
INTRODUCTION
Head and Neck Squamous Cell Carcinoma (HNSCC) is one of the most common solid tumors worldwide. The risk factors for HNSCC are well known and include consumption of alcohol and tobacco and infection by HPV. Recent technological advances in large-scale sequencing resulted in the identification of common genetic alterations in HNSCC, including mutations occurring in genes of the phosphoinositide 3-kinase (PI3K) pathway [1-5], as PIK3CA, GRB1, PIP5K3, AKT2, TSC1, TSC2, mTOR, Rictor, Raptor among others. Loss of function and mutations of key molecular regulators of PI3K signaling, including PTEN, are associated with the formation of various solid tumors, such as head and neck cancer [1, 6-8]. PTEN is a tumor suppressor gene that negatively regulates PI3K signaling and downstream PI3K family members, including AKT and mammalian target of Rapamycin (mTOR). Interestingly, PTEN mutations and protein loss are common events in the carcinogenesis and progression of HNSCC and other cancers such as glioblastomas and cancers of the breast, endometrium, and prostate [1, 9-12]. In HNSCC, PTEN protein loss is close to 31% and may reach 50% in advanced cases [2, 6, 7, 13, 14] (Wagner and Squarize, under submission).
Although clinically relevant [7, 13], the mechanisms associated with loss of PTEN function are largely unknown. Epigenetic events are likely the primary cause of PTEN loss of function in solid tumors, such as malignant melanomas, sporadic breast cancer, hepatocellular carcinoma, and thyroid cancer [15-18]. Inactivation of PTEN protein also occurs following oxidative inactivation that is induced by H2O2. The H2O2 targets the redox active Cys124 residue of PTEN, resulting in the formation of a disulfide bond between Cys71 and Cys124, as described in cervix tumor cell lines and fibroblasts [19]. However, little is known about how oxidation affects PTEN signaling in HNSCC.
The aim of the study was to investigate the effects of compromised PTEN function mediated by the reduction/oxidation mechanism in the regulation of core clock genes. We found that oxidation of PTEN in HNSCC resulted in the accumulation of the core circadian molecule BMAL1 (i.e., ARNTL - Aryl hydrocarbon receptor nuclear translocator-like protein 1, Homo sapiens [Human], UniProtKB - O00327), which aligns to our studies in the epidermis of PTEN conditional knockout mice (Zagni et al., submitted). Surprisingly, accumulation of BMAL1 was associated with upregulation of the mTOR pathway. In vivo inhibition of mTOR by Rapamycin causes reduced expression of BMAL1 in PTEN and Per2 knockout mice, two mouse models that constitutively express high levels of BMAL1 in the epidermis. Interfering with the mTOR pathway through siRNA technology aiming at Raptor and Rictor further strengthen our findings that mTOR plays a novel role in the control of the core clock gene BMAL1. These results suggest that pathological conditions leading to disruption of the PI3K/PTEN/mTOR axis are likely to deregulate the circadian clock in normal and epithelial-derived tumor cells.
RESULTS
PTEN oxidation in HNSCC activates mTOR signaling
Administration of hydrogen peroxidase caused oxidation of PTEN in HNSCC (Figure 1A and 1B; oxPTEN *p<0.05, **p<0.01, ***p<0.001), as previously described in HeLa and NIH 3T3 cells [19]. Interestingly, oxidation-mediated deactivation of PTEN resulted in time-dependent pS6 activation (Figure 1A and 1B; pS6 *p<0.05, **p<0.01, ***p<0.001), a molecular marker for mTOR activity and pharmacological inhibition efficacy [20]. Activation of pS6 becomes statistically significant after PTEN oxidation (Figure 1A and 1B). Used as a positive control or readout of oxidation, reactive oxygen species (ROS) was observed to build up on tumor cells along with the pS6 activation (Figure 1C, ***p<0.001) and the increased proliferation (Figure 1D, **p<0.01, ***p<0.001).
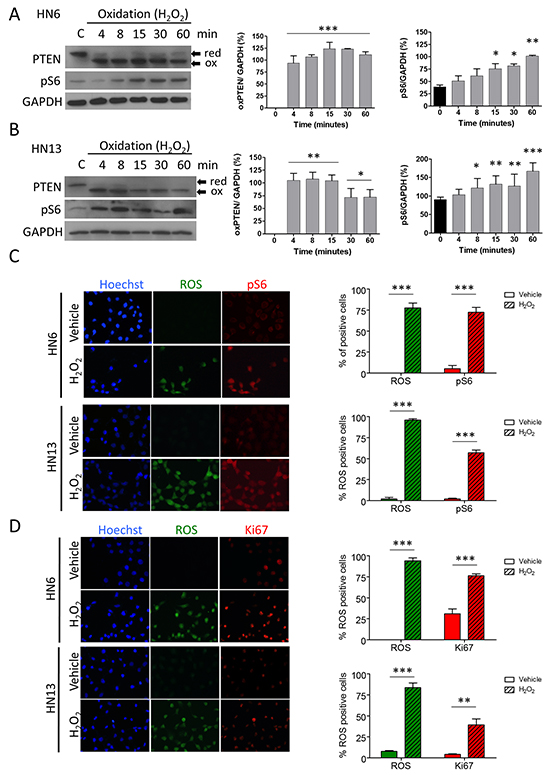
Figure 1: Oxidation of PTEN results in upregulation of mTOR signaling. Oxidation of PTEN and pS6 activation is observed after of oxidative stress (H2O2) in HN6 (A) and HN13 (B) (*p<0.05, **p<0.01, ***p<0.001). C. Oxidative stress induces abrupt nuclear accumulation of reactive oxygen species (ROS) in head and neck cancer cell lines (green channel). Note that accumulation of ROS is accompanied by pS6 activation (red channel) (***p<0.001). D. Tumor cells oxidation leads to increased proliferation (Ki-67, red channel) and simultaneous accumulation of ROS (green channel)(**p<0.01, ***p<0.001).
