
INTRODUCTION
Mitochondria are important ancient organelles present in nearly all eukaryotic cells. They play an essential role in energy metabolism [1] and other cellular processes such as the β-oxidation of fatty acids [2], maintaining proper concentration of mitochondrial matrix calcium [3], amino acids metabolism [4], heme- and iron-sulfur (Fe-S) cluster biogenesis [5, 6], control of cell death including apoptosis [7-9], steroid synthesis [10], and hormonal signaling [11, 12].
Mitochondria consist of outer and inner membranes separated by an intermembrane space. The outer mitochondrial membrane (OMM) contains porins, which mediate the exchange of small molecules and information between mitochondria and the rest of the cell [13]. The inner mitochondrial membrane (IMM) encloses the matrix space and has numerous invaginations called cristae. The number of cristae per mitochondrion is related to the energy requirement for the vital functions of certain cell type as well as the number of mitochondria per cell. Cristae extend the available working space of the inner membrane surface area [14]. IMM is enriched in the proteins involved in mitochondrial fusion, transport of nuclear-encoded proteins, oxidative phosphorylation (OXPHOS), iron-sulfur cluster biogenesis, protein synthesis and transport of mtDNA-encoded proteins [15, 16].
Mitochondrial genome is a small circular DNA molecule. There are multiple copies of mitochondrial DNA (mtDNA) in the matrix of each mitochondrion. Replication of mtDNA is not related to cell cycle and may be performed many times [17]. This leads to generation of mtDNA mutations by replication errors in addition to ones due to accumulated damage [18]. The point mutations or rearrangements of mtDNA are mainly related to the OXPHOS dysfunction and cause a variety of human mitochondrial diseases as well as mutations in nuclear genes involved in the maintenance of mitochondria [19-25].
The mitochondrial dysfunction is known to be associated with aging, age-related diseases and cancer. Herein, we review current studies in the area to show that oxidative stress, as the cause or consequence of defect mitochondrial function, could be a common phenomenon in these pathologies.
MITOCHONDRIA AND REACTIVE OXYGEN SPECIES
The first paper on free radical theory was published by Denham Harman in 1956 [26]. This theory suggests that free radicals, produced by mitochondria as by-products of their normal metabolism, later go on to attack cell constituents. A year after the publication of Harman’s paper, Mills described a factor that coacted with the glutathione to protect the hemoglobin in the erythrocyte from oxidative breakdown. It was termed glutathione peroxidase (GPX) and further was described as the main enzyme involved in hydrogen peroxide detoxification [27-29]. In 1969 McCord and Fridovich discovered the anti-free radical enzyme superoxide dismutase (SOD), which was widely distributed within mammalian organisms [30]. Following this discovery, a number of studies demonstrated that mitochondria isolated from different sources (e.g., cow and pigeon heart [31-33], rat liver, heart, and brain [34-38], and yeast [39]) could generate hydrogen peroxide. Finally, Harman proposed that the mammalian lifespan depends on the genetic regulation of oxygen utilization rate, and suggested the Mitochondrial Free Radical Theory of Aging (MFRTA) [40].
A product of mitochondrial oxidative metabolism is highly reactive and unstable oxygen, which can oxidize many molecules and form reactive oxygen species (ROS) [41]. ROS are generated intracellular in different compartments through multiple mechanisms (Table 1). Mitochondrial-derived reactive oxygen species (mtROS) include singlet oxygen (O2), superoxide anion (O2•−), hydrogen peroxide (H2O2), nitric oxide (NO•), hydroxyl radical (OH•), and hydroxyl ion (OH-). Initially, oxygen is converted to a superoxide anion with xanthine oxidase (XO) or mitochondrial respiratory chain complexes I (NADH dehydrogenase) and III (bc1 complex) [42-45]. Complex III produces a superoxide anion in both the matrix and the intermembrane space [46]. The concentration of these complexes in IMM varies with organism, tissue, state, age or hormonal status. The superoxide anion is then converted to hydrogen peroxide by SOD. Hydrogen peroxide can be detoxified to water and oxygen with glutathione peroxidase, catalase (CAT) or thioredoxin peroxidase (TPx) [42, 47]. It can be also converted to hydroxyl radical and hydroxyl ion via the Fenton reaction (Figure 1) [48].
