
INTRODUCTION
Non-Hodgkin’s lymphoma (NHL) is the 7th most common cancer in the US [1], with 530,919 reported cases [2]. 71,850 new cases (39,850 males and 32,000 females) were predicted for 2015 [3]. NHL is a group of heterogeneous diseases resulting from malignant transformation of lymphoid cells with different molecular pathogeneses [4]. 90% of NHLs involve B cells; of these, 30% are characterized as aggressive and 20% as indolent [5]. Diffuse large B-cell lymphoma (DLBCL), representative of aggressive NHLs, is often curable but follicular lymphoma (FL), the prototype indolent NHL, remains difficult to treat [6, 7].
BCL-2 (B-cell lymphoma 2), a key regulator of apoptosis first cloned from a human B-cell lymphoma, is expressed in a number of hematologic malignancies and solid tumors. BCL-2 is overexpressed in both FL and DLBCL, mostly as a result of a t(14;18) translocation [8–10]. The translocation, a cytogenetic hallmark of FL, is seen in 80%-90% of cases [11, 12] and in 30% of cases of DLBCL [13, 14]. 10% of FL tumors lack the translocation and do not express BCL-2 protein: this is common in grade 3b, which is dominated by centroblasts [15, 16].
To date, most methods aimed at interfering with the anti-apoptotic effects of BCL-2 have relied on inhibition via small molecules such as ABT-199 or on decreasing expression at the RNA level via RNAi [17–26]. In this article, we describe a novel approach, the use of a specific oligonucleotide to block transcription by DNA interference (DNAi) [27].
We recently reported on the development, physiochemical characterization and activity of the DNAi, PNT2258 [28]. PNT2258, a 24 base-single stranded, unmodified phosphodiester DNA oligonucleotide (PNT100) encapsulated in specialized liposomes (SMARTICLES®), was designed to target an untranslated region of BCL-2 to block transcription. The drug exhibited broad antitumor activity in vitro and in xenograft models [28]. PNT2258 was shown to be safe and well-tolerated at doses of up to150 mg/m2 in a phase I study [29] and to have activity in patients with relapsed or refractory NHL in a pilot phase II study [30]. We hypothesized that PNT2258 inhibits BCL-2 via direct inhibition of transcription, although it may affect promoter or transcription factor binding. BCL-2 contributes to the genesis of lymphomas, is critical for cancer cell survival, and promotes chemo-resistance [31]. Current chemotherapeutic treatment options are non-specific and cause significant off-target toxicity. Specific targeting of the BCL-2 family of proteins offers the opportunity to minimize off-target effects and control apoptotic and survival pathways directly.
In the current study, we used three lymphoma cell lines with distinct genetic characteristics, WSU-FSCCL (follicular small cleaved cell lymphoma; t(14;18) BCL-2 and t(8;11) c-myc rearrangements), WSU-DLCL2 (diffuse large cell lymphoma; t(14;18) BCL-2 rearrangement) and WSU-WM (Waldenstrom’s macroglobulinemia; t(8;14) c-MYC rearrangement), to investigate the mechanism of action of PNT2258 and its role as a DNAi. Our results show that the down regulation of BCL-2 mRNA and protein expression following PNT2258 exposure triggers cell death pathways in FSCCL cells. We conclude that DNAi is a novel gene-silencing strategy that could be applied to therapeutic targeting of a variety of genes important to different types of cancer.
RESULTS
PNT2258 represses the BCL-2 promoter
The DNAi sequences used here, complementary to the template strand of the DNA, were originally screened against the ENCODE database to ensure that the targeted areas did not encode mRNA or microRNA. The 25 mer PNT100 sequence of PNT2258 is complementary to a segment of the consensus sequence recognized by the Sp1 transcription factor in the BCL-2 P1 promoter. We first sought to determine whether a putative 2638-bp (-3934 to-1287) region in the BCL-2 promoter (P1) confers PNT2258 responsiveness by transiently transfecting K562 cells with a plasmid containing the BCL-2 promoter construct before treatment with or without PNT2258 for 48 h. As shown in Figure 1A, PNT2258 strongly down-regulated BCL-2 promoter activity.
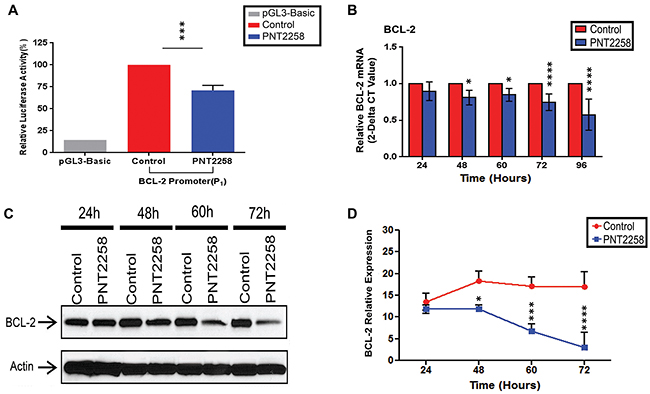
Figure 1: PNT2258 represses BCL-2 promoter activity and down regulates BCL-2 protein and mRNA expression. A. Luciferase activity of K562 cells transiently transfected with the BCL-2 promoter construct was significantly lower in the presence of PNT2258. Results are representative of three independent experiments (***P<0.001 by ANOVA). B-D. WSU-FSCCL cells were treatedwith 2.5μM PNT2258 for the indicatedtimes. (B) Quantitative RT-PCR analysis indicates that PNT2258 inhibition of BCL-2 occurs at the level of transcription. RPLPO was used as control. (C) Representative Western blots demonstrating time-dependent decreases in BCL-2 protein in PNT2258-treated cells; β-actin was used as loading control. (D) Densitometric analysis of BCL-2 band intensities (24 h-72 h) normalized to β-actin. Results are representative of three independent experiments. (* P<0.05, ***P<0.001, and ****P<0.0001 by ANOVA for B and D).
