
INTRODUCTION
Despite the significant improvement in survival rates of chronic phase (CP) chronic myeloid leukemia (CML) patients made possible by tyrosine kinase inhibitor (TKI) therapy, cures outside of allogeneic blood or marrow transplantation are rare[1-4]. This appears to be due to the resistance of leukemia stem cells (LSCs) in CML to the pro-apoptotic effects of TKI agents[5-8]. Accordingly, most CML patients who discontinue TKIs while in molecular remission eventually relapse[9]. Moreover, for most of the TKI-induced cytogenetic remissions that remain durable at least 7 years, CML LSCs in these patients can still acquire additional mutations with progression to blast crisis (BC)[10]. Thus, there remains a clear need to identify novel molecular targets specific to the CML LSCs[11].
The precise mechanisms of CML LSC resistance to TKIs are not fully defined. CML LSCs appear to share many biological properties with their normal counterparts[6, 12] that probably limit the effectiveness of therapeutic strategies targeting BCR-ABL signaling. Hematopoietic stem cells (HSCs) are largely quiescent and normally express high levels of the multidrug resistance-1 gene[13], two factors that may limit the cellular uptake of imatinib[14]. Moreover, BCR-ABL expression appears to be required for the survival of CML progenitors but not CML LSCs, where the BCR-ABL gene can be silent likely because HSCs already are long-lived and self-renew[12, 15].
Biologic studies on LSCs have been hampered by the relative rarity of these cells, as well as the lack of a consensus on their exact phenotype. LSCs are often phenotypically defined as simply the CD34+ leukemia cells or, more recently, the more enriched CD34+CD38- subset, but even the CD34+CD38- cells are a heterogeneous population of which the LSCs constitute only a fraction[12, 16]. Normal CD34+CD38- cells can be further refined for HSCs based on low side scatter and high aldehyde dehydrogenase (ALDH) 1 activity[17, 18]. As few as 1,000 normal CD34+CD38-ALDHhigh cells will reproducibly engraft NOD/SCID-IL2Rnull (NSG) mice[18]. The major biologic function of the ALDH1 family, also known as the retinaldehyde dehydrogenases, is the biosynthesis of retinoic acid, but they also participate in the detoxification of a variety of compounds such as ethanol and active metabolites of cyclophosphamide[19]. We previously reported that high ALDH expression also can distinguish CML cells capable of engrafting NSG mice (e.g. CML LSCs) from more differentiated CML progenitors within the CML CD34+CD38- population[20]. Importantly, expression of putative therapeutic targets by CML progenitor cells was not necessarily representative of that in the CML LSCs[20], highlighting the need to search for new targets in refined LSC populations. Here, we report a comprehensive transcriptional profile of CML LSCs as compared to normal HSCs and identify unique cell surface molecules and mechanistic pathways that may serve as potential CML LSC targets.
RESULTS
Identification of potential targets that can distinguish CML LSCs from normal HSCs
In order to characterize the expression profile of CP CML LSCs and identify potential therapeutic targets unique to this population, we sorted CD34+CD38+ and CD34+CD38-ALDHhigh cells to obtain highly enriched populations of progenitor and stem cells, respectively, from bone marrow of both healthy donors and CP CML patients (Figure 1A; Supplementary Table 1). As already discussed, HSCs are enriched in the CD34+CD38-ALDHhigh cells[17, 18], and these cells contain few of the more differentiated colony-forming unit or progenitor cells, which are enriched in the CD34+CD38+ cell fraction[26]. Likewise, CD34+CD38-ALDHhigh cells show enrichment for CML LSCs with enhanced engraftment capabilities in immune deficient mice compared to the remaining CD34+CD38- cells[20]. Whole transcriptome profiling of each population was carried out by microarray analysis using an Affymetrix Human Exon 1.0 ST array, allowing measurement of differential gene expression and analysis of alternative transcripts. Principal components analysis of the gene-level data revealed distinct clustering of the four populations and showed that global gene expression patterns between the normal and CML CD34+CD38-ALDHhigh cells are closer to each other than either are to their matched CD34+CD38+ cells (Figure 1B). Furthermore, the CML subsets displayed greater variability in the gene expression patterns than their normal counterparts. Part of this variability in the CML CD34+CD38-ALDHhigh fraction could be accounted for by the presence of residual BCR-ABL negative normal HSCs in this cell population; the two subjects with the highest fraction of residual normal HSCs clustered most closely with the normal HSCs (Figure 1B; Supplementary Table 1).
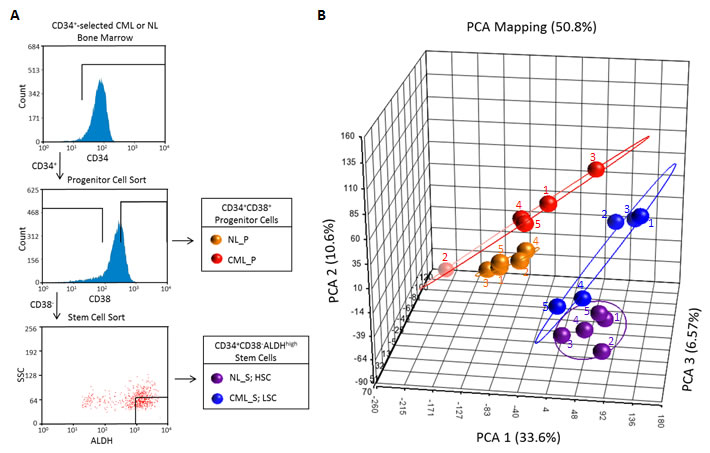
Figure 1: Global gene expression patterns in CML and normal stem and progenitor populations. (A) Cell sorting schematic for isolation of stem CD34+CD38-ALDHhigh and CD34+CD38+ cells. A representative CML sample is shown. An analogous strategy was used to sort normal (NL) samples. (B) Principal components analyses (PCA) were done on microarray gene-level expression data for CML and normal CD34+CD38-ALDHhigh and CD34+CD38+ cell populations. CML_S (blue symbols), chronic myeloid leukemic stem (CD34+CD38-ALDHhigh) cells; CML_P (red symbols), chronic myeloid leukemic progenitor (CD34+CD38+) cells; NL_S (purple symbols), normal stem (CD34+CD38-ALDHhigh) cells; NL_P (yellow symbols), normal progenitor (CD34+CD38+) cells. Sample IDs correspond to Supplementary Table 1.
