
INTRODUCTION
Among the four subtypes of breast cancer 15-20% of breast cancers are HER2+, which is associated with aggressive clinical course [1]. HER2+ breast cancers typically respond well to trastuzumab treatment in early stage diseases [2]. However, in metastatic HER2+ breast cancers the majority of patients either demonstrate de novo or acquired trastuzumab resistance after one to two year of treatment [3–5]. Numerous studies have investigated molecular mechanisms associated with trastuzumab resistance including HER2 degradation, overexpression of other tyrosine kinase receptors, and reduced expression of the PTEN tumor suppressor [6, 7]. Inactivation of PTEN has been shown to occur within 40% of HER2+ breast cancer patients and has been correlated with poor prognosis, as well as, adaption of mesenchymal characteristics in vitro[8, 9].
Korkaya et al. have previously demonstrated that trastuzumab treatment in HER2+ PTEN deficient cancer cells expands the breast cancer stem cell (BCSC) population [10]. The unique properties of self-renewal and differentiation of the BCSC population is suspected to be responsible for drug resistance [11–13]. Our recent study showed that continued use of trastuzumab in PTEN-deficient HER2+ breast cancer induces the epithelial-mesenchymal transition (EMT) and transform HER2+ to a triple negative like breast cancer, which requires unique treatment options [14]. Liu et.al. proposed that two states of BCSCs, mesenchymal-like BCSCs (CD44+CD24−) and epithelial-like BCSCs (ALDH+), may exist in equilibrium and can transition between states. mesenchymal-like BCSCs were reported to be primarily quiescent and highly invasive, whereas epithelial-like BCSCs are proliferative, and are localized centrally within hypoxic zones [15]. Conceptually, BCSCs plasticity could alter more differentiated cell morphology (epithelial vs mesenchymal), classical subtype makers, and result in distinct capacities for invasion, metastasis, and drug resistance due to the distinct epigenetic state from which those cells are derived. Identification of dramatic molecular changes following drug induced EMT in lung cancer has proven useful to identify potential new therapies following erlotinib resistance [16, 17]. However, the molecular signatures that are associated with the transition between cell states in PTEN-deficient trastuzumab resistant breast cancer has yet to be completely elucidated.
The purpose of this study is to identify the changes in BCSCs states and reveal novel cancer stem cell targets following the EMT in PTEN-deficient trastuzumab-resistant breast cancer. Our previous study demonstrated that parental HER2+ BT474 and the PTEN-deficient trastuzumab-resistant derivative (BT474-PTEN-LTT) exhibit epithelial and mesenchymal morphology respectively. Here we identify using traditional BCSC markers that while BT474 exhibits no CD44+/CD24- cells and high Aldefluor positive cell percentages the opposite is true following the generation of trastuzumab resistance. RNA-sequencing was employed for global gene expression analysis and to reveal novel targets which could be exploited for therapy following EMT and transition of CSC states. These results independently confirmed the bulk transition to a mesenchymal/basal like phenotype, and alteration in traditional BCSC marker expression.
Interestingly, differential response to the small molecule sulforaphane (SF) was observed in parental BT474 and BT474-PTEN-LTT. SF, a natural compound derived from cruciferous vegetables, has proven effective at abrogating CSCs in a host of cancers. Early evidence suggested it is capable of preventing tumor formation in chemically induced models of carcinogenesis [18]. Further, in breast cancer SF is able to decrease the Aldefluor-positive cell population, suppress mammosphere formation, and prevent secondary tumor formation in vivo [19]. Additional data suggests it can eliminate CSCs and enhance traditional chemotherapeutic efficacy in prostate and pancreatic cancer cell lines [20–22]. Together, these studies support the notion that SF may possess broad therapeutic potential against CSCs, which provides a unique secondary filter to identify potential gene candidates that regulate the mesenchymal state.
Functional gene set enrichment analysis and siRNA knockdown of several candidate genes revealed a set of homeobox transcription factors (specifically MEOX1) as novel potential targets in the PTEN-deficient trastuzumab-resistant breast cancer cells. In tumor biopsies MEOX1 is associated with poor patient survival, lymph node metastasis, and higher cancer stages. Reduced level of MEOX1 by siRNA or small molecule inhibitor could decrease mammosphere and colony formation in vitro, and decreased tumor growth and BCSC frequency in vivo. These findings suggest that unique molecular signatures may regulate mesenchymal and epithelial-like cell states in PTEN-deficient trastuzumab-resistant breast cancer, where MEOX1 is a clinically relevant target to regulating both BCSCs and mesenchymal-like cell proliferation.
RESULTS
Transition from epithelial to mesenchymal-like BCSCs and bulk characteristics of parental BT474 and PTEN-deficient trastuzumab-resistant BT474
Our previous studies have shown that continued use of trastuzumab in PTEN-deficient breast cancer cells induced the EMT in bulk cell lines, increased BCSC characteristics, and transformed HER2+ to a triple negative phenotype [10, 14]. However, recent evidence suggests BCSCs may exist in two distinct states, mesenchymal-like (CD44+/CD24-) and epithelial-like (ALDH+), which can interconvert in breast cancer [15]. Therefore, we sought to simultaneously characterize both populations of BCSCs using flow cytometry (Table 1). The HER2 amplified breast cancer cell line BT474 (HER2+ trastuzumab-sensitive) has a distinct population of epithelial-like BCSCs with undetectable mesenchymal-like BCSCs. However, when BT474 cells were genetically manipulated by PTEN inactivation through shRNA knockdown and cultured under long term treatment (LTT) of trastuzumab, the resulted PTEN-deficient trastuzumab-resistant cells (BT474-PTEN-LTT) express primarily mesenchymal-like BCSCs markers and low epithelial-like BCSCs characteristics (Figure 1A).