PNT2258 down-regulates BCL-2 protein and mRNA expression in WSU-FSCCL cells
We next examined whether PNT2258 inhibits BCL-2 expression in WSU-FSCCL cells. In this study, we compared PNT2258-treated and non-treated cells as we previously reported that three PNT2258 control sequences (scrambled, mismatched and reverse complement) had no anti-proliferative effects [28]. RT-PCR (Figure 1B) and Western blot (Figure 1C, 1D) analysis revealed that BCL-2 mRNA and protein levels were significantly decreased from 48 h through 72 h after exposure to 2.5μM PNT2258. Since BCL-2 is known to prevent apoptosis, we investigated whether PNT2258 treatment induced apoptosis in these cells and in two other lymphoma cell lines with different BCL-2 expression characteristics.
PNT2258 decreases lymphoma cell viability
We treated three lymphoma cell lines, two with t (14;18) and BCL-2 rearrangements (WSU-FSCCL and WSU-DLCL2) and one (WSU-WM) lacking these rearrangements, with different concentrations (2.5μM, 5.0μM, and 10μM) of PNT2258. Each of the cell lines expressed BCL-2 at baseline (Supplementary Figure S1). Cell viability was decreased in a dose dependent manner: growth inhibition between 2.5μM and 10 μM was statistically significant in all three cell lines at 48h-96h, but differences between 2.5 μM and 5 μM and 5 μM and 10 μM were variable. Cell viability was significantly lower in WSU-FSCCL cells at 24h. As predicted, NHL cell lines with the (14;18) translocation were more sensitive to PNT2258 than the cell line without these alterations (Figure 2A-2C). We chose to continue our investigations in the cell line with the greatest response, WSU-FSCCL (Figure 2A; p ≤ 0.0001).
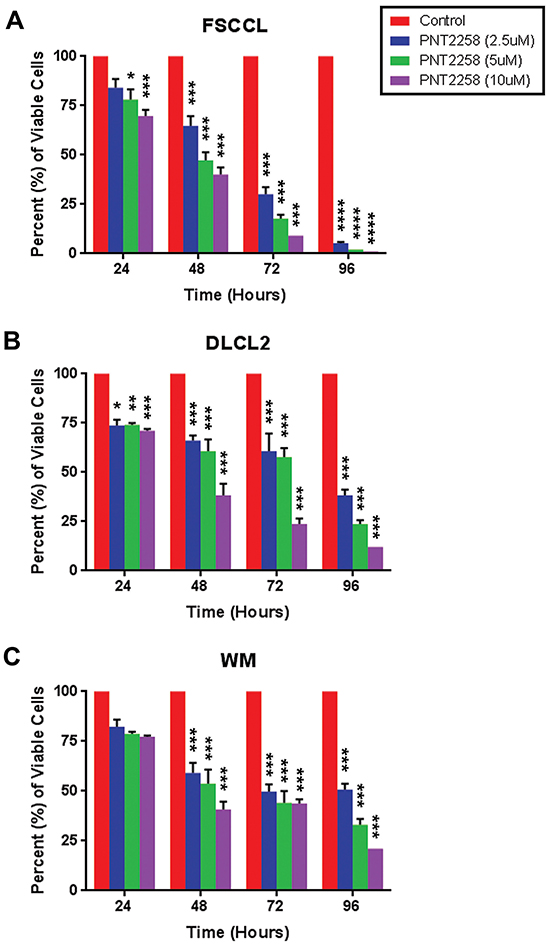
Figure 2: In vitro dose- and time-dependent lymphoma cell growth inhibition by PNT2258. Cell viability was measured by 0.4% trypan blue exclusion every 24 h over 96 h in A. WSU-FSCCL, B. WSU-DLCL2, and C. WSU-WM lymphoma cell lines exposed to PNT2258 (2.5μM, 5μM, and 10μM) from time 0. PNT2258 decreased cell viability in all cell lines but was most effective in WSU-FSCCL cells. All values represent mean ± SE of triplicate experiments. (* P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 by ANOVA).
PNT2258 induces cell cycle arrest and apoptosis
To determine whether PNT2258 induced cell death through on-target intrinsic apoptosis, WSU-FSCCL cells exposed to 2.5μM PNT2258 for up to 96 h were analyzed by flow cytometry. The S-phase population was increased (from 46% to 54%; Figure 3A and 3G) and the proportion of cells in G2-M was reduced (from 16% to 7%; Figure 3A and 3H) at 24 h. There was a reciprocal increase in the sub-G0 compartment (apoptosis) and G0/G1 (resting cells) starting at 48 h (Figure 3B and 3E) with a progressive increase in sub-G0 (from 3% to 35%; Figure 3C and 3E) and G0/G1 (from 34% to 55%; Figure 3C, 3D and 3F) at 72 and 96 h. There was a significant decrease in S-phase cells (from 56% to 28%; Figure 3D and 3G) at 96 h compared with control. The results are summarized in Supplementary Table S1. PNT2258 induces: 1) apoptosis, and 2) cell cycle arrest at G0/G1 (growth inhibition), indicating an effect on the G0/G1 checkpoint. Since current anti-BCL-2 drugs usually cause apoptosis without affecting the cell cycle, our results suggest that PNT2258 may control the cell cycle via non-BCL-2 mechanisms.