Table 1: Plasma Membrane-Associated Genes that are Differentially Expressed in CML versus Normal Stem Cells |
||||
Gene Symbol |
Fold Change Expression* |
P |
Genomic Location† |
Gene Name |
DPP4 |
9.77 |
1.23E-06 |
2q24.3 |
dipeptidyl-peptidase 4 |
IL2RA |
6.08 |
3.27E-07 |
10p15-p14 |
interleukin 2 receptor, alpha |
RAB31 |
5.13 |
7.92E-06 |
18p11.3 |
RAB31, member RAS oncogene family |
PTPRD |
5.01 |
5.02E-06 |
9p23-p24.3 |
protein tyrosine phosphatase, receptor type, D |
CACNA1D |
3.53 |
8.39E-07 |
3p14.3 |
calcium channel, voltage-dependent, L type, alpha 1D subunit |
IL1RAP |
2.90 |
7.69E-05 |
3q28 |
interleukin 1 receptor accessory protein |
SLC4A4 |
2.50 |
6.28E-05 |
4q21 |
solute carrier family 4, sodium bicarbonate cotransporter, member 4 |
KCNK5 |
2.06 |
5.58E-05 |
6p21 |
potassium channel, subfamily K, member 5 |
CADPS2 |
-2.29 |
2.74E-05 |
7q31.3 |
Ca++-dependent secretion activator 2 |
GEM |
-2.52 |
1.58E-05 |
8q13-q21 |
GTP binding protein overexpressed in skeletal muscle |
ANK3 |
-2.87 |
1.35E-05 |
10q21 |
ankyrin 3, node of Ranvier (ankyrin G) |
PGM5 |
-2.96 |
1.13E-04 |
9q13 |
phosphoglucomutase 5 |
IGF1R |
-2.96 |
1.28E-05 |
15q26.3 |
insulin-like growth factor 1 receptor |
EMCN |
-3.12 |
4.91E-05 |
4q24 |
endomucin |
CNTN1 |
-4.32 |
2.28E-05 |
12q11-q12 |
contactin 1 |
PERP |
-4.41 |
1.31E-04 |
6q24 |
PERP, TP53 apoptosis effector |
CDH2 |
-4.73 |
5.75E-07 |
18q11.2 |
cadherin 2, type 1, N-cadherin (neuronal) |
Functional annotation results by DAVID are represented in the table, showing genes enriched for gene ontology term “Plasma Membrane Part” (GO:0044459).
*Calculated between CML and normal stem (CD34+CD38-ALDHhigh) cell populations from five CML or normal marrow donors from log2 transformed, default RMA background corrected array intensities. Positive values (red) indicate upregulation of gene in CML compared to normal, and negative values (blue) indicate downregulation in CML.
†Genomic coordinates refer to the human reference genome hg19 (GRCh37).
Although global gene expression patterns in the CML and normal CD34+CD38-ALDHhigh cells were fairly similar, gene-level analysis allowed us to identify several genes with significant differential expression that may serve as therapeutic targets. Using ANOVA, we identified genes that were significantly differentially expressed between all CML vs. normal samples, regardless of sorted population, and also those that were significantly differentially expressed specifically between CD34+CD38-ALDHhigh cell populations of CML and normal samples (FDR = 0.05, |log2(Fold Change)| > 1). A total of 97 genes were identified through this analysis and a heatmap was created showing the expression patterns of each gene across the four cell populations (Figure 2A). Notably, expression of this gene set was able to distinguish CML stem and progenitor cells from their normal counterparts by hierarchical clustering. Thirty-one transcripts were found to be upregulated in CML CD34+CD38-ALDHhigh cells compared to normal CD34+CD38-ALDHhigh or CD34+CD38+ cells (Figure 2A), representing selective putative CML stem cell targets. These included BLM, FAS, KYNU, NCF4, PTPRD, RAB31, SCD, ABHD10, and HPGDS, genes known to be involved in key cell signaling and metabolic pathways. The most upregulated gene selectively expressed on CML CD34+CD38-ALDHhigh cells compared to their normal counterparts was GAS2 (p = 5.96 x 10-11, average fold change = 23.5; Figure 2B). To further analyze our list of potential LSC-specific targets, functional annotation by the Database for Annotation, Visualization, and Integrated Discovery (DAVID) of genes differentially expressed on the CML versus normal CD34+CD38-ALDHhigh cells (FDR = 0.05, |log2(Fold Change)| > 1); represented by blue dots in Figure 2A) was carried out and highlighted several plasma membrane-associated genes (GO:0044459, Plasma Membrane Part), including the most up- and down-regulated genes, DPP4 and CDH2, respectively (Table 1). From this list, DPP4, IL2RA, RAB31, PTPRD, CACNA1D, IL1RAP, SLC4A4, and KCNK5 were upregulated in the CML CD34+CD38-ALDHhigh population and exhibit a cell surface protein localization. Microarray expression levels were verified by quantitative RT-PCR for a few select interesting genes (Figure 2B). Microarray intensity values were highly correlated with relative expression levels determined by quantitative RT-PCR analysis.
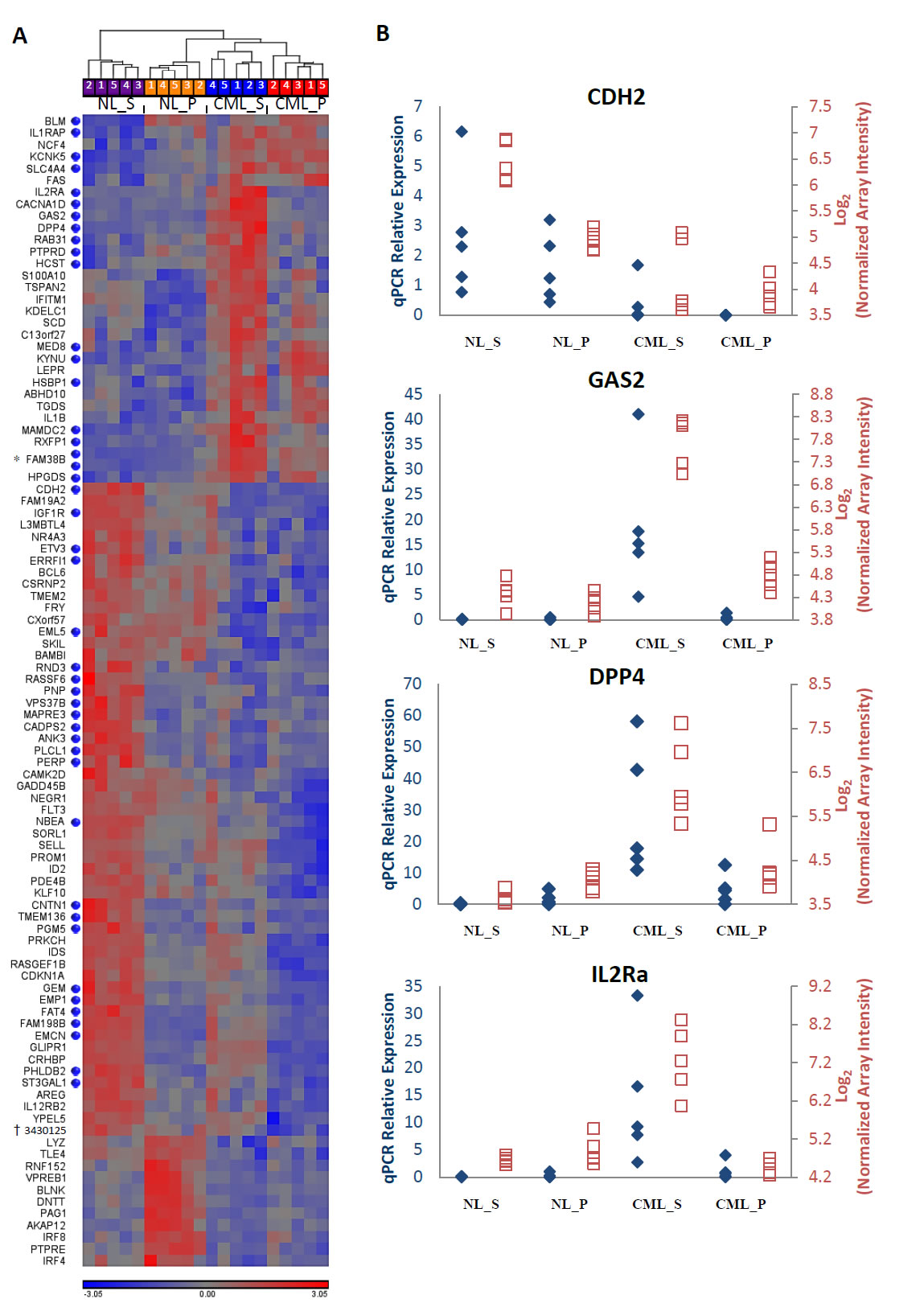
Figure 2: Differentially expressed genes between CML and normal stem and progenitor cells. CML_S, chronic myeloid leukemic stem (CD34+CD38-ALDHhigh) cells; CML_P, chronic myeloid leukemic progenitor (CD34+CD38+) cells; NL_S, normal stem (CD34+CD38-ALDHhigh) cells; NL_P, normal progenitor (CD34+CD38+) cells. A) Heatmap showing expression patterns of genes found by ANOVA to be differentially expressed between CML and normal CD34+CD38-ALDHhigh and CD34+CD38+ cells. Sample IDs correspond to Supplementary Table 1. Blue dots represent genes differentially expressed in CML versus normal CD34+CD38-ALDHhigh cells with FDR = 0.05 and |log2(Fold Change)| > 1. Upregulated and downregulated expression levels are indicated in red and blue, respectively. *FAM38B is represented by 2 separate Affymetrix transcript IDs (3798778; 3798829). †No gene name associated with Affymetrix transcript ID 3430125. B) Four candidate differentially expressed genes are shown. cDNA was prepared for each sample as described in Methods. To visualize quantitative RT-PCR (qPCR) results (blue axes labels,♦), the relative amount of the gene of interest was determined using the ΔΔCt method. Microarray expression was plotted using log2 transformed, default RMA background corrected array intensities (red axes labels,□).