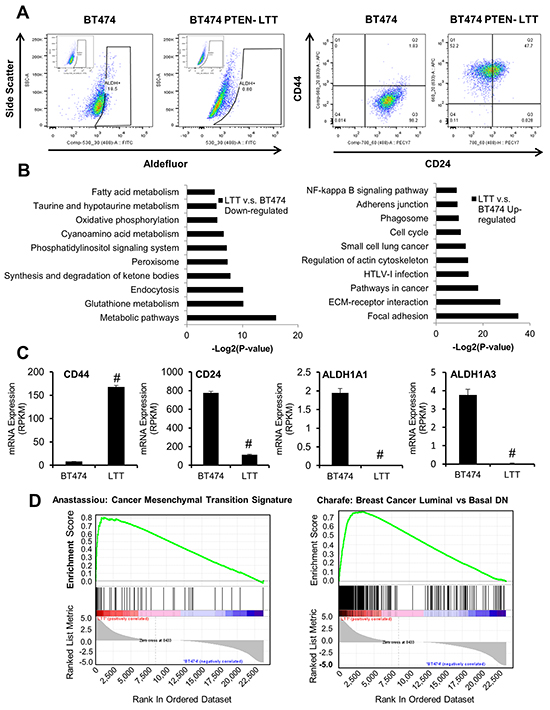
Figure 1: PTEN-deficient trastuzumab-resistant and parental BT474 breast cancer cells exhibit unique BCSC populations and distinct gene expression signatures. A. Representative flow cytometry analysis of aldefluor positive cells (left) and CD44/CD24 staining (right) in BT474 and BT474 PTEN- LTT cells. Inset, DEAB negative control. B. The top ranking gene ontology attributes down-regulated (left) or upregulated (right) following the generation of trastuzumab resistance in BT474 with associated –log2(P-values). C. mRNA expression of CD44, CD24, ALDH1A1, and ALDH1A3 in BT474 and BT474 PTEN- LTT cells expressed as reads/kilobase/million mapped reads as determined by RNA sequencing. N=4. Data shown as average ± SD. # p ≤ 0.01. D. Gene set enrichment analysis identifying enrichment of upregulated genes in BT474 PTEN- LTT cells (left) and those downregulated (right) pertaining to EMT and molecular subtype.
Table 1: Mesenchymal- and epithelial-like BCSCs in BT474 and PTEN-deficient trastuzumab-resistant BT474-PTEN
Surface markers |
PTEN status |
Trastuzumab sensitivity |
Sulforaphane sensitivity |
Mesenchymal-like BCSCs (CD44+CD24-) |
Epithelial-like BCSCs (ALDH+) |
|
|---|---|---|---|---|---|---|
BT474 |
HER2+/ER low /PR+ |
Wild-type |
Sensitive |
Resistant |
0.0-0.3% |
29.0-43.0% |
BT474 PTEN- LTT |
HER2- /ER-/PR- |
Low |
Resistant |
Sensitive |
30.7-39.0% |
0.7-2.0% |
In order to reveal the potential genes involved in the mesenchymal and epithelial phenotypes, we used an RNA-seq approach to compare the transcriptomes of BT474 and BT474 PTEN- LTT cells. While the majority of mRNA expression was consistent between the cell lines (19,811 genes, 83.5%), based on the RVM (Random variance model) algorithm (p-value<0.05, FDR<0.05) and fold change (fold change ≥2 or ≤0.5), we identified 3901 alterations in gene transcription including 2023 up-regulated and 1878 down-regulated genes. Gene Ontology (GO) pathway enrichment analysis was used to categorize the significant genes and related pathways. The results revealed up-regulated genes were significantly enriched for 3 pathways including focal adhesion, ECM-receptor interaction and pathway in cancer progression, whereas down-regulated genes were enriched for primarily metabolic pathways (Figure 1B). In agreement with two types of mesenchymal- and epithelial-like BCSCs status, the levels of ALDH and CD24 were reduced while CD44 was up-regulated in BT474 PTEN- LTT cells compared to parental BT474 cells (Figure 1C).
In order to further explore the global molecular changes in PTEN-deficient and trastuzumab resistant breast cancer cells, gene set enrichment analysis (GSEA) was performed on the differentially expressed genes (Figure 1D). GSEA reveals that the PTEN-deficient and trastuzumab resistant BT474 PTEN- LTT exhibit similar gene expression to cells which have transitioned to a mesenchymal cell type (ES=0.81, p-value<0.01). Further, expression of epithelial markers E-cadherin and EpCAM are specifically within parental BT474 whereas mesenchymal markers N-cadherin and vimentin expression is distinctly within the trastuzumab resistant derivative (Supplementary Figure S1). Interestingly, the genes which were down regulated in BT474-PTEN- LTT were enriched in a data set encompassing genes which are similar in basal breast cancer cells when compared to luminal (NES=0.76, p-value<0.01). These data were in agreement with our recent finding that trastuzumab resistance in PTEN-deficient breast cancer cells induced the conversion to a triple negative phenotype. Taken together, these results suggest that trastuzumab-resistance in PTEN-deficient breast cancer (BT474) induced a transition converting epithelial- to mesenchymal-like BCSCs ultimately converting the HER2+ cell line to basal/triple negative phenotype.
Distinct response of PTEN-deficient trastuzumab resistant BT474 PTEN- LTT to sulforaphane narrows down gene candidates following EMT of breast cancer cells
Since expression of a great number of genes were altered in BT474 PTEN- LTT and BT474, it is difficult to narrow down novel molecular targets following the EMT. Fortunately, our data showed that trastuzumab resistant BT474 PTEN- LTT responded selectively to sulforaphane (SF) in comparison to parental BT474. Using the MTS cell proliferation assay BT474 PTEN- LTT exhibited high sensitivity to SF (IC50 = 11.5 µM) relative to parental BT474 cells which exhibited no significant response up to 25 µM (Figure 2A). By coupling the treatment of SF with analysis of differentially expressed genes in BT474 and BT474 PTEN- LTT cells, it is feasible to narrow down the molecular targets. Therefore, both parental BT474 and BT474 PTEN- LTT cells were treated with increasing concentrations of SF (2 and 10 μM) to identify dose-dependent changes, and time-dependent gene expression changes at 8 and 24 hours.
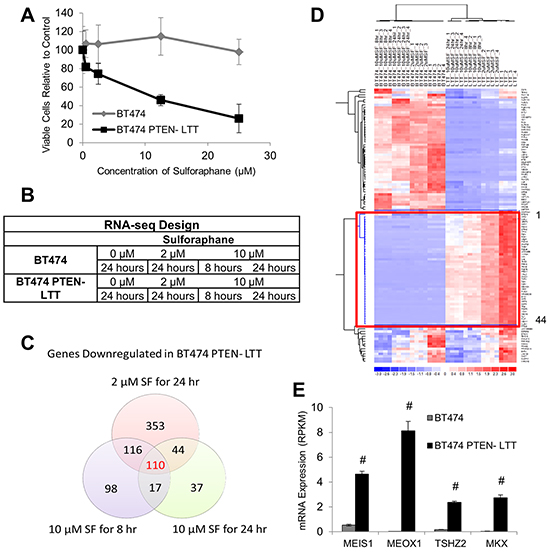
Figure 2: Sulforaphane elicits unique efficacy and gene expression changes in trastuzumab-resistant and parental BT474 cell lines. A. Cell viability of BT474 and BT474 PTEN- LTT cells following 72 hour treatment with SF as determined by the MTS assay. N=6. B. RNA sequencing experimental design for BT4747 and BT474-PTEN- LTT in response to SF to identify time-dependent (10 μM SF 8 hours v.s. 10 μM SF 24 hours) and dose-dependent (2 μM SF 24 hours v.s. 10 μM SF for 24 hours) gene expression changes. N=4. C. Venn diagram illustrating number of genes reduced 2-fold with P-value ≤ 0.05 in BT474 PTEN- LTT cells following each treatment. D. Heat map illustrating expression changes in BT474 and BT474 PTEN- LTT cells of the 110 genes which are inhibited in a dose and time dependent manner by 2-fold after SF treatment. Red box highlights the 44 genes which were also upregulated in the BT474 PTEN- LTT cell line. E. mRNA expression level of the 4 homeobox transcription factors which were functionally enriched from the 44 gene set. N=4. Data shown as average ± SD. # p ≤ 0.01.