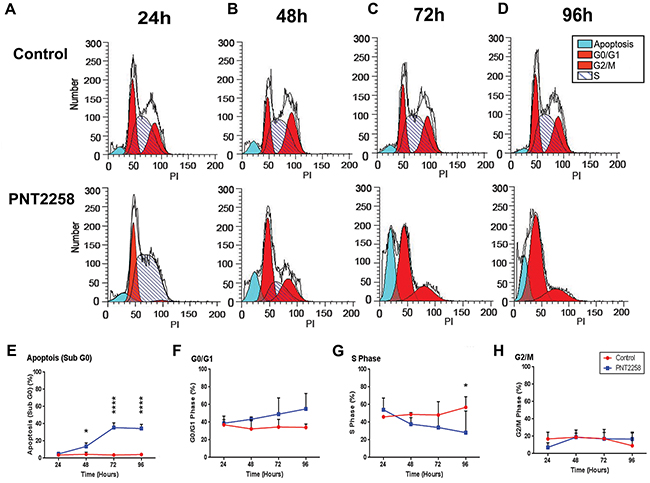
Figure 3: PNT2258 induces S phase cell cycle arrest in WSU-FSCCL cells. FSCCL cells weretreated for 24-96 h with 2.5μM PNT2258and hypotonically lysed in propidium iodide. Nuclei were analyzed for DNA content by flow cytometry. A-D. Original flow data for each time point. E-H. Percent of cells in each phase of the cell cycle (sub-G0, G0/G1, S, G2/M) at each time point. All values represent mean ± SE of triplicate experiments. (* P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001 by ANOVA).
Further examination of the PNT2258 effect on apoptotic cell death in WSU-FSCCL demonstrated that, at the lowest concentration used (2.5uM), PNT2258 elicited the formation of the distinct morphologic features of apoptosis, including cell shrinkage, nuclear chromatin condensation, and formation of membrane blebs and pyknotic bodies after 72h of treatment (data not shown). TUNEL assays show an increase in the FITC-TdT-positive population in PNT2258 treated cells compared with control (horizontal axis) (Figure 4A). Total FITC positive cells also increased with increasing time of incubation, from 20% at 48h to 40% at 72h (Figure 4B). Moreover, there was a progressive shift-to-the-right of the FITC positive cell population with increasing incubation time indicating increasing frequency of DNA breaks (apoptosis). Most of the FITC positive cells were in the sub-G0 phase; however, there were also positive cells in G0/G1, S and G2/M. Induction of apoptosis was confirmed independently using AnnexinV staining as shown in Figure 4C and 4D.
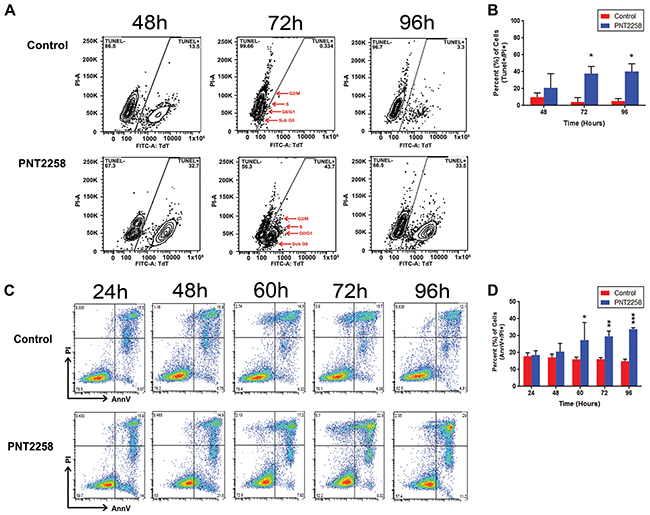
Figure 4: PNT2258 induces apoptosis. WSU-FSCCL cells, treated with 2.5 μM PNT2258 for 24-96 h, were analyzed for terminal transferase dUTP Nick End Labeling (TUNEL) staining by flow cytometry. A. Flow data of FITC positive cells by cell cycle phase (sub G0, G0/G1, S, G2/M). The largest FITC positive population in PNT2258-treated cells was in G0/G1 followed by sub-G0 (red arrows) at 72-96 h. B. Percent of apoptotic cells was significantly higher in PNT2258 treated cells (* P<0.05 by ANOVA). C. Complete time course (24 h -72 h) flow cytometric analysis of apoptosis in cells stained with Annexin V-FITC. D. PNT2258 increased the percent of apoptotic cells from 60- 96 h (* P<0.05, **P<0.01, and ***P<0.001 by ANOVA). Results are representative of three independent experiments.
PNT2258 increases cleaved caspase-3, and cleaved PARP and decreases Mcl-1 and cytochrome C protein levels
We then assessed expression of several BCL-2 family members and selected markers of apoptosis in our PNT2258-treated WSU-FSCCL cells. Protein levels of Mcl-1L and Mcl-1S were decreased (Supplementary Figure S2A, S2B, S2C) but expression of Bcl-xL, Bax, Bak, Bid, and p53, remained unchanged (Supplementary Figure S2A and S2C). However, there was activation of the apoptosis executioner (caspase-3) and induction of both caspase-3 and PARP cleavage (Figure 5A, 5B, 5C) as well as a reduction of Cytochrome C (Figure 5A and 5D), supporting flow cytometry data (Figures 3 and 4) that 2.5μM PNT2258 induces significant apoptosis.