Dysregulation of proliferation, differentiation and molecular pathways in CML LSCs
Characterization of the molecular mechanisms underlying malignant transformation of the normal HSCs to LSCs may aid in target discovery by uncovering pathways critical to initiation, self-renewal, and survival of the CML LSCs. Gene set enrichment analysis (GSEA)[23-25] of all Gene Ontology (GO)[27] and KEGG[28, 29] gene sets was used to identify pathways that show significant and coordinate up or down regulation of pathway components using all genes interrogated by the microarray platform. Significant terms with a q-value (multiple hypothesis testing corrected p-value) less than 0.01 indicated upregulated and downregulated gene sets that are putatively important to LSC biology; these terms were categorized by cellular functions (Supplementary Table 2 shows all significant gene sets). The top three GO and KEGG terms for each category are shown in Figure 3. Gene sets that were upregulated in CML versus normal CD34+CD38-ALDHhigh cells were involved in cell cycle and proliferation, mRNA processing, translation, DNA repair, oxidative metabolism, protein processing, immune response, and metabolic processes. Key downregulated gene sets in the CML CD34+CD38-ALDHhigh cells were associated with the cell surface and extracellular matrix, differentiation and developmental programs, cellular response to stimuli, and TGF-β and BMP signaling pathways.
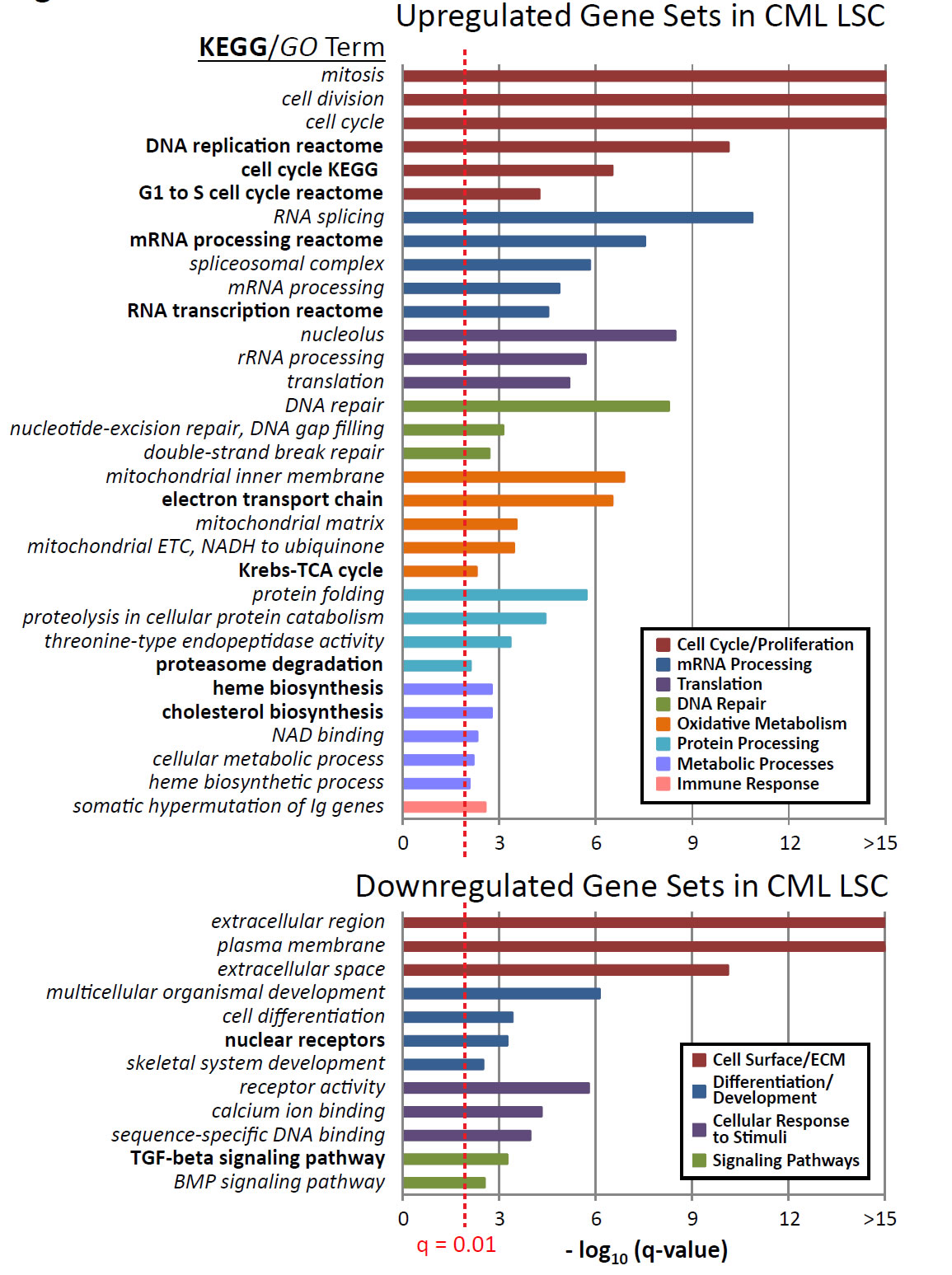
Figure 3: Altered cellular functions and pathways in CML LSCs compared to normal HSCs. Gene-set enrichment analyses (GSEA) were carried out to identify upregulated or downregulated GO and KEGG terms in CML versus normal CD34+CD38-ALDHhigh cells. Upregulated or downregulated GO and KEGG terms were categorized by common cellular function among a group of associated terms, indicated by bar color. Gene sets with a q-value < 0.01 (red dotted line) were considered significant. Q-value represents the false discovery rate of the p-value, as previously described.[50] Top three GO or KEGG terms in each category are shown. Bold text indicates KEGG terms. Italicized text indicates GO terms.