Increased oxidation leads to the accumulation of the core clock protein BMAL1
In the typical process, the cells in the body follow a circadian rhythm. The circadian rhythm oscillations orchestrate several biological processes intimately involved in the maintenance of tissue homeostasis and stem cell heterogeneity [21], and it may be dysregulated in cancer and aging. For example, disruption in the core clock protein BMAL1 results in severe premature aging [22] and overexpression is associated with decreased overall survival, particularly in colorectal cancer patients [23]. In cell culture, the circadian rhythm synchronizing agent Forskolin is a potent inducer of circadian gene expression including BMAL1 accumulation [24, 25]. Interesting, head and neck tumor cell lines present different levels of BMAL1 upon administration of forskolin. BMAL1 expression ranged from highly expressed protein observed in HN6 cells to extremely low levels as observed, for example, in HN13 cells (Figure 2A). We next examined whether exposure of HNSCC cells to H2O2 interferes with levels of BMAL1 in HNSCC cells. We found that administration of H2O2 resulted in the nuclear accumulation of BMAL1 in both HNSCC cell lines compared to vehicle, along with the accumulation of ROS (Figure 2B). Interestingly, HN13 cells, which had low levels of BMAL1 (Figure 2A), showed high accumulation of nuclear BMAL1 in response to oxidation (Figure 2B right panel). Accumulation of BMAL1 was also time dependent (Figure 2C and 2D)(*p<0.05, **p<0.01). Accumulation of BMAL1 in HN6 cells was statistically evident after 15 minutes of administration of H2O2 (Figure 2C) while HN13 cells showed statistical significance after 8 minutes of H2O2 administration (*p<0.05) (Figure 2D).
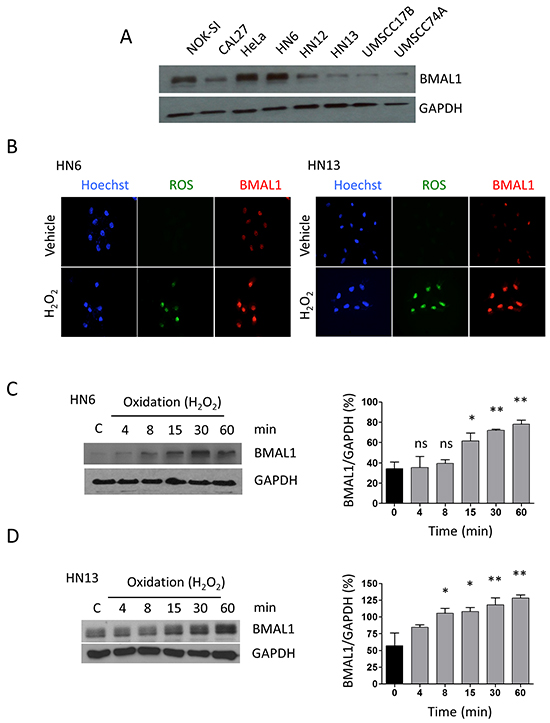
Figure 2: Oxidation causes accumulation of BMAL1. A. Head and neck cancer cells have different expression levels of the core clock protein BMAL1. B. Immunofluorescence assay depicts accumulation of ROS (green channel) and BMAL1 (red channel) upon oxidation. C. and D. Western blot assay demonstrates time-dependent accumulation of BMAL1 upon oxidative stress in HNSCC cells (*p<0.05, **p<0.01).
Targeted disruption of PTEN in HNSCC results in activation of mTOR and accumulation of BMAL1
We next examined whether loss of PTEN function caused accumulation of BMAL1. PTEN-targeted siRNA (Figure 3A) (Figure 3B) resulted in a reduction of PTEN protein. PTEN suppression was accompanied by accumulation of BMAL1 in a siRNA concentration-dependent manner. Similarly, downregulation of PTEN resulted in the activation of mTOR signaling, as detected by the increase in pS6 (Figure 3A and 3B). The correlation between PTEN loss and activation of mTOR signaling is well documented by our group and others [20, 26-28]; however, concurrent inactivation of PTEN protein and upregulation of clock genes, such as BMAL1, is a novel discovery. It is unclear whether compromised PTEN function that leads to accumulation of BMAL1 is specific to head and neck cancer or is a normal regulatory mechanism found in epithelial cells. To address this question, we excised PTEN from epithelial cells in vivo using epithelial-specific Pten conditional knockout mice generated by crossing mice that harbor a floxed Pten allele containing two loxP sites (Ptentm1Hwu or PtenF/F) with mice that express the Cre recombinase under the control of the K14 promoter (K14Cre)[20]. Through immunolocalization, we found that control mice present a mean of 61% of epithelial cells positive for nuclear BMAL1 localized in the interfollicular component of the epidermis (Figure 3C_Control arrowhead). Interestingly, deletion of PTEN from the basal layer of the epidermis resulted in in vivo accumulation of nuclear BMAL1 (mean 79%) in epithelial cells (Figure 3C_K14Cre PtenF/F, arrow). Of note, cornified layer of the epidermis (absent of nuclear structures) present typical unspecific staining for the secondary antibody. Collectively, our data suggest that PTEN-deficient HNSCC cells showing deregulated clock gene machinery can also be observed in normal epithelial cells presenting genetic excision of PTEN. These results suggest that PTEN/PI3K signaling pathway may play an unexpected role in the regulation of the core clock gene BMAL1 in epithelial cells.