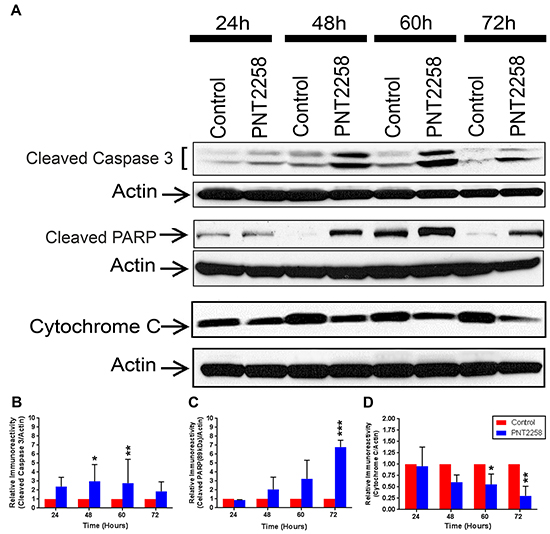
Figure 5: Cleavage of Caspase 3 and PARP protein and down regulation of Cytochrome C in PNT2258 treated WSU-FSSCL cells. A. Representative Western blots of cleaved Caspase 3 and cleaved PARP and Cytochrome C in WSU-FSCCL cells incubated with 2.5μM PNT2258 for 24-72 h; β-actin was used as loading control. B-D. Densitometric analysis indicates that cleaved Caspase 3 and PARP levels were significantly higher (B and C) and Cytochrome C levels were lower (D) in PNT2258 treated cells than in the control. Band intensities were normalized to β-actin. Results are representative of three independent experiments. (* P<0.05 and **P<0.01 by ANOVA for B, C and D).
BCL-2 overexpression represses PNT2258-induced cell death
To document that PNT2258 induces cell death via downregulation of BCL-2, K562 cells were transfected with a pcDNA3 expression vector containing BCL-2 cDNA or the control empty vector before treatment with PNT228. Cell viability, assessed by Trypan blue, was significantly lower in PNT2258-treated cells transfected with the empty vector at 96 hr but not in those expressing BCL-2 (Supplementary Figure S3).
DISCUSSION
DNA interference (DNAi) sequences are designed to target regions upstream of the ATG start site of genes that cause disease. These regions lie within CG-rich areas and, by design, coincide with (or are near) consensus motifs recognized by transcription factors or other cis-regulatory acting elements. Gene transcription is inhibited when sufficient DNAi molecules are available. The PNT100 sequence used for this study specifically targets a region recognized to be regulatory for BCL-2 that is translocated with (14;18) chromosomal rearrangement [28]. The results presented above indicate that PNT2258, a prototype DNAi, is most effective at decreasing cell viability in BCL-2 expressing lymphoma cell lines with the t(14;18) translocation. PNT2258 induced cell cycle arrest and increased apoptosis. BCL-2 expression was significantly inhibited at both the mRNA and protein levels, suggesting that this decrease in BCL-2 expression is responsible for PNT2258-induced apoptosis.
It is noteworthy that PNT2258 induced its therapeutic effects on lymphoma cells at concentrations achievable in patients. A 2.5 μM concentration of PNT2258 translates to 17.6 μg/mL (mol. weight of PNT2258 is 7221g/mol). According to the pharmacokinetic analysis conducted as part of the phase I clinical trial, a maximum plasma concentration of 29.222 μg/mL (cycle 1 day 1) and 42.125 μg/mL (cycle 1 day5) were achieved at a dose of 113 mg/ m2 [30]. The selected safe dose utilized in phase II trial was 120mg/m2.
BCL-2 is a logical target for the treatment of NHL and has been the focus of significant drug development efforts. Our group has developed and evaluated different anti-BCL-2 strategies including small molecule inhibitors (SMIs) [20, 42–45] and anti-sense oligonucleotides [23, 27]. RNAi often requires large doses and continuous exposure for effective suppression of gene function. SMI toxicity varies by type: pan BCL-2 homology domain 3 (BH3) mimetics (AT-101) tend to be more toxic, causing dose-dependent thrombocytopenia [17, 46, 47], selective BCL-2 BH3 mimetics, although platelet sparing, induce resistance and increase the risk of tumor lysis syndrome [17, 48, 49]. Side effects and host toxicity may limit the ability to deliver effective dosages of all these treatment regimens. These limitations can be overcome if one can target a gene of interest at the DNA level.
PNT2258 has shown great promise in preclinical and pilot phase II clinical evaluations (28, 30). The DNAi was designed to target the non-coding, non-transcribed regions 5’ upstream of the ATG start site in the BCL-2 promoter. Disruption of Sp1 results in transcriptional inhibition of BCL-2 [50]. Sp1 binds to double-stranded DNA and recognizes the consensus sequence 5’- G/T (GGGCGG) G/AA -3’, and, coincidentally, a complementary sequence within PNT100 (28). The DNAi sequence used here is unique and 100% homologous to the CpG region of BCL-2. Off-target binding to other Sp1 sites, and/or genomic DNA or RNA is unlikely. Our results confirm that the region between 1506 bp and 1737 bp of the BCL-2 promoter confers PNT2258 responsiveness. BCL-2 overexpression protected K562 cells from PNT2258-induced cell death. The fact that BCL-2 mRNA and protein expression were also inhibited support a DNAi mechanism of action.