There is a hypothesis that the nitric oxide is produced by mitochondrial NO synthase (mtNOS). This was suggested after the detection of a high rate of NO production and functionally active mitochondrial nitric oxide synthase (NOS) in rat liver mitochondria [49-51]. However, these data were not reproduced by other laboratories, implying that the NOS enzymes are not present at physiologically relevant levels in mitochondria [52, 53]. Today, NO production by mitochondria still remains an open question [54].
Table 1: Major intracellular sources of reactive oxygen species (ROS)
Reactive oxygen species |
Intracellular sources |
Compartment |
Singlet oxygen (O2) |
Fenton reaction Lipid peroxidation chain reactions Haber-Weiss reaction Superoxide Dismutase (SOD)-mediated reaction Catalase-mediated reaction Glutathione peroxidase-mediated reaction Xanthine oxidase (XO)-mediated reaction |
Mitochondria Cytosol Peroxisomes Nucleus Plasma membrane Endoplasmic reticulum Lysosome All membranes |
Hydroxyl radical (OH•) |
Proton-catalyzed decomposition of peroxynitrite Fenton reaction Haber-Weiss reaction Decomposition of ozone (O3) Beckman-Radi-Freeman pathway |
Mitochondria Cytosol Endoplasmic reticulum Lysosome |
Hydrogen peroxide (H2O2) |
Superoxide dismutase (SOD)-mediated reaction NADPH oxidase-mediated reaction Cytochrome P450-mediated reaction Xanthine oxidase (XO)-mediated reaction Monoamine oxidases (MAO)-mediated reaction Peroxisomal fatty acid oxidation Flavin adenine dinucleotide (FAD)-mediated reaction Antibody-catalyzed water (H2O) oxidation Electron-transfer flavoprotein pathway |
Mitochondria Cytosol Peroxisomes Plasma membrane Endosomes Endoplasmic reticulum Lysosome Nucleus |
Superoxide anion (O2•−) |
Fenton reaction NADH/NADPH oxidase (NOX)-mediated reaction Xanthine oxidase (XO)-mediated reaction Lipoxygenase pathway Cyclooxygenase pathway Cytochrome P450 monooxygenase reaction Mitochondrial oxidative phosphorylation Electron-transfer flavoprotein reaction Hemoglobin auto-oxidation (within erytrocyte) Nitric oxide synthases (NOS)-mediated reaction |
Mitochondria Cytosol Plasma membrane Peroxisomes Nucleus Endoplasmic reticulum |
Hypochlorous acid (HOCL) and related species (HOBr, HOI, and HOSCN) |
Eosinophil peroxidase (EPX)-mediated reaction (within eosinophil granulocytes) Myeloperoxidase (MPO)-dependent oxidation (within neutrophil granulocytes) |
Cytosol Endoplasmic reticulum Lysosome Vacuole Plasma membrane Mitochondria Nucleus |
Hydroxyl ion (OH-) |
Fenton reaction Haber-Weiss reaction Hydroperoxide (ROOH) decomposition |
Mitochondria Cytosol Endoplasmic reticulum Lysosome |
Peroxide (O2•2−) |
Peroxide is unstable molecule. Hydrogen peroxide is more stable one that is formed as described above. |
Mitochondria Cytosol Peroxisomes Plasma membrane Endosomes Endoplasmic reticulum Lysosome Nucleus |
Ozone (O3) |
Ozone (O3) is unstable molecule generated during antibody catalyzed oxidation of H2O to H2O2 |
Cytosol |
Nitric oxide radical (NO•) |
Nitric oxide synthases (NOS)-mediated nitrite (NO2-) reduction Xanthine oxidase (XO) reducing nitrates and nitrites |
Mitochondria Cytosol Peroxisomes Endoplasmic reticulum Plasma membrane Nucleus |
Peroxynitrite (ONOO-) |
Fenton reaction Rapid reaction of singlet oxygen (O2) and nitric oxide radical (NO•) The reaction of hydrogen peroxide (H2O2) with nitrite (NO2−) |