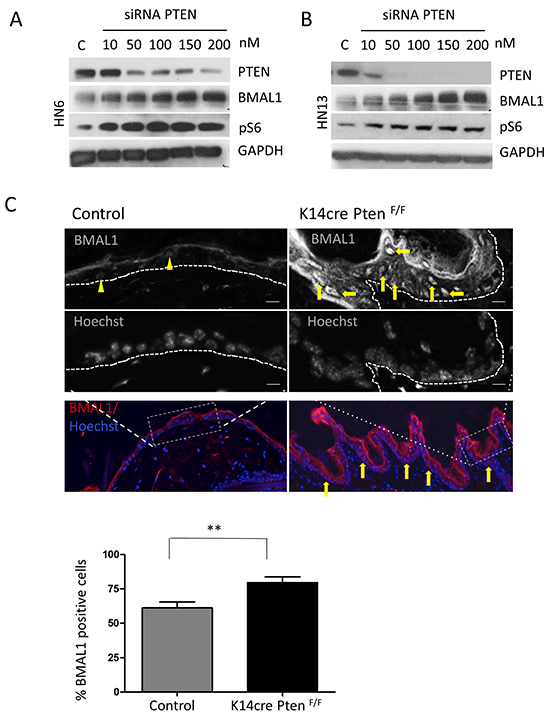
Figure 3: Targeted disruption of PTEN in vitro and in vivo induces activation of pS6 and BMAL1. A. and B. Targeted inhibition of PTEN using siRNA results in concentration-dependent inhibition of PTEN protein in HNSCC cells. HNSCC cells show accumulation of BMAL1 and pS6 in response to PTEN inhibition. C. Immunofluorescence assay to detect BMAL1 in PTEN conditional knockout mice (K14cre PtenF/F) and control littermates. Immunofluorescences and graphic show high accumulation of nuclear BMAL1 in K14cre PtenF/F mice compared to control mice (K14cre). Scale bars represent 50 μm. Black and white images depict cells positive for BMAL1 in K14cre PtenF/F mice (arrows) compared to few BMAL-positive cells in control mice (arrowhead). Scale bars represent 10 μm. (**p<0.01).
PTEN-mediated accumulation of BMAL1 requires activation of mTOR signaling
The progressive accumulation of BMAL1 and pS6 in HNSCC with compromised PTEN function suggests crosstalk between mTOR and clock signaling. We next examined the effect of mTOR on the accumulation of BMAL1. Our work and the work of others have previously shown that disruption of PTEN results in activation of mTOR signaling [20, 26, 29, 30]. We used Rapamycin, a well-known specific inhibitor of mTOR [31], to examine a potential role for mTOR in the accumulation of BMAL1 in K14Cre PtenF/F mice. We found that vehicle-treated K14Cre PtenF/F mice had an abrupt accumulation of nuclear BMAL1 compared to control littermates (Figure 4, *** p<0.001). Remarkably, administration of Rapamycin rescued BMAL1 accumulation by significantly diminishing the amount of BMAL1-positive cells in K14Cre PtenF/F(Figure 4) (*** p<0.001). Furthermore, rapamycin reduced BMAL1 expression in K14Cre PtenF/F mice to levels comparable to control littermates (Figure 4) (ns p>0.05).
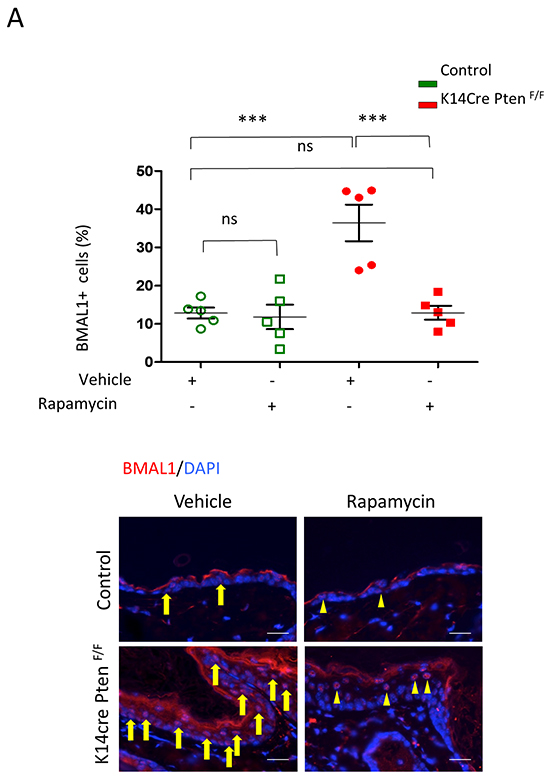
Figure 4: Inhibition of mTOR signaling with Rapamycin rescues PTEN-driven accumulation of BMAL1. A. Tissue samples and graphic show BMAL1 in K14cre PtenF/F mice and control (arrows). Note a significant amount of BMAL1 expression in PTEN mutant mice. K14cre PtenF/F mice and control littermates received Rapamycin or vehicle every other day for 15 days. Rapamycin treatment reduces the expression of nuclear BMAL1 in K14cre PtenF/F mice (arrowhead) (*** p<0.001) to similar levels found in control mice (ns: p>0.05). Scale bars -30 μm.
Nuclear BMAL1 increase requires mTOR signaling in vivo
The circadian rhythm molecule Period2 (Per2) plays a critical role as a tumor suppressor by controlling levels of BMAL1. To confirm that BMAL1 nuclear accumulation depends on mTOR activity, we test whether mTOR inhibition interferes with PER2-driven BMAL1 accumulation. For this, we used Per2 core negative transcriptional inhibitor of the circadian clock knockout mice that harbor constitutively active BMAL1 [32-34]. Accumulation of BMAL1 in mPer2 knockout mice occurs in the epidermis (Figure 5A). BMAL1 levels were more than three-fold higher in mPer2 knockout mice than wild-type littermate controls (*** p<0.001) (Figure 5A). We showed that mPer2 knockout mice treated with Rapamycin had a significant reduction in BMAL1 expression in the epidermis compared to mPer2 mice treated with vehicle (** p<0.01), resulting in BMAL1 levels similar to WT control mice (ns p>0.05)(Figure 5A). Our data suggests that mTOR plays a critical role in the control of BMAL1.