The mechanisms of action of this new DNAi are just beginning to be understood [51]. Induction of apoptosis by PNT2258 in WSU-FSCCL cells is predominantly the result of its actions on BCL-2 expression. Its additional effects on cell cycle arrest suggest PNT2258 may have secondary targets as BCL-2 is not involved in cell cycle arrest. It is conceivable that DNAi oligonucleotides may bind to other BCL-2 family proteins. In our hands, only Mcl-1 protein levels were lower in PNT2258-treated cells. However, we believe that this decrease is secondary to the cell cycle changes induced by PNT2258 as Mcl-1 down-regulation was recently shown to be associated with apoptosis and G1/S transition [52]. The possibility that PNT2258 may target both intended (BCL-2) and unintended genes, thus enhancing its effectiveness, remains to be investigated further.
Although some lymphomas can be competently treated with a combination of chemotherapy and immunotherapy, many patients eventually become refractory to these treatments and ultimately succumb to their disease. To our knowledge, this is the first systematic study showing the selective induction of apoptosis and cell cycle arrest in lymphoma cells treated with PNT2258, suggesting potential translational application of PNT2258 as a safe treatment for NHL, including recurrent disease. DNAi oligonucleotides may prove to be an essential tool for future therapeutic intervention, particularly in cancer.
MATERIALS AND METHODS
Cell culture
The cell lines used in this study (WSU-FSCCL, WSU-DLCL2, and WSU-WM), established at Wayne State University (WSU) [32–34] and EBV (Epstein-Barr virus) negative were cultured as previously described [35]. Molecular characterization, translocations and breakpoints, of each cell line have been published [32]. Human K562 cells were maintained according to a standard protocol (ATCC, Manassas, VA).
Transient transfections and luciferase assays
Constructs containing the P1 promoter (LB124: −3934 to −1287) in Bluescript were purchased from Addgene (Cambridge, MA). The P1 promoter construct was obtained by digesting LB124 with SpeI and HindIII and inserting the P1 promoter region into NheI (which has SpeI-compatible overhangs)/HindIII restricted pGL3-Basic (Promega, Madison, WI) that contains the reporter firefly luciferase gene. All transfections were carried out in 24 well-plates, Thermo Scientific, (Rockford, IL, USA). Briefly, plasmids (1 μg BCL-2 promoter construct, 0.1 μg Renilla luciferase-expressing reporter vector pRL-Null (Promega; Madison, WI), were introduced into < 50% confluent cells with Lipofectamine® LTX transfection reagent (Life Technologies; Grand Island, NY). Cells were lysed 48 h later, and promoter activity analyzed in a MicroLumat Plus LB96V (Berthold Technologies) using the Dual Luciferase® Reporter Assay System (Promega). The firefly luciferase values were normalized to those of Renilla luciferase; all transfections were repeated at least three times.
Antibodies and reagents
The following antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA): BCL-2 (2876, 1:1000), Caspase-3 (9662, 1:1000) and PARP (9532, 1:1000). The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA): Cytochrome C (7H8: sc-13560, 1:1000), Bcl-xL (H-5: sc-8392, 1:500), Mcl-1 (S-19: sc-819, 1:1000), Bax (N-20: sc-493, 1:500), Bak (G-23: sc-832, 1:500), Bid (FL-195: sc-11423, 1:1000), p53 (DO-1: sc-126, 1:500), and the actin antibody (ACTN05, 1:2000) from Thermo Scientific, (Rockford, IL, USA). Protein concentrations were determined using the Micro bicinchoninic acid (BCA) protein assay (Pierce Chemical Company, Rockford, IL, USA). PNT2258 and associated reagents were provided by ProNAi Therapeutics (Ann Arbor, MI).
Immunoblotting
PNT2258-treated and untreated cells were harvested, washed in PBS and lysed in M-PER lysis buffer containing a protease and phosphatase inhibitor cocktail (Thermo Scientific, Rockford, IL, USA) [36]. Equal amounts of protein lysates were subjected to SDS-PAGE followed by blotting with the indicated antibodies and detection by Western Super Signal West Pico Chemiluminescent substrate reagents (Thermo Scientific, Rockford, IL, USA). Select images were quantified using ImageJ densitometry software (Version 1.45, US National Institutes of Health) and normalized to the actin signal. Data are presented as relative band signal intensity compared to control.
Quantitative real-time PCR analysis
RNA was extracted from PNT2258 treated and untreated control lymphoma cells using the miRNeasy Micro Kit (Qiagen, Valencia, CA, USA). Total RNA was quantified by NanoDrop and 1μg of each sample was reverse-transcribed using the SuperScriptW VILOTM cDNA synthesis kit according to the manufacturer’s instructions (Life Technologies, Invitrogen). The BCL-2 and RPLPO genes were used for detection of gene amplification and normalization of each sample. Reverse transcription and PCR were performed using the qRT-PCR Primer Set (Life Technologies, Invitrogen). PCR product amplification was detected by the level of fluorescence emitted by SYBR Green I master (Roche, Pleasanton, CA, USA) according to manufacturer’s protocol. Reactions were carried out in a 384 well microtiter plate using the LightCyclerW 480 System (Roche). Three independent experiments were performed in triplicate and each reaction was repeated at least once to ensure accuracy. The PCR cycle number at threshold (Ct) was used for the comparison. Gene expression at baseline and post treatment was quantified by qRT-PCR relative to RPLPO using the ΔΔCt method and expressed as fold change of gene expression relative to that in untreated control [37]. Values represent mean ± SE of three independent experiments performed in triplicate.