Both BT474 and BT474-PTEN- LTT cells were treated with increasing SF concentration and duration, and RNA-seq of total isolated mRNA was performed (Figure 2B). A Venn diagram illustrating the number of genes down regulated by >2-fold in the BT474-PTEN- LTT cells under different treatment conditions reveals that only 110 gene candidates exhibit both time and dose dependent inhibition by SF (Figure 2C). Of these 110 genes only 44 are upregulated by >2-fold in BT474 PTEN- LTT cells relative to the parental BT474 cell line (Figure 2D).
Three methods were used to narrow down fewer candidate genes for further study. (1) Real-time PCR was used to confirm the gene expression differences between BT474 and BT474-PTEN-LTT identified by RNA-seq under identical treatment conditions with SF; (2) siRNA knockdown of the genes with known biological function to assess potential effects on cell proliferation and BCSC characteristics; (3) Functional GSEA using the bioinformatics toolkit DAVID using all identified 44 genes. From these three sets of experiments we selected one functional group, homeobox transcription factors, for additional focus within all candidates (Figure 2E). Among these genes MEOX1 displays the highest mRNA expression in BT474 PTEN- LTT cells and its expression was nearly undetectable in BT474 cells (2989-fold upregulated). Confirmation of RNA-seq results for MEOX1 was performed by real time PCR and similarly demonstrated a 2.5-fold reduction of expression by SF at 24 hours. Therefore, MEOX1 was identified as the top candidate gene for further analysis. A summary of results from functional studies for the top 5 gene candidates can be found in Supplementary Table S1.
Nuclear MEOX1 expression correlates with patient survival, lymph node metastasis and stage of breast cancers
In order to investigate the potential clinical implications of MEOX1 in breast cancer patients, we performed immunohistological staining of MEOX1 in a breast cancer tissue microarray (TMA) containing tumor samples and associated survival data (N=150). In this TMA 103 patients survived and 45 patients were deceased, with a median follow-up period of 123.5 months (range, 2-160 months). MEOX1 cytoplasmic staining was positive in 105 primary tumors (Figure 3A, top), whereas nuclear staining was positive in 72 tumor samples (Figure 3A, bottom). Interestingly, the vast majority of the tumor stroma did not show any MEOX1 staining.
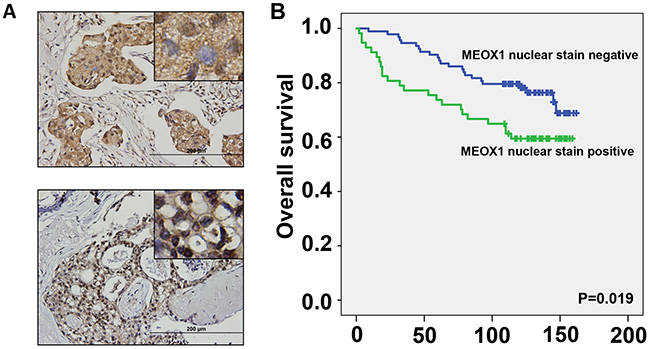
Figure 3: MEOX1 staining in breast cancer tissues correlates with poor patient survival. A. Representative immunohistochemical (IHC) staining of MEOX1 in patient tumor tissue, top: positive cytoplasmic staining with variable nuclear staining, bottom: strong nuclear staining. Image insets originally obtained using a 40X objective. B. Kaplan-Meier survival analysis of the MEOX1 stained TMA when patients were stratified by the presence of nuclear MEOX1 staining. N=150.
Statistical analysis indicated that positive MEOX1 nuclear staining was associated with poor survival of breast cancer patients compared to patients with negative nuclear staining (P =0.019) (Figure 3B). In addition, MEOX1 nuclear staining was significantly correlated with the presence of Lymph node metastasis and cancer stage (P<0.05, Table 2). Further, a Cox regression model was employed to perform multivariate statistical analysis. The analyses revealed that Nuclear MEOX1 expression level (p=0.042), with a Hazard Ratio (HR) of 2.19 and a 95% CI of 1.029-4.669, was an independent prognostic factor in breast cancer patients. No significant correlations were present between cytoplasmic MEOX1 staining and the clinical characteristics noted above.
Table 2: Association between MEOX-1 nuclear staining and pathological characteristics of the 150 breast cancer patients
Variable |
MEOX-1 staining score |
p-value |
|
|---|---|---|---|
0 |
1 |
||
Age |
53.3±13.3 |
53.7±13.3 |
0.845 |
Depth of invasion |
0.467 |
||
T1+T2 |
69 |
61 |
|
T3 |
6 |
8 |
|
Lymph node metastasis |
0.011 |
||
N0 |
35 |
18 |
|
N1+N2+N3 |
40 |
51 |
|
Grade |
0.275 |
||
I or I-II |
22 |
15 |
|
II, II-III or III |
54 |
56 |
|
Stage |
0.029 |
||
1 |
9 |
1 |
|
2 |
45 |
41 |
|
3 |
21 |
27 |
|
MEOX1 silencing suppresses the self-renewal of BCSC and mesenchymal-cell proliferation in vitro
To elucidate the function of MEOX1 in the mesenchymal BT474 PTEN- LTT cell line siRNA knockdown was employed, reducing expression by 88.3% (Figure 4A). The effect of MEOX1 knockdown on in vitro tumorigenicity was then evaluated using the colony formation assay in soft agar. Colony formation rates following MEOX siRNA treatment were reduced 8.9-fold relative to nontargeted siRNA control in BT474 PTEN- LTT cells over 14 days (Figure 4B). The role of MEOX1 in BCSC self-renewal was also determined using the mammosphere formation assay. Over the course of 7 days, MEOX siRNA treatment led to a 60.5% reduction in the number of mammospheres formed and the size of the average sphere was reduced by 90% (Figure 4C).