Mitochondria Cytosol Lysosome Endoplasmic reticulum Nucleus Peroxisomes |
Peroxyl radical (ROO•/RCOO•) (also denoted Lypid peroxyl radical (LOO•)) |
Lipid peroxidation chain reactions Synthesis of eicosanoids Hydroperoxide (ROOH) decomposition induced by heat or radiation ROOH reaction with transition metal ions and other oxidants capable of abstracting hydrogen |
Cytosol Plasma membrane Peroxisomes Endoplasmic reticulum Mitochondria Nucleus Lysosome All membranes |
Hydroperoxy radical (HOO•) |
Fenton reaction |
Mitochondria Cytosol Endoplasmic reticulum Lysosome |
Organic hydroperoxide (ROOH/RCOOH) |
Lipoxygenase-mediated reaction Oxidation of biomolecules, including lipids, proteins and DNA Cyclooxygenase reaction Cytochrome P450 monooxygenase reaction Heme-peroxidase turnover |
Cytosol Plasma membrane Nucleus Endoplasmic reticulum Mitochondria Peroxisomes Lysosome |
Organic radicals (R•, RO•, R-S•) |
Hydroperoxide (ROOH) decomposition induced by heat or radiation ROOH reaction with transition metal ions and other oxidants capable of abstracting hydrogen Lipid peroxidation chain reactions |
Cytosol Plasma membrane Mitochondria Lysosome Peroxisomes Endoplasmic reticulum Nucleus All membranes |
Carbonate Radical (CO3●-) |
The reaction between peroxynitrite and CO2 Superoxide Dismutase (SOD)-mediated reaction Xanthine oxidase (XO)-mediated reaction Metal-ion catalyzed decomposition of peroxymonocarbonate (HCO4-) |
Mitochondria Cytosol Peroxisomes Endoplasmic reticulum Peroxisomes Lysosome Vacuole |
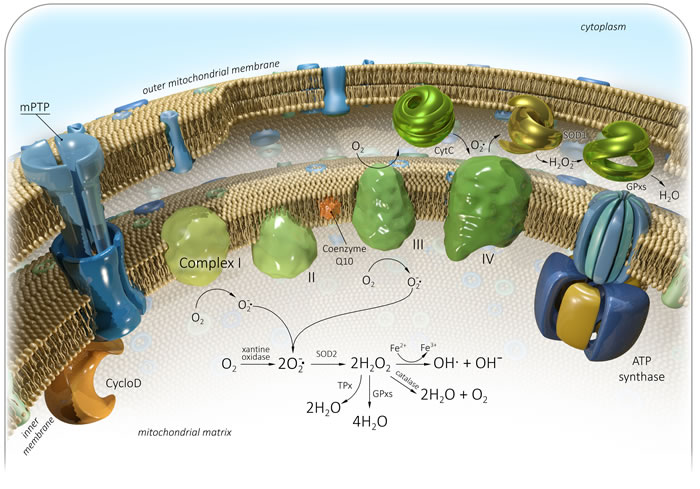
Figure 1: Generation of mitochondrial reactive oxygen species (mtROS). Complex I - NADH dehydrogenase, II - Succinate dehydrogenase, III - bc1 complex, IV - Cytochrome C oxidase, V - ATP synthase, Q - Ubiquinone, Cyt C - Cytochrome C, Cyclo D - Cyclophilin D, mPTP - Mitochondrial permeability transition pore, SOD - Superoxide dismutase, GPxs - Glutathione peroxidase, TPx - Thioredoxin peroxidase. See text for details.
LIPID PEROXIDATION
The oxidative stress leads to cell injury by three basic ways: lipid peroxidation of membranes, oxidative modification of proteins and DNA damage. Lipid peroxidation affects cell membranes and other lipid-containing structures [55]. β-oxidation of lipids is usually followed by a release of oxygen, which is reduced to water through the mitochondrial respiratory chain. At the same time, lipids can be oxidized with efficient ROS initiators, particularly hydroxyl radical and perhydroxyl radical (HO2•), forming water and a lipid radical. This initiates the reaction of lipid peroxidation, which constantly takes place in the cells. The lipid radical reacts directly with molecular oxygen and produces a lipid peroxyl radical. The lipid peroxyl radical is not a very stable molecule and can combine with another adjacent fatty acid to form a lipid hydroperoxide and different lipid radicals, or it can react with itself. Lipid hydroperoxide can be also broken down into a lipid alhoxyl radical and a hydroxyl radical. The lipid radicals formed at the previous stage can react with oxygen to produce another lipid peroxyl radical, and so on. Thus, this process is called “chain reaction of lipid peroxidation” (Figure 2). The main intermediate products of the reaction are lipid hydroperoxides (LOOHs). They can disturb membrane structure, an being dangerous for cells [56].