One challenge in interpreting the results of the gene-set enrichment analyses is that, for many molecular pathways, there may be a de-coupling between the transcriptional levels of the pathway components and the steady-state downstream output of the pathways, often due to complex feedback mechanisms. Therefore, it would be useful to directly examine whether the steady state transcriptional output of the pathway is consistent with overall pathway activation or inactivation. To carry out this type of vectoral analysis, we used the IPA Upstream Regulator analysis, which integrates literature-based information on the relationship between a given candidate upstream regulator and the direction of its influence on the transcriptional level of each of its downstream targets with the differential expression data generated in a given experiment to predict the activation (or inactivation) state of the upstream regulator (Ingenuity® Systems, www.ingenuity.com). Each candidate upstream regulator was assigned a Z-score, representing the confidence with which the regulator is activated or inactivated, with high positive Z-scores representing activation and high negative Z-scores representing inactivation of the function of each upstream regulator. We applied this analysis to our gene expression data from CML and normal CD34+CD38-ALDHhigh cells. A Z-score greater than 2 or less than -2 was considered to be activated or inhibited, respectively, in CML relative to normal CD34+CD38-ALDHhigh cells (Supplementary Table 3 shows all significant molecules, excluding all “chemical”-related upstream molecule types). The top upstream regulator molecules showed activation of several oncogenes, such as MYC, TBX2, and CCND1, and inflammatory chemokines, such as CCL2 and CXCL2, and inhibition of several tumor suppressors, including TP53 and CDKN2A (Figure 4A, excluding “chemical”-related and “other” upstream molecule types; Supplementary Table 3). Consistent with downregulation of the TGF-β/BMP pathways as observed by GSEA, we observed a strong inhibition of the transcriptional output of the TGF-β and BMP signaling pathway (Figure 4B), with inhibition of pathway agonists including TGFB1 itself, BMP2, GDF2, and activation of pathway antagonists, such as SMAD7 and NOG (Figure 4A,B).
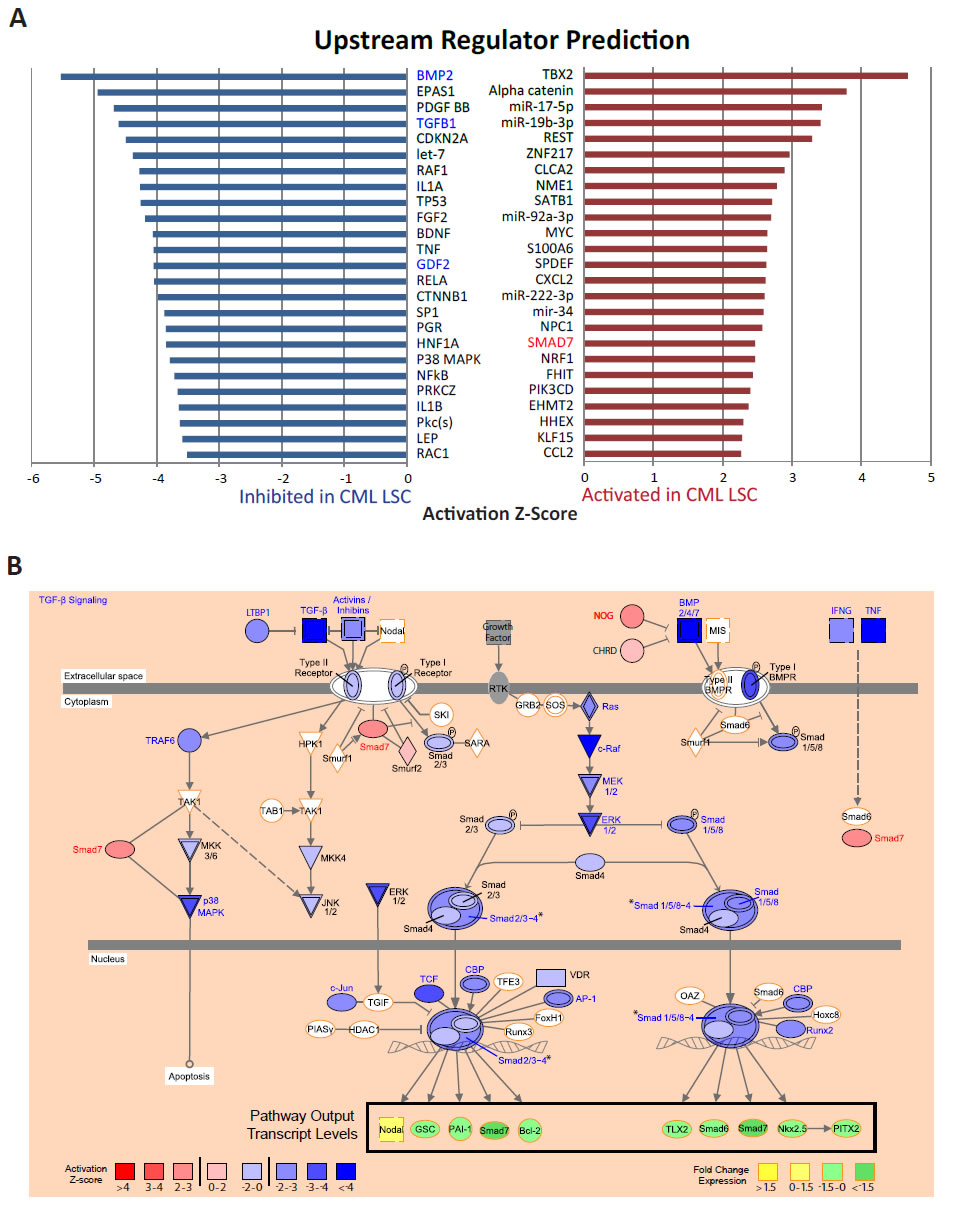
Figure 4: TGF-beta signaling pathway activity is altered in CML LSCs. A) The IPA Upstream Regulator analysis was used to identify key regulatory molecules predicted to explain the gene expression differences observed between CML and normal CD34+CD38-ALDHhigh cells. Activation Z-scores were calculated for each candidate regulator. Upstream regulators with Z-scores > 2 were considered to be activated (red bars) in CML CD34+CD38-ALDHhigh cells. Those with Z-scores < -2 were considered to be inhibited (blue bars) in CML CD34+CD38-ALDHhigh cells. Names of activated or inhibited TGF-β pathway members are distinguished in red or blue text, respectively. B) TGF-β signaling pathway. Activated upstream regulators are colored in red; inhibited, blue. Red to blue gradient denotes Z-score value. Activated molecules with a significant Z-score > 2 are distinguished with red text; Inhibited molecules with a significant Z-score < -2, blue text; upstream regulators with a Z-score between -2 and 2, black text. White molecules with orange outline are not considered upstream regulators by IPA. Shape of molecule corresponds to molecule type, as described (Ingenuity® Systems, www.ingenuity.com). A group of molecules with similar functions, depicted by a slash (/) in group name, is colored by a representative molecule with the greatest absolute value Z-score. *indicates a complex of upstream regulators where the activity of the complex is dependent on the activity of all molecules represented. In this case, a separate Z-score was assigned for the complex as a whole and is colored accordingly. Fold change expression values of pathway output transcripts are colored by yellow to green gradient. Yellow indicates upregulation and green, downregulation, of gene expression observed by differential expression analysis of microarray, as discussed in “Methods”.