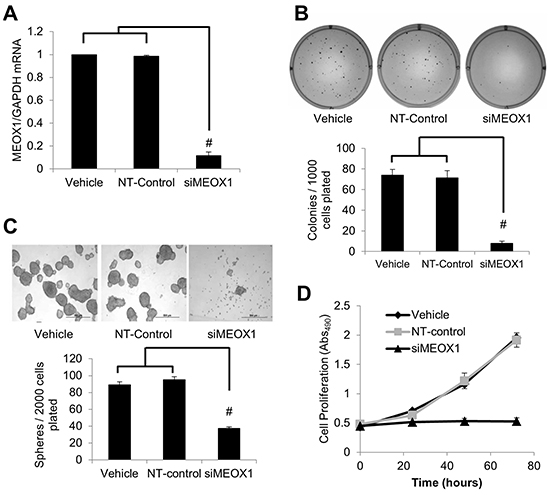
Figure 4: MEOX1 functionally regulates mesenchymal bulk cell proliferation and breast cancer stem cell characteristics in vitro. A. Real time PCR analysis of MEOX1 mRNA expression relative to GAPDH in transfection reagent (Vehicle), non-targeted siRNA control (NT-control), or siRNA for MEOX1 (siMEOX1) treated BT474 PTEN- LTT cells. B. Top, representative images of colonies formed 14 days after siRNA knockdown of MEOX1 in BT474-PTEN-LTT cells in comparison with vehicle and NT-control treatment. Bottom, quantification of number of colonies formed after culture in soft agar. C. Top, representative images of mammospheres formed after 7 days following siRNA knockdown of MEOX1 in BT474 PTEN- LTT cells in comparison with vehicle and NT-control. Bottom, quantification of number of mammospheres formed after 7 days of culture in serum free non-adherent conditions. D. Proliferation of cells following siRNA knockdown of MEOX1 over the course of 72 hours as determined by MTS proliferation in BT474 PTEN- LTT cells. N=3 in all experiments. Data shown as average ± SD. # p ≤ 0.01.
Given the importance of proliferation and invasion in cancer progression, we tested whether inhibiting the expression of MEOX1 in BT474 PTEN- LTT cells could affect cell growth by MTS assay and invasion into matrigel. Strikingly, down-regulation of MEOX1 in BT474 PTEN- LTT cells completely inhibited the cell proliferation (Figure 4D). Further, the matrigel invasion assay reveals a 63% reduction in cell invasion following siRNA knockdown (Supplementary Figure S2). Together, these results indicated that MEOX1 might play key roles to regulate BCSCs and mesenchymal cancer cell proliferation in the BT474 PTEN- LTT cells.
MEOX1 protein expression can be reduced in vitro and in vivo by sulforaphane, which is associated with a decreased frequency of BCSCs and reduction in tumor growth
MEOX1 mRNA is highly expressed in PTEN-deficient trastuzumab resistant BT474 PTEN- LTT, which can be inhibited by SF treatment in vitro. Next, we sought to determine if MEOX1 protein can also be downregulated by SF. MEOX1 protein resides primarily within the nucleus in BT474 PTEN- LTT cells in vitro, as evident by overlap between MEOX1 (Green) and DAPI (Blue) staining (Figure 5A). Treatment with SF (10 μM for 24 hours) led to a reduction in both overall intensity and number of MEOX1 nuclear foci, suggesting SF also reduces MEOX1 protein expression. In order to validate if SF was capable of reducing MEOX1 expression in vivo, immunohistochemistry (IHC) was carried after daily SF treatment (50 mg/kg) in mice bearing orthotopic mouse xenografts of parental BT474 and BT474 PTEN- LTT cell lines. The results demonstrates that SF reduced MEOX1 expression at the protein level in BT474 PTEN- LTT xenografts (Figure 5B, bottom), whereas parental BT474 tumors express no MEOX1 protein regardless of SF treatment (Figure 5B, top).
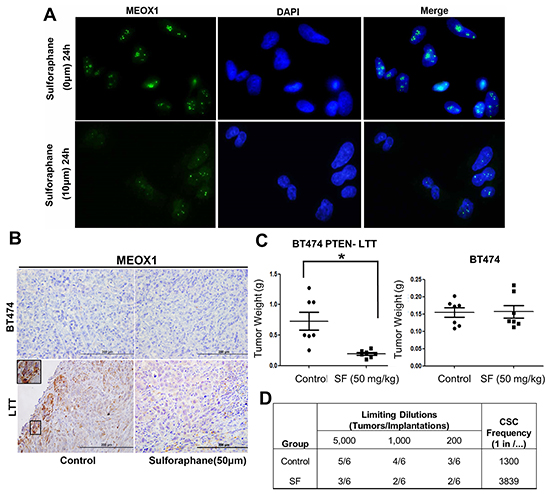
Figure 5: Down-regulation of MEOX1 by sulforaphane in vitro and in vivo is associated with reduced frequency of BCSCs and inhibition of tumor growth in vivo. A. Representative immunofluorescent staining of MEOX1 in the presence or absence of SF treatment for 24 hrs. MEOX1 (green) resides primarily within the nucleus as evident by overlap with DAPI (Blue) staining. Images obtained originally with 40X objective. B. Representative IHC staining of MEOX1 from control or SF treated primary xenografts (BT474 top, BT474 PTEN- LTT bottom) counter stained with hematoxylin and eosin. MEOX1 protein was down-regulated in BT474-LTT-PTEN-LTT by sulforaphane, while BT474 tumor did not express MEOX1. C. Left, final tumor mass of orthotopic mouse xenografts (BT474-PTEN-LTT) from mice treated with daily I.P. administration of 0.9% saline or 50 mg/kg SF. Right, final tumor mass of orthotopic mouse xenografts (BT474) treated with SF with the same dose regimen. Data shown as average ± SD. * p ≤ 0.05. D. Extreme limiting dilution analysis (ELDA) in secondary mice 8 weeks following implantation of residual cancer cells from primary BT474 PTEN- LTT tumors. Residual cells from SF treated mice exhibit a significantly reduced frequency of tumor initiating cells (P = 0.034).
As demonstrated previously, siRNA knockdown of MEOX1 in BT474 PTEN- LTT cells in vitro resulted in the inhibition of self-renewal of BCSC and bulk cell proliferation. In order to evaluate if the downregulation of MEOX1 was consistent with these observations in vivo, frequency of BCSC and tumor growth in vivo were measured after treatment with sulforaphane (50 mg/kg, I.P). Along with the accompanied MEOX1 downregulation, SF significantly reduced tumor volume by 54% (Figure 5C, left, p-value=0.028) in the BT474-PTEN-LTT xenograft model whereas no inhibitory effect was observed in BT474 xenografts. Further, extreme limiting dilution analysis (ELDA), performed with residual cancer cells from primary mice implanted into secondary recipients, showed that treatment with SF reduced the frequency of BCSCs within these tumors from 1 in 1300 cells to 1 in 3839 cells (Figure 5D, p-value=0.034). Taken together these results provide a proof of concept that inhibition of MEOX1 using a small molecule is a feasible approach to inhibit BCSCs and bulk tumor growth in vivo.
