, Travers J and Workman P. Shock about heat shock in cancer. Oncotarget. 2012; 3(8):741-743.
23. Pirkkala L, Nykanen P and Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001; 15(7):1118-1131.
24. Dai C, Whitesell L, Rogers AB and Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007; 130(6):1005-1018.
25. Whitesell L and Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert opinion on therapeutic targets. 2009; 13(4):469-478.
26. Jin X, Moskophidis D and Mivechi NF. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011; 14(1):91-103.
27. Dai C, Santagata S, Tang Z, Shi J, Cao J, Kwon H, Bronson RT, Whitesell L and Lindquist S. Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest. 2012; 122(10):3742-3754.
28. Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ and Whitesell L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res. 2000; 6(8):3312-3318.
29. Zaarur N, Gabai VL, Porco JA, Jr., Calderwood S and Sherman MY. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer research. 2006; 66(3):1783-1791.
30. Breinig M, Caldas-Lopes E, Goeppert B, Malz M, Rieker R, Bergmann F, Schirmacher P, Mayer M, Chiosis G and Kern MA. Targeting heat shock protein 90 with non-quinone inhibitors: a novel chemotherapeutic approach in human hepatocellular carcinoma. Hepatology. 2009; 50(1):102-112.
31. Heimberger T, Andrulis M, Riedel S, Stuhmer T, Schraud H, Beilhack A, Bumm T, Bogen B, Einsele H, Bargou RC and Chatterjee M. The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br J Haematol. 2012.
32. Powers MV, Clarke PA and Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer cell. 2008; 14(3):250-262.
33. Behl C. BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy. 2011; 7(7):795-798.
34. Alcivar A, Hu S, Tang J and Yang X. DEDD and DEDD2 associate with caspase-8/10 and signal cell death. Oncogene. 2003; 22(2):291-297.
35. Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L and Lindquist S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012; 150(3):549-562.
36. Zajac M, Gomez G, Benitez J and Martinez-Delgado B. Molecular signature of response and potential pathways related to resistance to the HSP90 inhibitor, 17AAG, in breast cancer. BMC medical genomics. 2010; 3:44.
37. Roth W, Stenner-Liewen F, Pawlowski K, Godzik A and Reed JC. Identification and characterization of DEDD2, a death effector domain-containing protein. The Journal of biological chemistry. 2002; 277(9):7501-7508.
38. Conde R, Belak ZR, Nair M, O’Carroll RF and Ovsenek N. Modulation of Hsf1 activity by novobiocin and geldanamycin. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2009; 87(6):845-851.
39. Lang SA, Moser C, Fichnter-Feigl S, Schachtschneider P, Hellerbrand C, Schmitz V, Schlitt HJ, Geissler EK and Stoeltzing O. Targeting heat-shock protein 90 improves efficacy of rapamycin in a model of hepatocellular carcinoma in mice. Hepatology. 2009; 49(2):523-532.
40. de Billy E, Powers MV, Smith JR and Workman P. Drugging the heat shock factor 1 pathway: exploitation of the critical cancer cell dependence on the guardian of the proteome. Cell cycle (Georgetown, Tex. 2009; 8(23):3806-3808.
41. Neznanov N, Gorbachev AV, Neznanova L, Komarov AP, Gurova KV, Gasparian AV, Banerjee AK, Almasan A, Fairchild RL and Gudkov AV. Anti-malaria drug blocks proteotoxic stress response: anti-cancer implications. Cell cycle (Georgetown, Tex. 2009; 8(23):3960-3970.
42. Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, Sellers WR and Benson JD. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell cycle (Georgetown, Tex. 2009; 8(3):498-504.