Growth inhibition by trypan blue
Treated and control cells were seeded at a density of 0.2x106 viable cells/ml per well in a 24-well plate (Costar, Cambridge, MA, USA) and treated with PNT2258 (at 2.5, 5, and 10 μM) for variable periods of time. The number of viable cells was determined by trypan blue exclusion (Sigma Chemical Co. St. Louis, MO, USA) at 24 hour intervals [38].
Cell cycle analysis
PNT2258-treated and control cells were collected and centrifuged twice in cold PBS. Cells were then fixed in 5 ml of 70% ethanol and stored at 4oC overnight [38, 39]. For analysis, cells were centrifuged and resuspended in 1 ml of staining buffer containing 50ug/ml propidium iodide, 100ug/ml of RNase A, and 0.1% of Triton X-100. The DNA content was then analyzed on a Coulter EPICS 753 flow cytometer and the different stages of the cell cycle were determined using a ModFit 5.2 computer program. The flow cytometry work was done at the Microscopy, Imaging and Cytometry Resources Core at Wayne State University.
Apoptosis assay
Apoptotic cell death was determined using Annexin V-FITC staining and TUNEL assay (Bio-Vision Mountain View, CA). The Annexin V-FITC studies were performed according to the manufacturer’s instructions but the protocol for ApoDIRECT (TUNEL assay) was slightly modified as previously published by our lab [38]. In brief, the fluorescein-labeled DNA was detected by flow cytometry. PI staining was simultaneously used to separate cells into G0/G1, S, G2/ M, and sub-G0 compartments based on DNA content. Since dUTP-positive cells are considered apoptotic, dual staining with dUTP and PI allowed us to assign dUTP-positive cells to a specific phase of the cell cycle [40]. The percentage of apoptotic cells was determined by flow cytometric analysis of the sub-G0/G1 DNA population in cell-cycle histograms as described in detail elsewhere [38].
Transfections and rescue experiment
The BCL-2 expression vector used in the study has been previously described [41]. The BCL-2 and control empty vectors (pcDNA3) were transfected into K562 cells at a density of 0.2×106 viable cells/ml per well in a 24-well plate. 24hrs after transfection, cells were treated with 10 μM PNT2258 for variable periods of time. The number of viable cells was determined by trypan blue exclusion (Sigma Chemical Co. St. Louis, MO, USA) at 24 hour intervals [38].
Data analysis and statistical significance
Statistical analyses were performed by one-way ANOVA using PRISM. ImageJ densitometry software (Version 1.45, US National Institutes of Health) was used for quantification of Western blot bands. Selected bands were quantified based on their relative integrated intensities, calculated as the product of the selected pixel area and the mean gray value for those pixels normalized to the internal control. Fold increase or decrease was calculated by standardizing each treatment as a ratio to the control. Statistical significance was set at p < 0.05 for all data comparisons.
ACKNOWLEDGEMENTS
The authors wish to thank ProNAi Therapeutics for providing PNT2258 and related agents used in this study. This study was funded by a grant from ProNAi Therapeutics, by the St John Hospital and Medical Center Foundation and by Michigan Corporate Relations Network’s (MCRN) Small Company Innovation Program (SCIP). The authors also wish to thank Dr. Mary Walsh for assistance in editing the manuscript and Dr. Wendi Rodrigueza of ProNAi Therapeutics for helpful discussion. The authors wish to thank Eric Van Buren and Dr. Jessica Back at the Karmanos Cancer Institute Flow Cytometry Core Facility for running the flow cytometric experiments. The Microscopy, Imaging and Cytometry Resources Core is supported, in part, by NIH Center grant P30CA022453 to The Karmanos Cancer Institute, Wayne State University, and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.REFERENCES
1. Evens AM, Winter JN, Gordon LL, Chiu BCH, Tsang R, Rosen ST. Cancer management: a multidisciplinary approach medical, surgical, & radiation oncology. Cancer Management. 2014. from http://www.cancernetwork.com/cancer-management/non-hodgkin-lymphoma.
2. SEER Cancer Statistics Review, 2012. Bethesda, MD: National Cancer Institute. http://seer.cancer.gov/statfacts/html/nhl.html.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: A Cancer Journal for Clinicians. 2015; 65: 5-29.
4. Nogai H, Dorken B, Lenz G. (2011). Pathogenesis of non-Hodgkin’s lymphoma. J Clin Oncol. 2011; 29: 1803-1811. doi: 10.1200/jco.2010.33.3252.
5. Cashen F, VanTine B. The Washington Manual of Hematology and Oncology Subspecialty Consult. 3 ed. Lippincott Williams & Wilkins. 2012.
6. Cabanillas F. Curability of advanced indolent or low-grade follicular lymphomas: time for a new paradigm? J Clin Oncol. 2013; 31: 14-16. doi: 10.1200/jco.2012.41.7527.
7. Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014; 11: 12-23. doi: 10.1038/nrclinonc.2013.197.