DISCUSSION
Identification of different breast cancer subtypes has led to significant advances in targeted therapy with unique molecular targets responsible for dramatically different efficacy across subtypes [23, 24]. For HER2+ breast cancers several targeted therapies are currently in use with the front line therapy being trastuzumab. While this antibody has proven extremely useful for early stage HER2+ breast cancer patients, the majority of late stage (metastatic) patients demonstrate de novo resistance or will develop acquired resistance within 1 to 2 years of trastuzumab treatment [3–5]. Several mechanisms have been associated with the generation of trastuzumab resistance including antigen masking, activation of non-canonical HER2 binding partners, or activation of downstream signaling nodes which bypass the requirement for HER2 [25–28]. While second line therapies have been developed for trastuzumab resistant patients, those with distant metastasis will likely succumb to this disease.
Our most recent study has shown that continued use of trastuzumab in PTEN-deficient breast cancer cells induced the EMT, expands breast cancer stem cells (BCSCs)[10], and transform HER2+ to a trastuzumab-resistant triple negative phenotype [14]. These transformed cancer cells show distinct sensitivity to the small molecule sulforaphane, which suggests that different treatment options need to be developed for PTEN-deficient trastuzumab-resistant breast cancer. However, it is still unclear what molecular targets can be exploited for therapy following the observed EMT and apparent subtype switch.
The cancer stem cell (CSC) hypothesis suggests that many types of cancer are sustained by a small population of CSCs, which seem to be responsible for the origin of cancers, tumor recurrence, and drug resistance [13]. In breast cancer, CD44+/CD24- cells or ALDH1+ cells have both been reported to retain CSC characteristics [13, 29]. Recent evidence building on these studies indicates that BCSCs exist in two distinct and dynamic states which may interconvert to form a steady state equilibrium: epithelial-like BCSCs (ALDH+) and mesenchymal-like BCSCs (CD44+CD24-). Mesenchymal-like BCSCs are reported to possess mesenchymal cell type characteristics and are located at the tumor’s invasive front. Epithelial-like BCSCs are more proliferative and are located more centrally within tumors [15]. It is possible that the cellular plasticity displayed by BCSCs, coupled with the asymmetric division of either state into more differentiated cells, may ultimately determine the epithelial or mesenchymal characteristics of the bulk cell line however additional studies are to determine this are required.
Accumulating evidence suggests that the induction of EMT and expansion of CSCs may be critical when cancer cells become resistance to trastuzumab. For instance, JIMT-1 cells that exhibit de novo trastuzumab resistance, express relatively high levels of EMT markers SLUG and SNAIL, and are primarily CD44+/CD24-. Conversely, trastuzumab sensitive SKBR3 cells are primarily CD24+ and lack expression of EMT markers [30]. In another report associated with acquired resistance, 3 month culture of SKBR3 cells with trastuzumab generated drug resistance and resulted in expression of EMT inducer TGF-β and downstream target ZEB1 [31]. Further, Lesniak et al. demonstrated rare colonies within the SKBR3 cell line had spontaneously undergone EMT to generated drug resistance and the cells derived from these colonies were primarily CD44+/CD24- with lower HER2 expression [32]. Numerous studies also have indicated that inactivation of PTEN may play important roles in the EMT and trastuzumab resistance. In line with these studies, our work and that by Korkaya et. al. have established that induction of trastuzumab resistance by long term trastuzumab culture in PTEN deficient cells rapidly induces EMT and expands BCSCs [10]. While initially attributed to the activation of the IL-6/STAT3/NF-kB positive feedback loop, it is possible that additional signaling pathways may be critical to proliferation of these cells as well as the self-renewal of the BCSC population.
To further characterize molecular pathways which may play a critical role in the mesenchymal and epithelial states, we investigated the transcriptional landscape using RNA-Seq in cancer cells with primarily epithelial-like BCSCs (BT474, HER2+, trastuzumab sensitive) or mesenchymal-like BCSCs in PTEN-deficient trastuzumab resistant cells (BT474-PTEN-LTT). These experiments identified 3901 differentially-expressed genes between BT4747 and BT474-PTEN-LTT. Utilizing bioinformatics analysis differentially expressed genes were categorized into distinct gene ontology classifications. From them, most up-regulated genes were significantly enriched for 3 pathways including focal adhesion, ECM-receptor interaction and pathway in cancer progression. These pathways in cancer progression included cytokine/NF-kB, HIF-1α, and WNT signaling, all of which are known to play a role in regulating BCSCs and have been associated with EMT [19, 33–35].
In a recent work by Martin-Castillo et.al. the authors propose that the intrinsic molecular subtypes of a breast cancer, coupled with the distribution of epithelial and mesenchymal-like CSC states, can be used to predict the response to trastuzumab in the wide spectrum of clinically diagnosed HER2+ malignancies. Within this framework it is suggested that adaption of mesenchymal traits (EMT) and presence of CD44+/24- cells would coincide with the generation of acquired trastuzumab resistance [36]. Our unbiased GSEA demonstrates the trastuzumab resistant BT474 PTEN- LTT cells appear to have undergone the EMT and adopted a basal like breast cancer phenotype. Furthermore, the RNA-seq data confirmed ALDH1 and CD24 were reduced, and CD44 was up-regulated in BT474-PTEN-LTT cells compared to BT474. These results indicate that continued use of trastuzumab in PTEN-deficient breast cancer cells leads to a BCSC transition from an epithelial- to mesenchymal-like state and support the model proposed by Martin-Castillo et. al.
Due to the number of up regulated genes following the induction of trastuzumab resistance, a knowledge based approach to determining druggable targets is difficult. However, the differential sensitivity of BT474 PTEN- LTT and parental BT474 cells to SF has provided another filter to narrow down the potential target genes. We and other groups have previously shown that SF is able to preferentially inhibit the CSC population in cancer cell lines and suppress secondary tumor formation in vivo [19–21, 37]. The efficacy of SF toward inhibiting CSCs and ability to elicit distinct effects in BT474 and BT474 PTEN- LTT cells make it a valuable small molecule to interrogate unique molecular targets in CSCs.
Since only PTEN-deficient trastuzumab resistant BT474 PTEN- cells are sensitive to SF, we chose to focus on genes that were decreased in both a dose and time dependent manner by SF. This filtering scheme allowed us to narrow our search to only 44 genes. Experimental confirmation (PCR and siRNA knockdown) and functional classification analysis identified four homeobox transcription factors as potential targets. Among them MEOX1 displays the highest expression and fold-change difference between parental BT4747 and BT474-PTEN- LTT cell lines and was therefore selected as the top candidate gene for further study. It is worth noting that many other genes may also be involved in EMT and MET, which require further investigation; but MEOX1 was one of the most specifically upregulated genes within mesenchymal cancer cells in this study.