PODUCTS OF LIPID PEROXIDATION AS COMMON MARKERS OF OXIDATIVE STRESS IN AGING AND CANCER
The major secondary products of lipid peroxidation are toxic and mutagenic aldehydes, malondialdehyde (MDA) and 4-hydroxynonenal/4-hydroxy-2-nonenal (HNE). They are considered markers of the oxidative stress [57-61]. These products have unique properties compared with ROS because the non-charged structure of aldehydes allows them to easily migrate through membranes and cytosol and, consequently, to cause far-reaching damaging effects inside or outside the cells [62, 63]. There is objective evidence that HNE and MDA can modify the amino acid residues and form stable adducts leading to protein damage [85, 86]. They can also form covalent adducts with nucleic acids, and membrane lipids. The MDA and HNE have been shown to be implicated in normal aging, age-related neurodegenerative diseases, and cancer [64-69]. Recent study showed that HNE-modified proteins (HNE-MP) were accumulated during aging in vitro and could be supposed to measure aging parameters. The middle-aged human fibroblasts were cultured and maintained by serial passaging throughout their proliferative lifespan. Four age points of the cells were analyzed. Aging cells showed a considerable increase in HNE-MP levels compared with young and middle-aged ones [70].
The HNE-production in the brain is induced by the amyloid-β peptide (Aβ), which plays a primary role in Alzheimer’s disease (AD) pathogenesis [63]. Conversely, the preincubation of cells with HNE increased the uptake of Aβ and its intracellular accumulation. This indicates that HNE and Aβ may interact to provide potentiation of Aβ’s cytotoxicity effects on neuron-like cells in vitro [71, 72]. HNE-crosslinking modifications accumulating in the lysosomal/proteasomal pathway and leading to protein inactivation and insolubility were detected in patients with Alzheimer’s disease [73]. Immunocytochemical studies have demonstrated that pyrrole adducts formed by reacting HNE with lysine amino groups were present in neurons of patients with AD cases [74]. An increase in MDA immunoreactivity was detected in the cytoplasm of neurons and astrocytes in both normal aged and AD brains, but not in brains of young subjects [75]. Moreover, increased plasmatic levels of MDA and its correlation with age were also observed in AD patients [76-80].
Parkinson’s disease (PD) is pathologically characterized by progressive destruction and death of neurons, that produce dopamine. HNE may alter dopamine uptake in rat striatal synaptosomes through binding to SH groups of the dopamine transporter and to Na+/K+ ATPase [81]. In rat striatal membranes, HNE have been registered as an effector of signaling pathway mediated by D1/D5 dopamine receptors [82]. In addition, it has been shown that HNE could modulate the activity of regulator G-protein signaling 4 (RGS4) involved in PD [83]. The concentration of HNE was increased in the cerebrospinal fluid and plasma of Parkinson’s patients [84]. Furthermore, HNE-modified proteins were positively stained in more than half the nigral neurons of PD patients, and the levels of MDA were also increased. The data indicate that, in Parkinson disease, oxidative stress can contribute to nigral cell death [85, 86].