Alternative transcriptional isoforms in CML LSCs
Gene sets associated with RNA processing and, more specifically, mRNA processing were shown by GSEA to be significantly differentially regulated in the CML compared to normal CD34+CD38-ALDHhigh cells (Figure 3). We, therefore, examined the exon array data to explore differential exon usage in the CML versus normal CD34+CD38-ALDHhigh cells. Evidence of alternative splicing, defined for a given gene as one or more exons displaying expression patterns different from the behavior of the other exons, was apparent in 236 genes (FDR = 0.01; Supplementary Table 4). The top two genes ranked by alternative spicing p-value that showed unique exon behavior were CACNA1D and PDE4D (Figure 5A). CACNA1D also was identified as a top upregulated gene in CML stem and/or progenitor populations compared to normal (Figure 2A). This differential expression was probably due to extensive alterations in exon usage across the gene, whereas PDE4D displayed preferential expression of specific alternative transcript isoforms in CML CD34+CD38-ALDHhigh cells compared to their normal counterparts. Functional annotation of this alternatively transcribed gene list by DAVID analysis was done to gain further insight into the biological processes affected by alternative exon usage/alternative splicing in CML CD34+CD38-ALDHhigh cells. This analysis revealed that alternative transcripts in the CML CD34+CD38-ALDHhigh cells, when compared to normal counterparts, were enriched in cellular proliferation genes, p53 signaling pathway, and kinase binding genes. There were 29 genes identified to be involved in regulation of cellular proliferation, including MYCN and TIMELESS (Figure 5Biii). Seven genes were involved in p53 signaling, including CDKN1A, which was also found in the cell proliferation category, and PERP (Figure 5Bi). Twelve genes had kinase binding functions, including MARCKS and DUSP12 (Figure 5Bii).
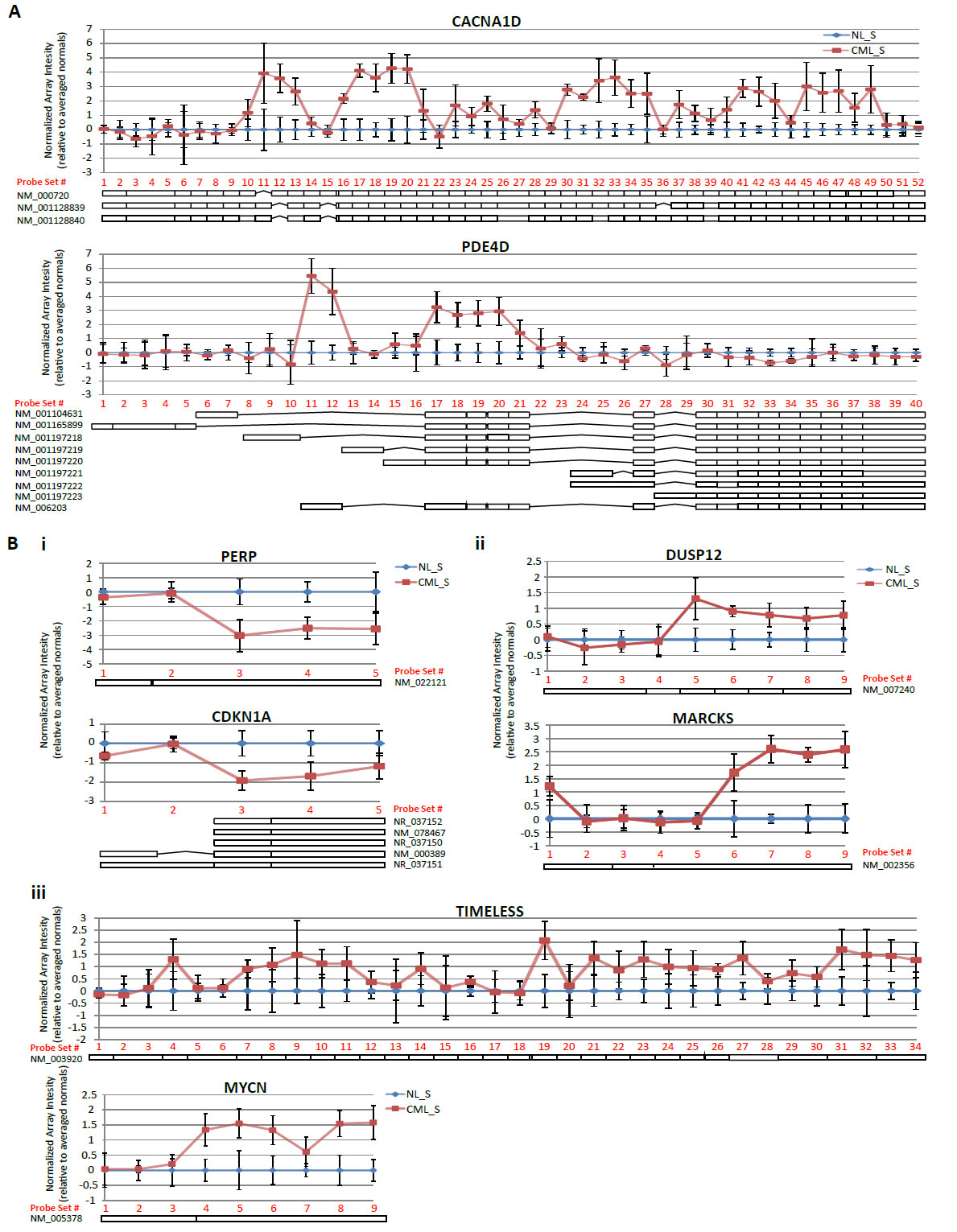
Figure 5: Exon-level analysis reveals evidence of alternative splicing in CML LSCs. A) Exon-level microarray data was analyzed for evidence of alternative transcript expression by ANOVA using the default conditions on the Partek alternative transcript workflow. Genes with an alternative splicing p-value < 0.01 were considered significant. The top two genes with the most significant alternative splicing p-values are shown. For each probeset within a gene, the log2(normalized intensities) for each sample was adjusted by the average normalized intensity of the normal samples. The resulting mean and standard deviation for CML or normal samples was plotted according to probeset number, assigned 5’-3’, for each representative gene. B) Functional annotation of genes with a p(alternative splicing) < 0.01 revealed that alternative transcripts in CML compared to normal CD34+CD38-ALDHhigh cells were enriched for genes involved in pathways commonly altered in cancer; (i) p53 signaling, (ii) kinase binding, (iii) cell proliferation. Plots were constructed based on the human reference genome hg19 (GRCh37). Schematics of known refseq transcriptional isoforms are positioned below each graph and are drawn with respect to location of probesets interrogating each exon/intron.
DISCUSSION
LSCs appear to persist in most CML patients on TKIs, and the persistence of these cells remains a major obstacle to cure [5, 7, 9]. We previously reported that ALDH expression enriched for CD34+CD38- cells capable of engrafting NSG mice from normal marrow[18] as well as CML[20], thus, presumably representing the primitive stem cell fractions in both. Moreover, expression of some putative targets by the CML LSCs differed significantly from that of the more prevalent progenitor cells[20], highlighting the need to study refined LSC populations. Additionally, other CML antigens were expressed at comparable levels to normal stem/progenitor cells, suggesting a lack of leukemia-specificity and a high likelihood that therapies targeting these candidates might cause undue toxicity to normal hematopoiesis[20].
We employed exon microarray technology to perform whole transcriptome analysis of highly enriched CP CML and normal stem and progenitor cell populations with the goal of identifying unique putative LSC targets. Interestingly, principal components analysis revealed that expression patterns were remarkably similar between the CD34+CD38-ALDHhigh cells from CML patients and those from normal donors. In fact, the similarities were greater than those observed between the CML LSCs (CD34+CD38-ALDHhigh cells) and the CML progenitors (CD34+CD38+ cells), underpinning the challenge in selectively targeting LSCs without injuring normal HSCs. Nonetheless, the comprehensive approach and highly refined populations utilized in this analysis allowed identification of important new putative LSC targets that were more highly expressed by the CML LSC and/or progenitor cell fractions compared to normal stem/progenitor cell fractions.