8. Scarfo L, Ghia P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol Lett. 2013; 155: 36-39. doi: 10.1016/j.imlet.2013.09.015.
9. Vogler M. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Advances in Medicine. 2014; 14. doi: 10.1155/2014/943648.
10. Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985; 228: 1440-1443.
11. Leich E, Salaverria I, Bea S, Zettl A, Wright G, Moreno V, Rosenwald A. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood. 2009: 114: 826-834. doi: 10.1182/blood-2009-01-198580.
12. Ross CW, Ouillette PD, Saddler CM, Shedden KA, Malek SN. Comprehensive analysis of copy number and allele status identifies multiple chromosome defects underlying follicular lymphoma pathogenesis. Clin Cancer Res. 2007; 13: 4777-4785. doi: 10.1158/1078-0432.ccr-07-0456.
13. Lin P, Medeiros LJ. High-grade B-cell lymphoma/leukemia associated with t(14;18) and 8q24/MYC rearrangement: a neoplasm of germinal center immunophenotype with poor prognosis. Haematologica. 2007; 92: 1297-1301. doi: 10.3324/haematol.11263.
14. Chang CM, Wang SS, Dave BJ, Jain S, Vasef MA, Weisenburger DD, Morton LM. Risk factors for non-Hodgkin lymphoma subtypes defined by histology and t(14;18) in a population-based case-control study. Int J Cancer. 2011; 129: 938-947. doi: 10.1002/ijc.25717.
15. Schraders M, de Jong D, Kluin P, Groenen P, van Krieken H. Lack of Bcl-2 expression in follicular lymphoma may be caused by mutations in the BCL2 gene or by absence of the t(14;18) translocation. J Pathol. 2005; 205: 329-335. doi: 10.1002/path.1689.
16. Bosga-Bouwer AG, van Imhoff GW, Boonstra R, van der Veen A, Haralambieva E, van den Berg A, Poppema S. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: t(14;18) and 3q27 are mutually exclusive. Blood. 2003; 101: 1149-1154. doi: 10.1182/blood.V101.3.1149.
17. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2014; 19: 202-208. doi: 10.1038/nm.3048.
18. Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, Wang S. Structure-Based Design of Potent Small-Molecule Inhibitors of Anti-Apoptotic Bcl-2 Proteins. Journal of Medicinal Chemistry. 2006; 49: 6139-6142. doi: 10.1021/jm060460o.
19. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Elmore SW. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008; 68: 3421-3428. doi: 10.1158/0008-5472.can-07-5836.
20. Al-Katib AM, Sun Y, Goustin AS, Azmi AS, Chen B, Aboukameel A, Mohammad RM. SMI of Bcl-2 TW-37 is active across a spectrum of B-cell tumors irrespective of their proliferative and differentiation status. J Hematol Oncol. 2009; 2: 8. doi: 10.1186/1756-8722-2-8.
21. Pro B, Leber B, Smith M, Fayad L, Romaguera J, Hagemeister F, Younes A. Phase II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in combination with rituximab in patients with recurrent B-cell non-Hodgkin lymphoma. Br J Haematol. 2008; 143: 355-360. doi: 10.1111/j.1365-2141.2008.07353.x.
22. Herbst RS, Frankel SR. Oblimersen sodium (Genasense bcl-2 antisense oligonucleotide): a rational therapeutic to enhance apoptosis in therapy of lung cancer. Clin Cancer Res. 2004; 10: 4245s-4248s. doi: 10.1158/1078-0432.ccr-040018.
23. Smith MR, Abubakr Y, Mohammad R, Xie T, Hamdan M, al-Katib A. Antisense oligodeoxyribonucleotide down-regulation of bcl-2 gene expression inhibits growth of the low-grade non-Hodgkin’s lymphoma cell line WSU-FSCCL. Cancer Gene Ther. 1995; 2: 207-212.
24. Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK. (2003). RNA interference: biology, mechanism, and applications. Microbiol Mol Biol Rev. 2003; 67: 657-685.
25. Izquierdo, M. Short interfering RNAs as a tool for cancer gene therapy. Cancer Gene Ther. 2004; 12: 217-227.
26. Mohammad R, Abubakr Y, Dan M, Aboukameel A, Chow C, Mohamed A, Al-Katib A. Bcl-2 antisense oligonucleotides are effective against systemic but not central nervous system disease in severe combined immunodeficient mice bearing human t(14;18) follicular lymphoma. Clin Cancer Res. 2002; 8: 1277-1283.
27. Sheikhnejad R. MicroDNAs and Transcriptional Regulation. 2009. Available from Nature Precedings http://hdl.handle.net/10101/npre.2009.3931.1
28. Rodrigueza WV, Woolliscroft MJ, Ebrahim AS, Forgey R, McGovren PJ, Endert G, Al-Katib A. Development and antitumor activity of a BCL-2 targeted single-stranded DNA oligonucleotide. Cancer Chemother Pharmacol. 2014; 74: 151-166. doi: 10.1007/s00280-014-2476-y.
29. Harb W, Lakhani N, Logsdon A, Steigelman M, Smith-Green H, Gaylor S, Messmann RA. The BCL2 targeted deoxyribonucleic acid inhibitor (DNAi) PNT2258 is active in patients with relapsed or refractory non-Hodgkin’s lymphoma; 2013. Paper presented at the American Society of Hematology Annual Meeting.