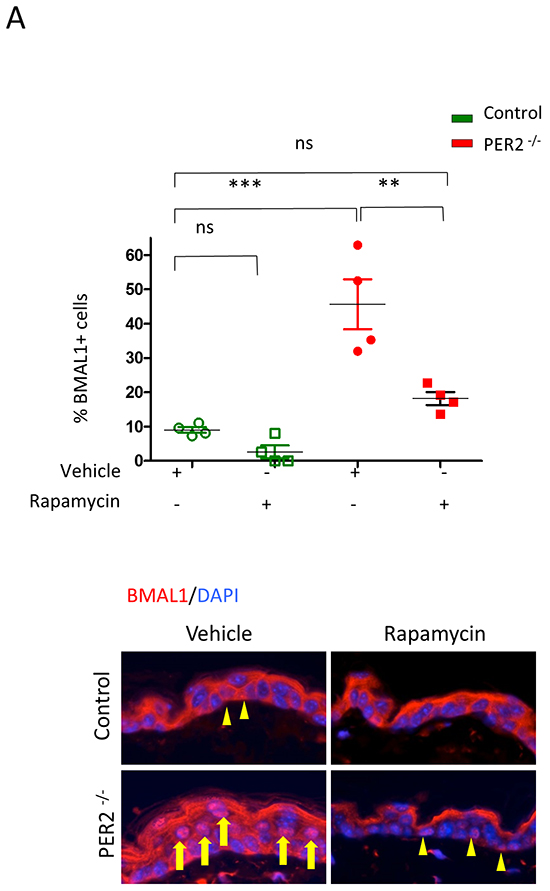
Figure 5: Rapamycin reduces the accumulation of BMAL1 in Per2 knockout mice. A. As shown in the left panel, tissue samples from Per2 knockout mice (mPER-/-) depict robust accumulation of nuclear BMAL1 (arrow) compared to control littermates (arrowhead)(*** p<0.001). Administration of Rapamycin reduces the accumulation of BMAL1 in the epidermis of mPer-/- mice (arrowhead) compared to mPer-/- mice receiving vehicle alone (** p<0.01) to levels comparable to wild-type mice receiving vehicle alone (ns: p>0.05).
mTORC1 and mTORC2 are involved in regulating BMAL1 in HNSCC
We have shown that downregulation of the tumor suppressor gene PTEN in HNSCC results in activation of the mTOR signaling pathway [7] and accumulation of BMAL1 (Figure 3A and 3B). Using genetically defined animal models; we also showed here that inhibition of mTOR reestablished the normal expression levels of BMAL1 protein in K14Cre PtenF/F and mPer2 knockout mice, suggesting a critical role for mTOR in controlling and maintaining BMAL1. We next examined whether HNSCC requires mTORC1 and mTORC2 signaling to drive BMAL1 activation. Using siRNA technology, we interfered with the mTORC1 and mTORC2 complexes by disrupting their scaffold proteins, Raptor [35, 36] and Rictor [37, 38] (Figure 6A–6D). Silencing Raptor and Rictor resulted in reduced BMAL1 expression. Overall, Raptor knockdown led to augmented downregulation of BMAL1 levels (Figure 6A and 6B) when compared to Rictor (Figure 6C and 6D). Nonetheless, small interference RNAs against Raptor and Rictor efficiently downregulated BMAL1. Collectively, our findings suggest that loss of function of PTEN in HNSCC results in constitutive accumulation of BMAL1 in an mTOR-dependent manner. Similarly, we also observed that accumulation of BMAL1 resulting from disruption of PER2 requires mTOR signaling. Our findings suggest that mTOR can act as a master regulatory mechanism of BMAL1 in normal and malignant epithelial cells (Figure 6E).
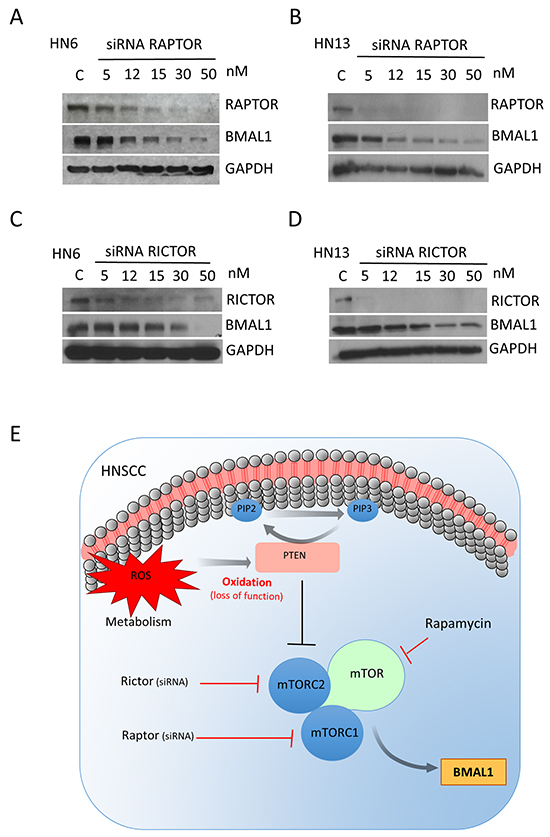
Figure 6: Small interference RNA targeting Raptor and Rictor disrupts BMAL1 accumulation in HNSCC. Targeted disruption of Raptor (A-B) and Rictor (C-D) using siRNA results in a dose-dependent downregulation of BMAL1 in HNSCC cells. E. Disruption of PTEN by protein oxidation causes activation of mTOR signaling, resulting in accumulation of BMAL1. Notably, inhibition of mTOR signaling, particularly mTORC1 and mTORC2, results in restoration of normal BMAL1 levels in the epidermis of mice and head and neck cancer cells. These results demonstrate a novel role for mTOR in regulating nuclear levels of the core clock gene BMAL1.
DISCUSSION
Dysfunction in circadian rhythm is associated with an increased incidence of solid tumors. Working the night shift was linked with high prostate-specific antigen (PSA) levels and increased risk of developing prostate, as well as, breast cancers [39-43]. Indeed, increased incidence of prostate cancer was also found in individuals living in countries with high light exposure at night [44] and those who sleep less [45-47]. Breast cancer patients who experience altered sleep-wake cycles or work night shifts also have acquired resistance to Tamoxifen therapy [48]. Although the circadian rhythm clearly influences the behavior of solid tumors, a better understanding of the molecular events that control clock gene expression in cancer is needed.