A significant number of genes over-expressed in CML LSCs compared to their normal counterparts encoded cell surface proteins, including, IL2Rα, DPP4, PTPRD, CACNA1D, IL1RAP, SLC4A4, and KCNK5. The surface location of these candidates may render them particularly vulnerable to targeting by immune-based strategies. DPP4, also known as CD26, encodes dipeptidyl peptidase 4 and is especially interesting as a possible target for LSC-directed therapy. One of the known targets of its peptidase cleavage activity is CXCL12[30], and upregulation of DPP4 on the surface of CML LSCs may allow these cells to escape the homing/niche interactions imposed by the CXCL12/CXCR4 chemokine-receptor system[31], leading to dysregulated LSC growth and survival. Therefore, drugs capable of inhibiting the DPP4 dipeptidyl peptidase catalytic activity, which are currently FDA-approved for treatment of diabetes[32], may have utility in targeting CML LSCs. IL2RA is also a particularly attractive LSC target since multiple biologic agents directed against it are currently under clinical investigation[33]. IL1RAP has been identified previously as a putative therapeutic stem cell-specific target in CML[34], as well as in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) patients, with high expression correlating with poor overall survival in AML[35]. Similarly, in this study, we identified IL1RAP upregulation on CML LSCs; the availability of IL-1 receptor antagonists or decoy receptors that are currently FDA-approved for the treatment of several inflammatory disorders[36] may allow effective targeting of the CML LSC. Among the other genes found to be upregulated in LSCs compared to normal HSCs, BLM, KYNU, PTPRD, RAB31, and HPGDS are known to have enzymatic activities involved in key signaling and metabolic pathways; development of inhibitors of these enzymes may allow LSC targeting.
Taking advantage of the comprehensive coverage of the exon array platform, we also identified several genes that were dysregulated in LSCs at the level of alternative transcriptional isoforms and alternative exon usage. Interestingly, genes showing alternative splicing were enriched in p53 signaling, protein kinase binding, and cell proliferation. Therefore, alternative splicing may account, in part, for the increased cell proliferation, resistance to apoptosis, and dysregulated kinase signaling characteristic of CML[37]. It is expected that these pathways and their components are susceptible to pharmacologic inhibition. Of particular interest, the cyclic-AMP specific phosphodiesterase, PDE4D, was found to be upregulated in CML LSCs compared to normal HSCs by preferential expression of a specific alternative transcript isoform. Likewise, the dual specificity phosphatase, DUSP12, and the voltage-dependent L-type calcium channel, CACNA1D, appear to become upregulated in CML LSCs via alternative exon usage. It is possible that alternative splicing of DUSP12 in CML LSCs could underlie immunogenic responses that seem to correlate with improved survival after donor lymphocyte infusion[38]. Although PDE4 inhibitors and L-type calcium channel blockers are available, development of isoform specific inhibitors may aid in CML LSC targeting. Therefore, such alternative transcription analyses could be used to identify functionally critical exons and their corresponding protein domains for development of targeted and immunomodulatory therapies.
Using these comprehensive transcriptome data, we were able to identify key pathways that were altered in the LSCs compared to normal HSCs. Consistent with previous findings of Bruns et al [39], we observed upregulation of several pathways involved in cell proliferation/cell cycle and downregulation of pathways involved in cell surface interactions, development, and differentiation. These pathway alterations may underlie the increased cell proliferation and resistance to apoptosis that are characteristic of CML and may also play a significant role in recognized resistance mechanisms of LSCs, such as dysregulation of niche interactions, cell cycle, survival, self-renewal, and metabolism. Interestingly, and somewhat unexpectedly, we also observed upregulation of pathways involved in oxidative metabolism, suggesting that LSCs may not be as metabolically quiescent as previously thought[40]. The accompanying upregulation of DNA repair pathways in the CML LSCs may indicate a requirement for guarding against DNA damage induced by a potential increase in production of reactive oxygen species during oxidative metabolism. Additionally, we identified a number of signaling pathways that showed evidence of activation in the LSCs. Particularly interesting are the targets with specific inhibitors already under clinical investigation, including a neutralizing monoclonal antibody to CCL2[41] and cyclin dependent kinase 4/6 inhibitors, inhibiting activation by partnering cyclin CCND1[42, 43]. Additionally, we found that the TGF-β/BMP pathway was coordinately downregulated in the CML LSC compared to normal HSCs, and pathway antagonists, such as SMAD7 were highly activated. The likely contribution of SMAD7 activation to the observed TGF-β pathway inhibiton in CML LSCs compared to normal HSCs and the current clinical investigation of antisense oligonucleotides for SMAD7 inhibition in Crohn’s disease[44] make it an attractive target for CML LSC-directed therapy. Although previous reports have shown that the TGF-β pathway is critical for CML LSC survival[45-47], it also has been suggested that TGF-β has a dual role in tumor progression, acting as a tumor suppressor in the very early stages of tumorigenesis[48, 49].
We have developed an important resource for identifying the gene expression changes, pathway alterations, and alternative exon usage that can allow selective targeting of CP CML LSCs. Some of these targets, such as IL2RA and DPP4, may be amenable to immediate clinical translation with currently available therapies. While this work requires further functional validation and target credentialing, it offers the promise of LSC-targeted therapies, which may prove curative in CML while minimizing harm to normal hematopoiesis.
METHODS
Patient and normal donor bone marrow specimens, enrichment of stem and progenitor cell populations, and nucleic acid extraction
Bone marrow was obtained from 5 patients with newly-diagnosed and untreated CP CML, as well as from 5 healthy bone marrow donors. Informed consent was obtained from all patients and healthy donors prior to sample collection in accordance with the Declaration of Helsinki, under a research protocol approved by the Johns Hopkins Institutional Review Board. CD34+CD38-ALDHhigh stem cells and CD34+CD38+ progenitor cells were collected from each marrow specimen as described previously[20]. Briefly, CD34+ cells were selected using Miltenyi Biotec (Auburn, CA) microbeads (binding the class II CD34 epitope) followed by column enrichment per the manufacturer’s recommendations. These cells were then stained with Aldefluor (Aldagen, Durham, NC) to assess ALDH activity, phycoerythrin-conjugated anti-CD34 antibodies (binding the class III CD34 epitope), and allophycocyanin-conjugated anti-CD38 antibodies (BD Biosciences, San Jose, CA), and sorted using a MoFlo cell sorter (Beckman Coulter) into CD34+CD38-ALDHhigh and CD34+CD38+ fractions. DNA and RNA were extracted from at least 50,000 cells from each population using the AllPrep DNA/RNA micro kit (Qiagen, Valencia, CA, USA).
Fluorescence in situ hybridization (FISH)
Isolation of leukemic cells was confirmed by FISH for BCR-ABL on cytospins of each sorted cell fraction, fixed in 3:1 Methanol: Glacial Acetic acid (Sigma-Aldrich, St. Louis, MO, USA). FISH was performed by the Johns Hopkins Cytogenetics Core, using the Vysis LSI BCR-ABL Dual Color, Dual Fusion translocation probe (Abbot Molecular, Des Plaines, IL, USA) per manufacturer’s instructions. Slides were analyzed on a fluorescence microscope with a triple-band pass filter for DAPI, Spectrum Orange, and Spectrum Green.