30. Tolcher AW, Rodrigueza WV, Rasco DW, Patnaik A, Papadopoulos KP, Amaya A, Messmann RA. A phase 1 study of the BCL2-targeted deoxyribonucleic acid inhibitor (DNAi) PNT2258 in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014; 73: 363-371. doi: 10.1007/s00280-013-2361-0.
31. Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008; 27: 6398-6406.
32. Mohammad RM, Mohamed AN, Smith MR, Jawadi NS, Al-Katib A. A unique EBV-negative low-grade lymphoma line (WSU-FSCCL) exhibiting both t(14;18) and t(8;11). Cancer Genet Cytogenet. 1993; 70: 62-67.
33. Al-Katib AM, Smith M, Kamanda WS, Pettit GR, Hamdan M, Mohamed AN, Mohammad RM. Bryostatin 1 down-regulates mdr1 and potentiates vincristine cytotoxicity in diffuse large cell lymphoma xenografts. Clin Cancer Res. 1998; 4: 1305-1314.
34. Al-Katib A, Mohammad R, Hamdan M, Mohamed AN, Dan M, Smith MR. Propagation of Waldenstrom’s macroglobulinemia cells in vitro and in severe combined immune deficient mice: utility as a preclinical drug screening model. Blood. 1993; 81: 3034-3042.
35. Al-Katib AM, Aboukameel A, Mohammad R, Bissery MC, Zuany-Amorim C. Superior antitumor activity of SAR3419 to rituximab in xenograft models for non-Hodgkin’s lymphoma. Clin Cancer Res. 2009; 15: 4038-4045. doi: 10.1158/1078-0432.ccr-08-2808.
36. Sosin AM, Burger AM, Siddiqi A, Abrams J, Mohammad RM, Al-Katib AM. HDM2 antagonist MI-219 (spiro-oxindole), but not Nutlin-3 (cis-imidazoline), regulates p53 through enhanced HDM2 autoubiquitination and degradation in human malignant B-cell lymphomas. J Hematol Oncol. 2012; 5: 57. doi: 10.1186/1756-8722-5-57.
37. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001; 25: 402-408. doi: http://dx.doi.org/10.1006/meth.2001.1262.
38. Al-Katib A, Arnold AA, Aboukameel A, Sosin A, Smith P, Mohamed AN, Mohammad RM. I-kappa-kinase-2 (IKK-2) inhibition potentiates vincristine cytotoxicity in non-Hodgkin’s lymphoma. Mol Cancer. 2010; 9: 228. doi: 10.1186/1476-4598-9-228.
39. Mohammad RM, Wu J, Azmi AS, Aboukameel A, Sosin A, Wu S, Al-Katib AM. An MDM2 antagonist (MI-319) restores p53 functions and increases the life span of orally treated follicular lymphoma bearing animals. Mol Cancer. 2009; 8: 115. doi: 10.1186/1476-4598-8-115.
40. Darzynkiewicz Z, Galkowski D, Zhao H. Analysis of apoptosis by cytometry using TUNEL assay. Methods. 2008; 44: 250-254. doi: http://dx.doi.org/10.1016/j.ymeth.2007.11.008.
41. Kandouz M, Haidara K, Zhao J, Brisson ML, Batist G. The EphB2 tumor suppressor induces autophagic cell death via concomitant activation of the ERK1/2 and PI3K pathways. Cell Cycle. 2010; 9: 398-407.
42. Mohammad RM, Wang S, Aboukameel A, Chen B, Wu X, Chen J, Al-Katib A. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) [(-)-gossypol] against diffuse large cell lymphoma. Mol Cancer Ther. 2005; 4: 13-21.
43. Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, Al-Katib A. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007; 13: 2226-2235. doi: 10.1158/1078-0432.ccr-06-1574.
44. Arnold AA, Aboukameel A, Chen J, Yang D, Wang S, Al-Katib A, Mohammad RM. Preclinical studies of Apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in Follicular Small Cleaved Cell Lymphoma model. Mol Cancer. 2008; 7: 20. doi: 10.1186/1476-4598-7-20.
45. Sun Y, Wu J, Aboukameel A, Banerjee S, Arnold AA, Chen J, Mohammad RM. Apogossypolone, a nonpeptidic small molecule inhibitor targeting Bcl-2 family proteins, effectively inhibits growth of diffuse large cell lymphoma cells in vitro and in vivo. Cancer Biol Ther. 2008; 7: 1418-1426.
46. Liu Q, Wang HG. Anti-cancer drug discovery and development: Bcl-2 family small molecule inhibitors. Communicative & integrative biology. 2012; 1;5:557-65.
47. Davids MS, Letai A. ABT-199: a new hope for selective BCL-2 inhibition. Cancer cell. 2013;23:139.
48. Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell death & disease. 2015; 6:e1593.
49. Pandey MK, Prasad S, Tyagi AK, Deb L, Huang J, Karelia DN, Amin SG, Aggarwal BB. Targeting Cell Survival Proteins for Cancer Cell Death. Pharmaceuticals. 2016;9:11.
50. Duan H, Heckman CA, Boxer LM. (2005). Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol. 2005; 25: 1608-1619. doi: 10.1128/mcb.25.5.1608-1619.2005.
51. Ebrahim AS, Sabbagh H, Liddane A, Raufi A, Kandouz M, Al-Katib A. Hematologic malignancies: newer strategies to counter the BCL-2 protein. Journal of Cancer Research and Clinical Oncology. 2016. 1-10.
52. Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M, Pinton P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Molecular biology of the cell. 2016. 27: 20-34.