The circadian rhythm is a complex molecular circuitry comprised of transcriptional activators and negative regulators with expression levels that oscillate in a day/night fashion. Positive signaling of the clock machinery is mediated largely by CLOCK and BMAL1, which induce clock-controlled genes. Other members of the clock regulatory machinery are negative feedback regulators, which are activated when CLOCK and BMAL1 are downregulated. PER2 is a member of the cryptochrome and period negative feedback regulators of the clock machinery. PER2 downregulation is present in leukemia, glioma, breast cancer [49-52]; and its deletion results in the development of lymphomas [34]. These findings suggest that PER2 is a tumor suppressor gene, a role that has been demonstrated in mice [34]. Notably, PER2 downregulation leads to BMAL1 activation. Indeed, we observed this activation in the epidermis of mPer2 knockout mice. Likewise, excision of PTEN, a tumor suppressor gene, unexpectedly led to the accumulation of BMAL1 in the epidermis of mice. The PTEN tumor suppressor is the major negative regulator of the PI3K/mTOR pathway, which is one of the most frequently active pathways in human cancers, including HNSCC [53-55].
We have previously demonstrated that pharmacological inhibition of the mTOR signaling using Rapamycin results in rescue of the skin phenotype observed in PTEN deficient mice [20]. Based on our previous findings, we asked whether Rapamycin would inhibit BMAL1 accumulation in PTEN conditional knockout and PER2 knockout mice. Remarkably, administration of Rapamycin blocked BMAL1 accumulation in both animal models, suggesting that mTOR serves as a key regulatory mechanism underlying epithelial circadian rhythm. Indeed, our results aligned with mTOR being directly associated with the control of organismal metabolism, similar to CLOCK and BMAL1 (reviewed in [56, 57]). Accumulation of pS6, readout of mTOR activation, in the frontal cortex, heart, and liver was also observed in Bmal1 knockout mice [58]. BMAL1-dependent regulation of the mTOR signaling pathway also affects aging and cellular senescence [58]. We also showed that accumulation of BMAL1 requires mTOR activity in vitro and in vivo. Our findings indicate BMAL1 as a readout of compromised PTEN and PER2 function and suggest that BMAL1 is involved in the progression of cancer. Likewise, if BMALl is removed, it leads to activation of senescence [58].
The correlation between the compromised function of tumor suppressors and the circadian rhythm is also poorly understood. PTEN mutations are observed in a fraction of HNSCC [3, 4], whereas the vast majority of HNSCC have functional PTEN gene but downregulated protein expression [6-8]. We found that H2O2 caused transient inactivation of PTEN protein in HNSCC. Accumulation of H2O2 is commonly observed in cancer as a by-product of deregulated metabolism, and its effects on PTEN have been shown in HeLa and NIH3T3 cells [19, 59]. Our data showed that oxidation-mediated inactivation of PTEN in HNSCC cells also leads to unexpected accumulation of the core clock protein BMAL1.
The connection between the activation of reduction/oxidation pathways and the circadian rhythm has been shown in zebrafish [60], in which the light modulates the circadian clock in Z3 light-responsive cells through the production of intracellular H2O2 that acts as an ROS signaling molecule. Notably, our data revealed that H2O2 induces activation of intracellular ROS and nuclear BMAL1 in HNSCC cells. Nevertheless, PTEN, rather than H2O2 alone, is a critical regulator of clock genes. Our results lead to meaningful findings. First, oxidation-driven and genetically defined inhibition of PTEN results in BMAL1 accumulation in normal and malignant epithelial cells. Second, PTEN-induced accumulation of BMAL1 requires a functional mTOR signaling. Third, PTEN-independent and PER2-driven accumulation of BMAL1 also requires a functional mTOR signaling. Fourth, maintenance of levels of BMAL1 in cancer cells requires functional mTORC1 and mTORC2 complexes. Although the requirements for mTOR signaling to induce BMAL1 expression in HNSCC are demonstrated in this manuscript, it’s still unknown if a similar mechanism would be existent during normal epithelial homeostasis. This study is also limited in the identification of different physiological mechanisms and conditions that could lead to a transient downregulation of PTEN in HNSCC tumors. Future studies are needs to further contribute to our knowledge on how deregulated molecules contribute to HNSCC.
In summary, we showed that downregulation of PTEN leads to activation of mTOR signaling and accumulation of BMAL1. Notably, inhibition of mTOR signaling (mTORC1 and mTORC2) results in restoration of normal BMAL1 levels in the epidermis in vivo, and also in head and neck cancer cells. In addition, these results suggest that BMAL1 activation in conjunction with mTOR is required for skin phenotype found in PER2 and PTEN mice. These results demonstrated a novel role for mTOR in regulating nuclear levels of the core clock gene BMAL1.
MATERIALS AND METHODS
Cell lineages and reagents
HNSCC cell lines (Cal27 (tongue), WSU-HN6 (base of tongue), WSU-HN12 (lymph node/metastasis), WSU-HN13 (tongue), UMSCC17B (lymph node/metastasis), and UMSCC74A (tongue) [61-65], HeLa cervical cells [66], and NOK-SI (spontaneously immortalized normal oral keratinocytes) cells [67] were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B. Cells were maintained in a 5% CO2-humidified incubator at 37°C. Cells were treated with hydrogen peroxide (1.5 mM-Sigma) and Rapamycin (50 nM – LC Laboratories). Cells were synchronized with forskolin (10μM; Sigma-Aldrich, USA) before each assay. All cells were previously authenticated by PCR amplification of short tandem repeats to ensure cell identity. Cal27 and HeLa cells were acquired from the American Type Culture Collection (ATCC-Manassas).
Immunofluorescence (IF)
Paraffin-embedded tissues were sectioned (3-5 μm), and a standard procedure was used to dewax and hydrate the tissues through graded alcohol followed by antigen retrieval and an endogenous peroxidase block [68]. IF was performed using primary antibodies BMAL1 (NB100-2288, Novus Biological, Littleton, CO), pS6 (S235/236, Cell Signaling Technology, Danvers, MA), and Ki-67 (Cell Signaling Technology, Danvers, MA) (Supplementary Table S1). After incubation overnight, slides were washed with PBS, incubated with a secondary antibody conjugated with either fluorescein (Jackson ImmunoResearch Labs 1:100) or rhodamine (Jackson Immuno Research Labs 1:100) and mounted with media containing DAPI (Vector Laboratories). Images were taken using a QImaging ExiAqua monochrome digital camera attached to a Nikon Eclipse 80i Microscope (Nikon, Melville, NY) and by a color QImaging Publisher attached to a Leica CTR5000 microscope. Images were visualized with QCapturePro software.