Gene expression microarrays and analysis
Total RNA from sorted cell populations was subjected to cDNA synthesis and linear amplification using the Ovation RNA Exon Module amplification system (NuGEN, San Carlos, CA) according to the manufacturer’s protocols. The resulting material was then fragmented and biotin-end-labeled using the Encore Biotin Module (NuGEN) and hybridized to Human Exon 1.0 ST whole genome gene expression microarrays (Affymetrix, Santa Clara, CA) according to the manufacturer’s protocols at the Johns Hopkins Microarray facility. The microarray gene expression data was analyzed with Partek Genomic Suite software (http://www.partek.com/partekgs) using the exon array workflow with default conditions (data imported and normalized using log2 transformation, default RMA background correction and normalization of core meta-probe sets) unless otherwise specified. Gene expression summaries from the imported normalized intensity data was subjected to principal components analysis. Two-way analysis of variance (ANOVA) of gene summary data was performed to find differentially expressed genes between all cell populations, focusing on the contrasts between CML versus normal samples and CML CD34+CD38-ALDHhigh versus normal CD34+CD38-ALDHhigh populations. Genes with |log2(fold-change)| > 1 and false discovery rate (FDR) of 0.05 were identified as significantly differentially expressed. A gene list specifically focusing on contrasts between CML and normal CD34+CD38-ALDHhigh cells with |log2(fold-change)| > 1 and false discovery rate (FDR) of 0.05 was uploaded to the Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7 (http://david.abcc.ncifcrf.gov/) for functional annotation analyses[21, 22] of enriched gene ontology (GO) and KEGG pathway terms. Lists comprised of all arrayed genes with expression data from the CML versus normal CD34+CD38-ALDHhigh comparison were subjected to gene-set enrichment analysis (GSEA), as described previously[23-25], or directly uploaded into Ingenuity Pathway Analysis (IPA) software (Ingenuity® Systems, June 2012, www.ingenuity.com). For GSEA, all GO and KEGG terms with a q-value less than 0.01 were considered significant. Core analysis was run in IPA utilizing all default settings, with exception of the Human Exon 1.0 ST array as the reference gene set. This analysis generated a list of potential upstream transcriptional regulators and predicted the activity of each by calculation of overlap p-value using a Fisher’s Exact test and the activation Z-score as described (Ingenuity® Systems, www.ingenuity.com). Calculations were based on known interactions between the predicted upstream transcriptional regulators and their downstream target gene set according to the Ingenuity® Knowledge Base and measured expression changes in the array data set. Upstream regulators with |z-score| > 2.00 were nominated as significant, with a positive Z-score representing activation and a negative value, inhibition. The list of upstream regulators and activation z-score values were also utilized to assign the activation state of each component of the TGF-β pathway, which was defined using the IPA and KEGG pathway map data (http://www.genome.jp/kegg/pathway.html). The raw and normalized data are available from the Gene Expression Omnibus (GEO) with accession number GSE43754. For alternative transcript analysis, exon level microarray data from the CML and normal CD34+CD38-ALDHhigh RNA was subjected to ANOVA analysis using the default conditions on the Partek alternative transcript workflow. Genes with alternative transcript p-value < 0.01 were subjected to analysis with DAVID v.6.7 as described above. For each probeset within a gene, the log2(normalized intensities) for each sample was adjusted by the average normalized intensity of the normal samples. The resulting mean and standard deviation for CML or normal samples was plotted according to probeset number, assigned 5’-3’, for each representative gene.
Real-time reverse transcriptase polymerase chain reaction
Excess extracted RNA from patient samples was used to synthesize cDNA using SuperScript® III Reverse Transcriptase (RT) (Invitrogen, Carlsbad, CA). Newly synthesized cDNA from unamplified RNA or excess amplified cDNA prior to labeling and array hybridization from each CML and normal patient sample was used to validate array results by quantitative RT-PCR of GAS2, DPP4, CDH2, IL2RA, GAPDH and ACTB using the iQ Supermix (Bio-Rad, Hercules, CA) and gene-specific TaqMan® assays (Life Technologies Co., Carlsbad, CA). The relative amount of the gene of interest was determined using the ΔΔCt method, relative to the average expression of all samples for that gene and GAPDH expression for GAS2, DPP4, and CDH2 or ACTB for IL2RA. Quantitative RT-PCR results from amplified starting material or SuperScript® III converted unamplified cDNA were compared for the gene IL2RA and showed consistent results. The remaining genes were verified using amplified starting material only. All quantitative PCR experiments were done in duplicate.
ACKNOWLEDGEMENTS
We wish to acknowledge the services of Dr. Haiping Hao and the members of the Johns Hopkins Microarray Core Facility for carrying out the microarray processing. We also wish to acknowledge the services of Dr. Hao Zhang of the Johns Hopkins Bloomberg School of Public Health Flow Cytometry Core Facility for carrying out the cell sorting. This work was supported by the Maryland Stem Cell Research Fund Exploratory Research Award (to S.Y.), the Masenhimer Fellowship (to S.Y.), American Society of Hematology Research Training Award for Fellows (to J.M.G.), Maryland Stem Cell Research Fund Post-doctoral Fellow Award (to J.M.G.), the Passano Foundation Johns Hopkins Clinician Scientist Award (to J.M.G.), the Madeline Conti Cancer Cure Fund, the There Goes My Hero Foundation, the Commonwealth Foundation for Cancer Research, the Irving A. Hansen Memorial Foundation, and NIH Grant P01 CA15396-26 (R.J.J.).
Contribution:
J.M.G, D.E., M.C.H, and M.V. prepared samples and performed experiments; J.M.G., J.L.G., D.E., M.G., S.Y. analyzed results and made the figures; J.M.G, W.G.N., R.J.J, and S.Y. designed the research; J.M.G, J.L.G., R.J.J, and S.Y. wrote the paper.
Conflict-of-interest disclosure:
R.J.J. holds the patent for Aldefluor and, under a licensing agreement between Aldagen and the Johns Hopkins University, is entitled to a share of royalties received by the University. The terms of this arrangement are being managed by the Johns Hopkins University in accordance with its conflict of interest policies.
References
1. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S and Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001; 344(14):1031-1037.
2. Kantarjian H, Talpaz M, O’Brien S, Garcia-Manero G, Verstovsek S, Giles F, Rios MB, Shan J, Letvak L, Thomas D, Faderl S, Ferrajoli A and Cortes J. High-dose imatinib mesylate therapy in newly diagnosed Philadelphia chromosome-positive chronic phase chronic myeloid leukemia. Blood. 2004; 103(8):2873-2878.
3. Cortes J, O’Brien S and Kantarjian H. Discontinuation of imatinib therapy after achieving a molecular response. Blood. 2004; 104(7):2204-2205.
4. Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O, Blanchet O, Marit G, Gluckman E, Reiffers J, Gardembas M and Mahon FX. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007; 109(1):58-60.
5. Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L and Holyoake TL. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002; 99(1):319-325.
6. Angstreich GR, Matsui W, Huff CA, Vala MS, Barber J, Hawkins AL, Griffin CA, Smith BD and Jones RJ. Effects of imatinib and interferon on primitive chronic myeloid leukaemia progenitors. British journal of haematology. 2005; 130(3):373-381.
7. Copland M, Hamilton A, Elrick LJ, Baird JW, Allan EK, Jordanides N, Barow M, Mountford JC and Holyoake TL. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006; 107(11):4532-4539.
8. Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, Arber DA, Slovak ML and Forman SJ. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003; 101(12):4701-4707.
9. Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, Varet B, Etienne G, Reiffers J and Rousselot P. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. The lancet oncology. 2010; 11(11):1029-1035.