The MEOX1 (previously MOX1) homeobox transcriptional factor represents a critical mediator of normal somite formation in a developing embryo, a process which requires both the EMT and MET process [38–41]. In patients, homozygous truncation mutations in the MEOX1 gene cause an autosomal-recessive form of Klippel-Feil Syndrome, a disease characterized by fusion of cervical vertebrae [42, 43]. Recent evidence in zebra fish demonstrates MEOX1+ cells also regulate normal hematopoietic stem cell formation in a cytokine dependent manner [44]. Further, MEOX1 has been shown to mediate Hedgehog signaling by regulating Gli1/2 expression during cardiomyogenesis [45].
While the role of MEOX-1 in cancer has largely been unexplored, one study by Thiaville et. al. suggests that this transcription factor partially mediates PBX1 signaling in ovarian cancers [46]. PBX1 has been shown to be a downstream target of NOTCH signaling in breast cancer, and NOTCH itself is a known regulator of CSCs [47, 48]. Initial evidence from our laboratory indicates knockdown of PTEN via shRNA is capable of increasing, while HER2 overexpression suppresses, MEOX1 expression (MCF7 and SUM159, data not shown). Together, this data suggests that there may be direct regulation of MEOX1 by the HER2 signaling cascade beyond the context of trastuzumab resistant HER2+ breast cancers.
In order to study the potential clinical application of MEOX1, TMA staining of a diverse range of breast tumor samples was carried out. Interestingly, cytoplasmic expression of MEOX1 was present in the majority of tumors but limited particularly to cancer cells. Within the cancer cells nuclear protein localization of MEOX1 was correlated with poorer overall survival, an advanced tumor stage, and the presence of lymph node metastasis.
To elucidate the function of MEOX1, its expression was reduced by siRNA knockdown in the PTEN-deficient trastuzumab resistant cell line, which resulted in a significant reduction of BCSC self-renewal and proliferation of bulk BT474 PTEN- LTT cells. Further, knockdown of MEOX1 by siRNA was capable of reducing proliferation of the basal like SUM149PT cell line by 45% (data not shown). Downregulation of MEOX1 by SF in the BT474 PTEN- LTT cell line in vitro and in an orthotopic mouse xenograft model in vivo was able to effectively inhibit bulk tumor volume and reduce frequency of BCSCs, as evident by ELDA in secondary mice. These results demonstrate the feasibility of regulating MEOX1 using small molecule inhibition in vivo. However, additional studies may be necessary to identify the upstream and downstream targets of MEOX1 and its role during the epithelial and mesenchymal transitions.
In summary, these data showed that continued use of trastuzumab in PTEN-deficient breast cancer induces a transition between mesenchymal- and epithelial-like BCSC states and transforms luminal HER2+ cells to a basal like phenotype. Using these cell lines we identify novel cancer stem cell targets in PTEN-deficient trastuzumab-resistant breast cancers. MEOX1 was identified as a clinically relevant molecular target regulating both BCSCs and mesenchymal bulk cell line proliferation. These results may provide a framework for future development of novel therapeutics for the treatment of PTEN-deficient and trastuzumab resistant breast cancers.
MATERIALS AND METHODS
Cell lines and reagents
BT474 was cultured in DMEM supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic under a 5% CO2 environment. BT474 PTEN- LTT cells were generated by lentiviral infection to introduce PTEN shRNA, followed by single cell colony formation to establish a genetically identical cell line. Long term treatment with trastuzumab was carried out for greater than 3 months as previous described [10, 49], and were otherwise maintained in the same media as the parental cell line. Sulforaphane was obtained from LKT Laboratories. Polyclonal antibodies against MEOX1 were purchased from Abcam (ab75895).
RNA-seq data analysis
Total RNA was extracted using RNeasy Mini kit (Qiagen) and mRNA library was prepare for RNA-seq (poly A selection based) using Illumina TruSeq technology (Illumina). The generated libraries were sequenced on Illumina Hi-Seq 2000 with 50 cycle single ended reads. RNA-Seq reads were aligned to annotated RefSeq transcripts using Bowtie. Only uniquely mapped reads were used for further analysis. Gene expression is expressed as reads/kilobase/million mapped reads (RPKM) and differences in gene expression were estimated using rSeq. Gene set enrichment analysis was performed using the GSEA Java desktop software application (Broad Institute). Finally, gene functional classification was performed using the DAVID Bioinformatics Resources v6.7.
Knockdown by siRNA
Small interfering RNAs for gene MEOX1 were purchased from Qiagen (validated FlexiTube siRNA, SI00630266). Transfection of BT474 PTEN- LTT cells was carried out using Lipofectamine® RNAiMAX vehicle according to the manufacturer’s instruction following optimization. As a negative control, a non-targeting sequence siRNA was utilized (Qiagen, catalog number 1027281). Knockdown at mRNA level was confirmed by isolating total RNA (RNeasy Mini kit, Quigen) and performing real-time quantitative RT-PCR in triplicate. Real-time PCR was carried out on an ABI PRISM 7900HT sequence detection system (Applied Biosystems).
MTS cell proliferation assay
Cell lines were seeded at a density of 3,000 cells per well in 96-well plates and allowed to adhere overnight. Cells were then incubated with SF in increasing concentrations for a period of 48 hours. Proliferation was determined by MTS assay according to manufacturer’s instruction by measuring the absorbance at 490 nm on a Synergy 2 plate reader (Biotek).
Advanced tumor model
The use of vertebrate animals in this study was conducted in accordance with a standard animal protocol approved by the University Committee on the Use and Care of Animals at the University of Michigan. 5 week old female non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice were obtained from Jackson Laboratory. BT474 or BT474 PTEN- LTT cells (500,000) mixed with Matrigel (BD Biosciences) were injected to the mammary fat pads of NOD/SCID mice. Tumors were measured by caliper and the volume was calculated using V = 1/2 (width2 × length). When tumor volume reached approximately 40 mm3, the mice were randomly separated into two groups, once receiving daily i.p. injected with 0.9% saline solution and the other receiving 50 mg/kg sulforaphane daily. Final tumor mass was measured on an analytical balance after primary control treated tumor volume reached an average of 500 mm3.
Secondary reimplantation
Isolated primary tumors were mechanically dissociated by mincing with scalpels and suspended in Media 199 and single cell suspensions generated by incubation with collagenase and hyaluronidase (Stem Cell Technologies). Human tumor cells with DsRed label were then isolated using FACS on a SY3200 (Sony Biotechnology) flow cytometer. Secondary female, 5 week old, NOD/SCID mice were inoculated with 5,000, 1,000, or 200 cells for BT474 PTEN- LTT xenografts as described above. Tumor formation rate in secondary mice was assessed 8 weeks following implanting cells by direct palpitation and used to assess cancer stem cell frequency by extreme limiting dilution analysis.