ROS assay
The ROS assay was performed as previously reported [69]. Intracellular levels of ROS were analyzed using chloromethyl CM-H2DCFDA (Molecular Probes/Life Technologies, Grand Island, New York) and detection of positive cells using a Nikon fluorescent microscope. ROS was detected after intracellular esterases removed the acetate groups upon cellular oxidation.
Experimental mice
The in vivo study was performed according to the University of Michigan Committee on Use and Care of Animals (UCUCA) approved protocol and in compliance with the Guide for the Care and Use of Laboratory Animals. Animals were housed in 12-hrs light/dark cycles, and they received standard rodent chow and water ad libitum in compliance with AAALAC guidelines. PtenF/F mice (The Jackson Laboratory) were crossed with K14Cre mice to generate K14Cre Pten mutant mice [20]. Briefly, K14Cre PTENF/+ mice were crossed with PtenF/F mice to generate K14Cre PtenF/F, K14Cre PtenF/+ and control mice in the same litter. Genotyping was performed on tail biopsies using a PCR assay with primers previously described [20]. mPer2 knockout mice (Per2tm1Drw) were genotyped as previously described [32].
Administration of rapamycin
Rapamycin (LC Laboratories) was reconstituted in absolute ethanol at 10 mg/mL and stored at −20°C. Rapamycin was diluted in 5.2% Tween 80 (Sigma) and 5.2% polyethylene glycol (PEG-400; Hampton Research) and injected i.p. (1 mg/kg) every other day for 15 days [20, 68]. Only vehicle solution was administered as controls.
Western blotting
Tumor cells were lysed with cell lysis buffer containing protease inhibitors and briefly sonicated. Total protein was run in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to an Immobilon membrane (Millipore, Billerica, MA, USA). Membranes were blocked in 5% nonfat dry milk containing 0.1 M Tris (pH 7.5), 0.9% NaCl and 0.05% Tween-20 for 1 hour at room temperature. Membranes were incubated with PTEN (Cell Signaling Technology, Danvers, MA), p-S6 (S235/236, Cell Signaling Technology, Danvers, MA), BMAL1 (NB100-2288, Novus Biological, Littleton, CO), Raptor (53A2 - Cell Signaling Technology), and Rictor (24C12 - Cell Signaling Technology) primary antibodies at 4°C overnight (Supplementary Table S1). Membranes were then incubated with appropriate secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Sta. Cruz, CA, USA). The signal was developed using the ECL SuperSignal West Pico Substrate (Pierce Biotechnology, Rockford, IL, USA). GAPDH served as a loading control (Calbiochem, Gibbstown, NJ, USA). Identification of reduced and oxidized forms of PTEN was performed as previously described [19]. Briefly, cells were scraped into an ice-cold solution containing 50% trichloroacetic acid and sonicated. After centrifugation, cell lysate was washed with buffer containing 0.2 ml of Tris-HCL (100 nM, PH 6.8), 2% SDS, and 40 nM of MEM followed by polyacrylamide gel electrophoresis.
Raptor and rictor knockdown
Knockdown of Raptor and Rictor was performed as previously described [70-72]. In detail, cells were seeded in 24-well plates and transfected with HiPerFect (Qiagen) using 15 nM double-stranded RNA oligonucleotides directed against human Raptor (forward: 5′- GGA CAA CGG CCA CAA GUAdTdT-3′ and reverse: 5′- UAC UUG UGG CCG UUG UCCdTdT-3′), or 5 nM double-stranded RNA oligonucleotides against Rictor (forward: 5′- CCU AAU GAA UAU GGC UGC AUC CUU UdTdT-3′ and reverse: 5′- AAA GGA UGC AGC CAU AUU CAU UAG GdTdT-3′) (Invitrogen). Optimal concentrations and time points were determined by dilution curves of siRNA for each target and immunoblot analysis. The sequences of the control negative siRNA (Invitrogen) oligonucleotides were as follows: 5′-UUC UCC GAA CGU GUC ACG UdTdT-3′ and 5′- ACG UGA CAC GUU CGG AGA AdTdT-3′[73]. The sequences of the PTEN siRNA were 5′-CCAAUGGCUAAGUGAAGAUGACAAUdTdT-3′ and 5′-AUUGUCAUCUUCACUUAGCCAUUGGdTdT-3′.
Statistical analysis
Statistical analyzes were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA). Statistical analyzes of positive cells for BMAL1 in K14Cre Pten and Per2 knockout mice and others were performed using one-way ANOVA followed by the Tukey’s multiple comparison tests. Asterisks denote statistical significance (*p<0.05; **p<0.01; ***p<0.001; and NS p>0.05).
ACKNOWLEDGMENTS
The authors thank Dr. J. Silvio Gutkind and Dr. Thomas Carey for the HNSCC cell lines. (Dr. Gutkind kindly provided NOK-SI, HN6, HN12, and HN13 cell lines; and Dr. Carey provided UMSCC17B and UMSCC74A HNSCC cell lines).
This work was supported by the National Institutes of Health (NIH/NCI) P50-CA97248 (University of Michigan Head and Neck SPORE), the University of Michigan School of Dentistry startup, the University of Michigan-OVPR funds #U038018 (PP), the Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior - CAPES, FAPESP (Sao Paulo Research Foundation), and the Robert Wood Johnson Foundation.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, Lu Y, Zhang Q, Du Y, Gilbert BR, Freilino M, Sauerwein S, Peyser ND, Xiao D, Diergaarde B, Wang L, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer discovery. 2013; 3:761-769.
2. Giudice FS and Squarize CH. The determinants of head and neck cancer: Unmasking the PI3K pathway mutations. Journal of carcinogenesis & mutagenesis. 2013; pii: 003
3. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, Weinstein JN, Trevino L, Drummond JA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333:1154-1157.
4. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011; 333:1157-1160.
5. Pullos AN, Castilho RM and Squarize CH. HPV Infection of the Head and Neck Region and Its Stem Cells. Journal of dental research. 2015; 94:1532-1543.