10. Hughes TP, Hochhaus A, Branford S, Muller MC, Kaeda JS, Foroni L, Druker BJ, Guilhot F, Larson RA, O’Brien SG, Rudoltz MS, Mone M, Wehrle E, Modur V, Goldman JM and Radich JP. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS). Blood. 2010; 116(19):3758-3765.
11. O’Hare T, Zabriskie MS, Eiring AM and Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nature reviews Cancer. 2012; 12(8):513-526.
12. Bedi A, Zehnbauer BA, Collector MI, Barber JP, Zicha MS, Sharkis SJ and Jones RJ. BCR-ABL gene rearrangement and expression of primitive hematopoietic progenitors in chronic myeloid leukemia. Blood. 1993; 81(11):2898-2902.
13. Raaijmakers MH, van Emst L, de Witte T, Mensink E and Raymakers RA. Quantitative assessment of gene expression in highly purified hematopoietic cells using real-time reverse transcriptase polymerase chain reaction. Experimental hematology. 2002; 30(5):481-487.
14. Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, Goldman JM and Melo JV. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003; 101(6):2368-2373.
15. Jiang G, Yang F, Li M, Weissbecker K, Price S, Kim KC, La Russa VF, Safah H and Ehrlich M. Imatinib (ST1571) provides only limited selectivity for CML cells and treatment might be complicated by silent BCR-ABL genes. Cancer biology & therapy. 2003; 2(1):103-108.
16. Bonnet D. Normal and leukaemic stem cells. British journal of haematology. 2005; 130(4):469-479.
17. Christ O, Lucke K, Imren S, Leung K, Hamilton M, Eaves A, Smith C and Eaves C. Improved purification of hematopoietic stem cells based on their elevated aldehyde dehydrogenase activity. Haematologica. 2007; 92(9):1165-1172.
18. Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, Galkin S, Collector MI, Perkins B, Levis MJ, Griffin CA, Sharkis SJ, Borowitz MJ, Karp JE and Jones RJ. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood. 2012; 119(15):3571-3577.
19. Vasiliou V, Pappa A and Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug metabolism reviews. 2004; 36(2):279-299.
20. Gerber JM, Qin L, Kowalski J, Smith BD, Griffin CA, Vala MS, Collector MI, Perkins B, Zahurak M, Matsui W, Gocke CD, Sharkis SJ, Levitsky HI and Jones RJ. Characterization of chronic myeloid leukemia stem cells. American journal of hematology. 2011; 86(1):31-37.
21. Huang da W, Sherman BT and Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009; 4(1):44-57.
22. Huang da W, Sherman BT and Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009; 37(1):1-13.
23. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES and Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(43):15545-15550.
24. Irizarry RA, Wang C, Zhou Y and Speed TP. Gene set enrichment analysis made simple. Statistical methods in medical research. 2009; 18(6):565-575.
25. Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, Esopi D, Irizarry RA, Getzenberg RH, Nelson WG, Luo J, Xu J, Isaacs WB, Bova GS and Yegnasubramanian S. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Science translational medicine. 2013; 5(169):169ra110.
26. Cicuttini FM, Welch K and Boyd AW. Characterization of CD34+HLA-DR-CD38+ and CD34+HLA-DR-CD38- progenitor cells from human umbilical cord blood. Growth Factors. 1994; 10(2):127-134.
27. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000; 25(1):25-29.
28. Kanehisa M, Goto S, Sato Y, Furumichi M and Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic acids research. 2012; 40(Database issue):D109-114.
29. Kanehisa M and Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000; 28(1):27-30.
30. Shioda T, Kato H, Ohnishi Y, Tashiro K, Ikegawa M, Nakayama EE, Hu H, Kato A, Sakai Y, Liu H, Honjo T, Nomoto A, Iwamoto A, Morimoto C and Nagai Y. Anti-HIV-1 and chemotactic activities of human stromal cell-derived factor 1alpha (SDF-1alpha) and SDF-1beta are abolished by CD26/dipeptidyl peptidase IV-mediated cleavage. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95(11):6331-6336.
31. Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, Luster AD, Scadden DT and Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005; 435(7044):969-973.
32. Deacon CF, Carr RD and Holst JJ. DPP-4 inhibitor therapy: new directions in the treatment of type 2 diabetes. Frontiers in bioscience : a journal and virtual library. 2008; 13:1780-1794.
33. Rose JW. Anti-CD25 immunotherapy: regulating the regulators. Science translational medicine. 2012; 4(145):145fs125.
34. Jaras M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, Lassen C, Olofsson T, Bjerrum OW, Richter J and Fioretos T. Isolation and killing of candidate chronic myeloid leukemia stem cells by antibody targeting of IL-1 receptor accessory protein. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(37):16280-16285.
35. Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, Ben-Neriah S, Montagna C, Parekh S, Pellagatti A, Boultwood J, Paietta E, Ketterling RP, Cripe L, Fernandez HF, Greenberg PL, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012; 120(6):1290-1298.
36. Dinarello CA, Simon A and van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nature reviews Drug discovery. 2012; 11(8):633-652.
37. Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nature reviews Cancer. 2005; 5(3):172-183.
38. Biernacki MA, Marina O, Zhang W, Liu F, Bruns I, Cai A, Neuberg D, Canning CM, Alyea EP, Soiffer RJ, Brusic V, Ritz J and Wu CJ. Efficacious immune therapy in chronic myelogenous leukemia (CML) recognizes antigens that are expressed on CML progenitor cells. Cancer research. 2010; 70(3):906-915.
39. Bruns I, Czibere A, Fischer JC, Roels F, Cadeddu RP, Buest S, Bruennert D, Huenerlituerkoglu AN, Stoecklein NH, Singh R, Zerbini LF, Jager M, Kobbe G, Gattermann N, Kronenwett R, Brors B, et al. The hematopoietic stem cell in chronic phase CML is characterized by a transcriptional profile resembling normal myeloid progenitor cells and reflecting loss of quiescence. Leukemia. 2009; 23(5):892-899.
40. Flis K, Irvine D, Copland M, Bhatia R and Skorski T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leukemia & lymphoma. 2012; 53(12):2474-2478.
41. Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, Elsayed Y, Dawkins F and de Bono JS. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investigational new drugs. 2012.
42. Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R and O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). British journal of cancer. 2011; 104(12):1862-1868.
43. Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ and Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(2):568-576.
44. Monteleone G, Fantini MC, Onali S, Zorzi F, Sancesario G, Bernardini S, Calabrese E, Viti F, Monteleone I, Biancone L and Pallone F. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn’s disease. Molecular therapy : the journal of the American Society of Gene Therapy. 2012; 20(4):870-876.
45. Zhu X, Wang L, Zhang B, Li J, Dou X and Zhao RC. TGF-beta1-induced PI3K/Akt/NF-kappaB/MMP9 signalling pathway is activated in Philadelphia chromosome-positive chronic myeloid leukaemia hemangioblasts. Journal of biochemistry. 2011; 149(4):405-414.
46. Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N and Hirao A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010; 463(7281):676-680.
47. Moller GM, Frost V, Melo JV and Chantry A. Upregulation of the TGFbeta signalling pathway by Bcr-Abl: implications for haemopoietic cell growth and chronic myeloid leukaemia. FEBS letters. 2007; 581(7):1329-1334.
48. Roberts AB and Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100(15):8621-8623.
49. Bierie B and Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nature reviews Cancer. 2006; 6(7):506-520.
50. Storey JD. A direct approach to false discovery rates. J R Statist Soc B. 2002; 64(Part 3):479-498.