Colony formation assay
Colony formation was carried out in six-well plates layered with 1.5 mL of 0.5% agar (Difco Agar Noble) in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Subsequently, 1000 cells mixed with 0.35% agar and allowed to set in each well of the six-well plates in order to form the upper gel. After 2 weeks, pictures of colonies were taken using a digital camera after staining with 0.005% crystal violet. Each treatment was performed in triplicate.
Immunofluorescence
Cells were seated in glass chambers slides and allowed to adhere overnight. After rinsing with PBS, cells were fixed in PBS containing 4% paraformaldehyde for 30 minutes and permeabilized with 0.1% Triton X-100 (Roche Diagnostics) for 10 minutes at 4°C. Cells were then incubated with MEOX1 (1:200) primary antibodies, followed by FITC-conjugated anti-rabbit IgG (1:300) secondary antibodies. Samples were then mounted and visualized using fluorescent microscopy with a Nikon Eclipse TE2000-S microscope and the photos were acquired with MetaMorph 7.6.0.0.
Mammosphere formation assay
Mammosphere culture was done as previously described in a serum-free mammary epithelium basal medium (Lonza, Inc.) supplemented with B27 (Invitrogen), 1% antibiotic-antimycotic, 5 μg/mL insulin, 1 μg/mL hydrocortisone, 4 μg/mL gentamicin, 20 ng/mL EGF (Sigma-Aldrich), 20 ng/mL basic fibroblast growth factor (Sigma-Aldrich), and 1:25,000,000 β-mercaptoethanol (Sigma-Aldrich). Single cells prepared from mechanical and enzymatic dissociation were plated in six-well ultralow attachment plates (Corning) at a density of 500 cells/ml. After 7 days of culture, the number of mammospheres was counted on a Nikon Eclipse TE2000-S microscope and the photos were acquired with MetaMorph 7.6.0.0.
Immunohistochemistry and tissue microarray staining
Immunohistochemical analysis was performed on isolated tumors from mouse xenografts and patients tissues on a tissue micro array (TMA) obtained from US Biomax (HBre-Duc150Sur-01). Standard Envision method was performed. Briefly, after removal of paraffin in xylene and rehydration in graded alcohols, heated antigen retrieval was performed in citrate buffer (10 mmol/L pH 6.0) by microwave heating for 20 min. Endogenous peroxidase activity was prevented by incubation in 0.3% hydrogen peroxide for 10 min. Nonspecific binding was blocked by incubation in 10% normal animal serum for 30 min. Sections were incubated at 4 °C for 24 h with MEOX1 (Abcam, ab23279) antibody, diluted 1:100. For blank controls, the primary antibody was replaced with PBS solution (100 mM, PH 7.4). The percentages of MEOX1 positive cells were scored using the following scale: 0= no staining or less than 5%; 1=5-25%; 2=26-50%; 3=51-75%; 4=more than 75%. The intensity of staining was evaluated as 1 (low), 2 (moderate), and 3 (strong). MEOX1 in nucleus was defined as negative (combined score from 0 to 6) or positive (combined score from 8 to 12).
Statistics
For IHC staining univariate survival analyses were performed and survival curves were drawn using Kaplan-Meier method. The differences between curves were tested by the log-rank test. Comparison of the variables was made by χ2 test. Correlation coefficient was calculated by spearman rank correlation test. For additional studies comparing parental to the drug resistant cell line, siRNA, or sulforaphane treatment statistical differences were determined using two-tailed Student’s t test or ANOVA followed by Dunnett post hoc analysis. Data are presented as mean ± SD (n ≥ 3).
ACKNOWLEDGMENTS
The authors wish to thank Drs. Dai Chen (Novel Bioinformatics Ltd., Co, Shanghai, China) for their technical assistance in bioinformatics analysis.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest related to this work.
GRANT SUPPORT
This work was partially supported by NIH grant R01-CA120023, R21-CA143474, University of Michigan Rackham merit fellowship, and National High-tech R&D Program of China for Young Scholars (No: 2014AA020537).
REFERENCES
1. Carr J, Havstad S, Zarbo RJ, Divine G, Mackowiak P and Velanovich V. THe association of her-2/neu amplification with breast cancer recurrence. Archives of Surgery. 2000; 135:1469-1474.
2. Spiridon CI, Ghetie M-A, Uhr J, Marches R, Li J-L, Shen G-L and Vitetta ES. Targeting Multiple Her-2 Epitopes with Monoclonal Antibodies Results in Improved Antigrowth Activity of a Human Breast Cancer Cell Line in Vitro and in Vivo. Clinical Cancer Research. 2002; 8:1720-1730.
3. Gajria D and Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011; 11:263-275.
4. Bartsch R, Wenzel C and Steger GG. Trastuzumab in the management of early and advanced stage breast cancer. Biologics. 2007; 1:19-31.
5. Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ and Press M. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. Journal of Clinical Oncology. 2002; 20:719-726.
6. Bailey TA, Luan H, Clubb RJ, Naramura M, Band V, Raja SM and Band H. Mechanisms of Trastuzumab resistance in ErbB2-driven breast cancer and newer opportunities to overcome therapy resistance. J Carcinog. 2011; 10:28.
7. Nagata Y, Lan K-H, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung M-C and Yu D. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004; 6:117-127.
8. Mulholland DJ, Kobayashi N, Ruscetti M, Zhi A, Tran LM, Huang J, Gleave M and Wu H. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012; 72:1878-1889.
9. Song LB, Li J, Liao WT, Feng Y, Yu CP, Hu LJ, Kong QL, Xu LH, Zhang X, Liu WL, Li MZ, Zhang L, Kang TB, Fu LW, Huang WL, Xia YF, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009; 119:3626-3636.
10. Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK, Chung S, Luther T, Paholak HJ, Liu S, Hassan KA, Zen Q, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012; 47:570-584.
11. Hirsch HA, Iliopoulos D, Tsichlis PN and Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Research. 2009; 69:7507-7511.
12. Abdullah LN and Chow EK-H. Mechanisms of chemoresistance in cancer stem cells. Clinical and Translational Medicine. 2013; 2:3.
13. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ and Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100:3983-3988.
14. Burnett JP, Korkaya H, Ouzounova MD, Jiang H, Conley SJ, Newman BW, Sun L, Connarn JN, Chen CS, Zhang N, Wicha MS and Sun D. Trastuzumab resistance induces EMT to transform HER2(+) PTEN(-) to a triple negative breast cancer that requires unique treatment options. Scientific reports. 2015; 5:15821.
15. Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott Sean P, Landis Melissa D, Hong S, Adams A, D’Angelo R, Ginestier C, Charafe-Jauffret E, Clouthier Shawn G, et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Reports. 2014; 2:78-91.
16. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD and Rho JK. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature genetics. 2012; 44:852-860.
17. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S and Cosper AK. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine. 2011; 3:75ra26-75ra26.
18. Zhang Y, Kensler TW, Cho CG, Posner GH and Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci U S A. 1994; 91:3147-3150.
19. Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, Yu Y, Clouthier SG, Schwartz SJ, Wicha MS and Sun D. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010; 16:2580-2590.
20. Kallifatidis G, Rausch V, Baumann B, Apel A, Beckermann BM, Groth A, Mattern J, Li Z, Kolb A, Moldenhauer G, Altevogt P, Wirth T, Werner J, Schemmer P, Büchler MW, Salnikov AV, et al. Sulforaphane targets pancreatic tumour-initiating cells by NF-κB-induced antiapoptotic signalling. Gut. 2009; 58:949-963.
21. Rausch V, Liu L, Kallifatidis G, Baumann B, Mattern J, Gladkich J, Wirth T, Schemmer P, Büchler MW, Zöller M, Salnikov AV and Herr I. Synergistic Activity of Sorafenib and Sulforaphane Abolishes Pancreatic Cancer Stem Cell Characteristics. Cancer Research. 2010; 70:5004-5013.
22. Kallifatidis G, Labsch S, Rausch V, Mattern J, Gladkich J, Moldenhauer G, Büchler MW, Salnikov AV and Herr I. Sulforaphane Increases Drug-mediated Cytotoxicity Toward Cancer Stem-like Cells of Pancreas and Prostate. Molecular Therapy. 2011; 19:188-195.
23. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M and Jeffrey SS. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences. 2001; 98:10869-10874.
24. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
25. Nagy P, Friedländer E, Tanner M, Kapanen AI, Carraway KL, Isola J and Jovin TM. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Research. 2005; 65:473-482.
26. Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP and Maiese K. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002; 2:127-137.
27. Nahta R, Yuan LX, Zhang B, Kobayashi R and Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Research. 2005; 65:11118-11128.
28. Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA and Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Molecular Cancer Therapeutics. 2003; 2:1093-1103.
29. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer C, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS and Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007; 1:555-567.
30. Oliveras-Ferraros C, Corominas-Faja B, Cufí S, Vazquez-Martin A, Martin-Castillo B, Iglesias JM, López-Bonet E, Martin ÁG and Menendez JA. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle. 2012; 11:4020-4032.
31. Bai WD, Ye XM, Zhang MY, Zhu HY, Xi WJ, Huang X, Zhao J, Gu B, Zheng GX and Yang AG. MiR-200c suppresses TGF-β signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. International Journal of Cancer. 2014; 135:1356-1368.
32. Lesniak D, Sabri S, Xu Y, Graham K, Bhatnagar P, Suresh M and Abdulkarim B. Spontaneous epithelial-mesenchymal transition and resistance to HER-2-targeted therapies in HER-2-positive luminal breast cancer. PLoS One. 2013; 8:e71987.
33. Kim G, Ouzounova M, Quraishi A, Davis A, Tawakkol N, Clouthier S, Malik F, Paulson A, D’Angelo R and Korkaya S. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene. 2014.
34. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG and Wicha MS. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proceedings of the National Academy of Sciences. 2012; 109:2784-2789.
35. Turashvili G, Bouchal J, Burkadze G and Kolar Z. Wnt signaling pathway in mammary gland development and carcinogenesis. Pathobiology: journal of immunopathology, molecular and cellular biology. 2005; 73:213-223.
36. Martin-Castillo B, Lopez-Bonet E, Cuyas E, Vinas G, Pernas S, Dorca J and Menendez JA. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget. 2015; 6:32317-32338. doi: 10.18632/oncotarget.6094.
37. Xu C, Shen G, Chen C, Gelinas C and Kong A-NT. Suppression of NF-kB and NF-kB-regulated gene expression by sulforaphane and PEITC through IkBa, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005; 24:4486-4495.
38. Kirilenko P, He G, Mankoo BS, Mallo M, Jones R and Bobola N. Transient activation of meox1 is an early component of the gene regulatory network downstream of hoxa2. Molecular and cellular biology. 2011; 31:1301-1308.
39. Jukkola T, Trokovic R, Maj P, Lamberg A, Mankoo B, Pachnis V, Savilahti H and Partanen J. Meox1Cre: a mouse line expressing Cre recombinase in somitic mesoderm. Genesis. 2005; 43:148-153.
40. Petropoulos H, Gianakopoulos PJ, Ridgeway AG and Skerjanc IS. Disruption of Meox or Gli activity ablates skeletal myogenesis in P19 cells. Journal of Biological Chemistry. 2004; 279:23874-23881.
41. Umeda K, Zhao J, Simmons P, Stanley E, Elefanty A and Nakayama N. Human chondrogenic paraxial mesoderm, directed specification and prospective isolation from pluripotent stem cells. Scientific reports. 2012; 2.
42. Bayrakli F, Guclu B, Yakicier C, Balaban H, Kartal U, Erguner B, Sagiroglu MS, Yuksel S, Ozturk AR and Kazanci B. Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype. BMC genetics. 2013; 14:95.
43. Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA and Alkuraya FS. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. The American Journal of Human Genetics. 2013; 92:157-161.
44. Nguyen PD, Hollway GE, Sonntag C, Miles LB, Hall TE, Berger S, Fernandez KJ, Gurevich DB, Cole NJ and Alaei S. Haematopoietic stem cell induction by somite-derived endothelial cells controlled by meox1. Nature. 2014; 512:314-318.
45. Candia AF, Hu J, Crosby J, Lalley PA, Noden D, Nadeau JH and Wright CV. Mox-1 and Mox-2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development. 1992; 116:1123-1136.
46. Thiaville MM, Stoeck A, Chen L, Wu RC, Magnani L, Oidtman J, Shih Ie M, Lupien M and Wang TL. Identification of PBX1 target genes in cancer cells by global mapping of PBX1 binding sites. PLoS One. 2012; 7:e36054.
47. Magnani L, Stoeck A, Zhang X, Lánczky A, Mirabella AC, Wang T-L, Gyorffy B and Lupien M. Genome-wide reprogramming of the chromatin landscape underlies endocrine therapy resistance in breast cancer. Proceedings of the National Academy of Sciences. 2013:201219992.
48. Zhao D, Mo Y, Li M-T, Zou S-W, Cheng Z-L, Sun Y-P, Xiong Y, Guan K-L and Lei Q-Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. The Journal of Clinical Investigation. 2014; 124:5453-5465.
49. Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, Clouthier SG and Wicha MS. Regulation of Mammary Stem/Progenitor Cells by PTEN/Akt/β-Catenin Signaling. PLoS Biol. 2009; 7:e1000121.