6. Squarize CH, Castilho RM and Santos Pinto D, Jr. Immunohistochemical evidence of PTEN in oral squamous cell carcinoma and its correlation with the histological malignancy grading system. Journal of oral pathology & medicine. 2002; 31:379-384.
7. Squarize CH, Castilho RM, Abrahao AC, Molinolo A, Lingen MW and Gutkind JS. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia. 2013; 15:461-471.
8. Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V and Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral oncology. 2009; 45:324-334.
9. Poetsch M, Lorenz G and Kleist B. Detection of new PTEN/MMAC1 mutations in head and neck squamous cell carcinomas with loss of chromosome 10. Cancer genetics and cytogenetics. 2002; 132:20-24.
10. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997; 275:1943-1947.
11. Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, Li J, Parsons R and Ellenson LH. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer research. 1997; 57:3935-3940.
12. Djordjevic B, Hennessy BT, Li J, Barkoh BA, Luthra R, Mills GB and Broaddus RR. Clinical assessment of PTEN loss in endometrial carcinoma: immunohistochemistry outperforms gene sequencing. Modern pathology. 2012; 25:699-708.
13. Lee JI, Soria JC, Hassan KA, El-Naggar AK, Tang X, Liu DD, Hong WK and Mao L. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch Otolaryngol Head Neck Surg. 2001; 127:1441-1445.
14. Shao X, Tandon R, Samara G, Kanki H, Yano H, Close LG, Parsons R and Sato T. Mutational analysis of the PTEN gene in head and neck squamous cell carcinoma. International journal of cancer. 1998; 77:684-688.
15. Zhou XP, Gimm O, Hampel H, Niemann T, Walker MJ and Eng C. Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. The American journal of pathology. 2000; 157:1123-1128.
16. Garcia JM, Silva J, Pena C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, Espana P and Bonilla F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes, chromosomes & cancer. 2004; 41:117-124.
17. Wang L, Wang WL, Zhang Y, Guo SP, Zhang J and Li QL. Epigenetic and genetic alterations of PTEN in hepatocellular carcinoma. Hepatology research. 2007; 37:389-396.
18. Alvarez-Nunez F, Bussaglia E, Mauricio D, Ybarra J, Vilar M, Lerma E, de Leiva A, Matias-Guiu X and Thyroid Neoplasia Study G. PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid. 2006; 16:17-23.
19. Lee SR, Yang KS, Kwon J, Lee C, Jeong W and Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. The Journal of biological chemistry. 2002; 277:20336-20342.
20. Squarize CH, Castilho RM and Gutkind JS. Chemoprevention and treatment of experimental Cowden’s disease by mTOR inhibition with rapamycin. Cancer research. 2008; 68:7066-7072.
21. Janich P, Pascual G, Merlos-Suarez A, Batlle E, Ripperger J, Albrecht U, Cheng HY, Obrietan K, Di Croce L and Benitah SA. The circadian molecular clock creates epidermal stem cell heterogeneity. Nature. 2011; 480:209-214.
22. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV and Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes & development. 2006; 20:1868-1873.
23. Karantanos T, Theodoropoulos G, Pektasides D and Gazouli M. Clock genes: their role in colorectal cancer. World journal of gastroenterology. 2014; 20:1986-1992.
24. Yagita K and Okamura H. Forskolin induces circadian gene expression of rPer1, rPer2 and dbp in mammalian rat-1 fibroblasts. FEBS letters. 2000; 465:79-82.
25. Ripperger JA and Schibler U. Circadian regulation of gene expression in animals. Current opinion in cell biology. 2001; 13:357-362.
26. Squarize CH, Castilho RM, Bugge TH and Gutkind JS. Accelerated wound healing by mTOR activation in genetically defined mouse models. PloS one. 2010; 5:e10643.
27. Morgensztern D and McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anti-cancer drugs. 2005; 16:797-803.
28. Seront E, Pinto A, Bouzin C, Bertrand L, Machiels JP and Feron O. PTEN deficiency is associated with reduced sensitivity to mTOR inhibitor in human bladder cancer through the unhampered feedback loop driving PI3K/Akt activation. British journal of cancer. 2013; 109:1586-1592.
29. DeGraffenried LA, Fulcher L, Friedrichs WE, Grunwald V, Ray RB and Hidalgo M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Annals of oncology. 2004; 15:1510-1516.
30. Ozes ON, Akca H, Mayo LD, Gustin JA, Maehama T, Dixon JE and Donner DB. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:4640-4645.
31. Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G and Abraham RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. The Journal of biological chemistry. 1995; 270:815-822.
32. Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM and Weaver DR. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron. 2001; 30:525-536.
33. Hoogerwerf WA, Shahinian VB, Cornelissen G, Halberg F, Bostwick J, Timm J, Bartell PA and Cassone VM. Rhythmic changes in colonic motility are regulated by period genes. American journal of physiology Gastrointestinal and liver physiology. 2010; 298:G143-150.
34. Fu L, Pelicano H, Liu J, Huang P and Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002; 111:41-50.
35. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J and Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002; 110:177-189.
36. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002; 110:163-175.
37. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A and Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature cell biology. 2004; 6:1122-1128.
38. Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P and Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current biology. 2004; 14:1296-1302.
39. Hansen J. Increased breast cancer risk among women who work predominantly at night. Epidemiology. 2001; 12:74-77.
40. Schernhammer ES, Laden F, Speizer FE, Willett WC, Hunter DJ, Kawachi I and Colditz GA. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. Journal of the National Cancer Institute. 2001; 93:1563-1568.
41. Flynn-Evans EE, Mucci L, Stevens RG and Lockley SW. Shiftwork and prostate-specific antigen in the National Health and Nutrition Examination Survey. Journal of the National Cancer Institute. 2013; 105:1292-1297.
42. Kubo T, Ozasa K, Mikami K, Wakai K, Fujino Y, Watanabe Y, Miki T, Nakao M, Hayashi K, Suzuki K, Mori M, Washio M, Sakauchi F, Ito Y, Yoshimura T and Tamakoshi A. Prospective cohort study of the risk of prostate cancer among rotating-shift workers: findings from the Japan collaborative cohort study. American journal of epidemiology. 2006; 164:549-555.
43. Conlon M, Lightfoot N and Kreiger N. Rotating shift work and risk of prostate cancer. Epidemiology. 2007; 18:182-183.
44. Kloog I, Haim A, Stevens RG and Portnov BA. Global co-distribution of light at night (LAN) and cancers of prostate, colon, and lung in men. Chronobiology international. 2009; 26:108-125.
45. Kakizaki M, Inoue K, Kuriyama S, Sone T, Matsuda-Ohmori K, Nakaya N, Fukudo S, Tsuji I and Ohsaki Cohort S. Sleep duration and the risk of prostate cancer: the Ohsaki Cohort Study. British journal of cancer. 2008; 99:176-178.
46. Gozal D, Farre R and Nieto FJ. Obstructive sleep apnea and cancer: Epidemiologic links and theoretical biological constructs. Sleep medicine reviews. 2016; 27:43-55.
47. Gozal D, Farre R and Nieto FJ. Putative Links Between Sleep Apnea and Cancer: From Hypotheses to Evolving Evidence. Chest. 2015; 148:1140-1147.
48. Dauchy RT, Xiang S, Mao L, Brimer S, Wren MA, Yuan L, Anbalagan M, Hauch A, Frasch T, Rowan BG, Blask DE and Hill SM. Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen therapy in breast cancer. Cancer research. 2014; 74:4099-4110.
49. Gery S, Gombart AF, Yi WS, Koeffler C, Hofmann WK and Koeffler HP. Transcription profiling of C/EBP targets identifies Per2 as a gene implicated in myeloid leukemia. Blood. 2005; 106:2827-2836.
50. Xia HC, Niu ZF, Ma H, Cao SZ, Hao SC, Liu ZT and Wang F. Deregulated expression of the Per1 and Per2 in human gliomas. The Canadian journal of neurological sciences. 2010; 37:365-370.
51. Chen ST, Choo KB, Hou MF, Yeh KT, Kuo SJ and Chang JG. Deregulated expression of the PER1, PER2 and PER3 genes in breast cancers. Carcinogenesis. 2005; 26:1241-1246.
52. Yang X, Wood PA, Oh EY, Du-Quiton J, Ansell CM and Hrushesky WJ. Down regulation of circadian clock gene Period 2 accelerates breast cancer growth by altering its daily growth rhythm. Breast cancer research and treatment. 2009; 117:423-431.
53. Engelman JA, Luo J and Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature reviews Genetics. 2006; 7:606-619.
54. Cully M, You H, Levine AJ and Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature reviews Cancer. 2006; 6:184-192.
55. Hennessy BT, Smith DL, Ram PT, Lu Y and Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nature reviews Drug discovery. 2005; 4:988-1004.
56. Shimobayashi M and Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nature reviews Molecular cell biology. 2014; 15:155-162.
57. Bellet MM and Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. Journal of cell science. 2010; 123:3837-3848.
58. Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP and Kondratov RV. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014; 6:48-57. doi: 10.18632/aging.100633.
59. Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER and Rhee SG. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:16419-16424.
60. Hirayama J, Cho S and Sassone-Corsi P. Circadian control by the reduction/oxidation pathway: catalase represses light-dependent clock gene expression in the zebrafish. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:15747-15752.
61. Cardinali M, Pietraszkiewicz H, Ensley JF and Robbins KC. Tyrosine phosphorylation as a marker for aberrantly regulated growth-promoting pathways in cell lines derived from head and neck malignancies. International journal of cancer. 1995; 61:98-103.
62. Gioanni J, Fischel JL, Lambert JC, Demard F, Mazeau C, Zanghellini E, Ettore F, Formento P, Chauvel P, Lalanne CM and et al. Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: establishment, characterization and response to cytotoxic treatment. European journal of cancer & clinical oncology. 1988; 24:1445-1455.
63. Carey TE, Van Dyke DL, Worsham MJ, Bradford CR, Babu VR, Schwartz DR, Hsu S and Baker SR. Characterization of human laryngeal primary and metastatic squamous cell carcinoma cell lines UM-SCC-17A and UM-SCC-17B. Cancer research. 1989; 49:6098-6107.
64. Bradford CR, Zhu S, Ogawa H, Ogawa T, Ubell M, Narayan A, Johnson G, Wolf GT, Fisher SG and Carey TE. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head & neck. 2003; 25:654-661.
65. Basile JR, Castilho RM, Williams VP and Gutkind JS. Semaphorin 4D provides a link between axon guidance processes and tumor-induced angiogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:9017-9022.
66. Scherer WF, Syverton JT and Gey GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. The Journal of experimental medicine. 1953; 97:695-710.
67. Castilho RM, Squarize CH, Leelahavanichkul K, Zheng Y, Bugge T and Gutkind JS. Rac1 is required for epithelial stem cell function during dermal and oral mucosal wound healing but not for tissue homeostasis in mice. PloS one. 2010; 5:e10503.
68. Castilho RM, Squarize CH, Chodosh LA, Williams BO and Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell stem cell. 2009; 5:279-289.
69. Dillenburg CS, Almeida LO, Martins MD, Squarize CH and Castilho RM. Laser phototherapy triggers the production of reactive oxygen species in oral epithelial cells without inducing DNA damage. Journal of biomedical optics. 2014; 19:048002.
70. Rosselli-Murai LK, Almeida LO, Zagni C, Galindo-Moreno P, Padial-Molina M, Volk SL, Murai MJ, Rios HF, Squarize CH and Castilho RM. Periostin responds to mechanical stress and tension by activating the MTOR signaling pathway. PloS one. 2013; 8:e83580.
71. Peltzer N, Vanli G, Yang JY and Widmann C. Role of mTOR, Bad, and Survivin in RasGAP Fragment N-Mediated Cell Protection. PloS one. 2013; 8:e68123.
72. Verreault M, Weppler SA, Stegeman A, Warburton C, Strutt D, Masin D and Bally MB. Combined RNAi-mediated suppression of Rictor and EGFR resulted in complete tumor regression in an orthotopic glioblastoma tumor model. PloS one. 2013; 8:e59597.
73. Squarize CH, Castilho RM, Sriuranpong V, Pinto DS, Jr. and Gutkind JS. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 2006; 8:733-746.