Recently, it was found that the by-products of lipid peroxidation can induce carcinogenesis. Cell membranes contain a high concentration of polyunsaturated fatty acids, which are frequently subjected to peroxidation. This leads to an inhibition of growth and death of cells. The oxidation of phospholipids in the IMM can trigger the mitochondria-mediated pathway of apoptosis (Figure 3). ROS or lipid peroxidation by-products primarily react to cardiolipin molecules, the IMM phospholipids, which are bound to cytochrome c [87-89]. This induces disturbances of cytochrome c-cardiolipin interaction and dissociation of cytochrome c from the IMM [90-92]. The release of cytochrome c into the cytoplasm induces a series of biochemical reactions, resulting in caspase activation and subsequent cell death [9]. At this point, a major regulator of mitochondrion-dependent apoptosis is Bcl-2 family of proteins, which show both pro- and anti-apoptotic activities. The proteins belonging to the Bcl-2 family are bound to the OMM and can modulate its permeabilization [93]. Bax and Bak are anti-apoptotic proteins of the Bcl-2 family, which can be activated in two ways: through disturbance of their bond with anti-apoptotic proteins (e.g., Bcl-2, Bcl-xL and Mcl-1) [94], or interaction with activator proteins (e.g., BH3/tBid, Puma, BIM, NOXA, and p53), which induce their conformational changes [95-98]. Inactivated Bax proteins can be localized as monomers in the cytosol or closely associated with the OMM. Then, during the process of its activation, Bax forms homo-oligomers and inserts itself into the OMM as well as into Bak. This leads to membrane pore formation and permeabilization, which promotes the release of cytochrome c in cytosol [99]. Anti-apoptotic proteins prevent mitochondria-mediated apoptosis through their interaction with pro-apoptotic ones. The studies show that overexpression of Bcl-2 inhibits the release of cytochrome c from mitochondria and the subsequent apoptotic response is blocked. For example, HNE-induced caspase activation is suppressed in Bcl-2 transfected colorectal carcinoma cells [100]. The cytosol cytochrome c binds to the adapter protein apoptotic protease activating factor 1 (Apaf-1), and induces an apoptosome assembly in the presence of ATP/dATP. This activates pro-caspase-9 directly within the apoptosome complex [101]. Then the pro-caspase-9 is cleaved to the active caspase-9, which, in turn, activates the caspases-3, -6 and -7, leading to DNA fragmentation and cell death [101, 102]. If the cellular ATP/dATP level is depleted, the caspase activation is blocked and the cell death is re-directed from apoptosis to necrosis. The release of cytochrome c and apoptosome formation can be also triggered though the extrinsic pathway of apoptosis [103, 104]. Additionally, the accumulation of damage directly in mitochondria may also cause enhanced oxidant production and a cascade of degenerative events. It should be noted that HNE could be generated directly through the oxidation of mitochondrial phospholipid cardiolipin as well as other oxidation products. In this case, HNE reacts with surrounding molecules near the site of its formation, thereby promoting chain-reactions of the mitochondria-derived apoptosis again [105]. This process appears to be involved in atherosclerosis and cancer [106, 107]. Thus, it has been shown that HNE could induce mitochondria-mediated apoptosis in the pheochromocytoma (PC12) cell line and colorectal carcinoma cells [100, 108]. A statistically significant increase in MDA and HNE levels was detected in primary colorectal cancer, implying the association of colorectal carcinogenesis with serious oxidative stress [109]. Immunohistochemical staining of HNE adducts was demonstrated in animal models of liver cancer [110]. HNE treatment of MG63 human osteosarcoma cells could activate caspase-3 and altered the Bax/Bcl-2 ratio, thereby inducing cell death [111]. A recent research showed that HNE increased the growth of breast cancer cells and promoted their angiogenesis and invasion [112]. Elevated levels of MDA were observed in plasma and blood serum of patients with breast, lung, ovarian, thyroid, and oral cancer, and precancer states [113-122]. The MDA levels in patients with lung cancer correlated with the cancer stage [123]. In addition, significantly higher levels of salivary MDA were determined in squamous cell carcinoma and pre-cancer patients [124].
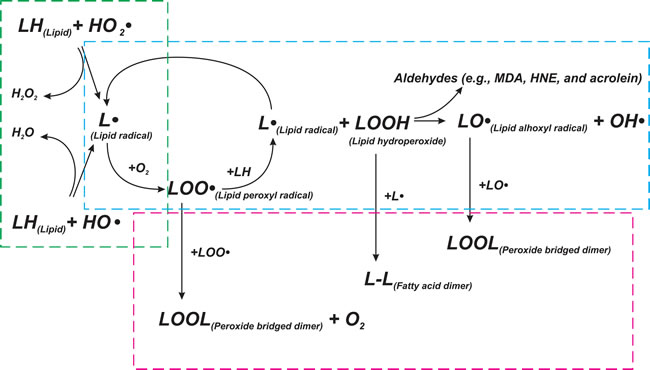
Figure 2: Scheme of lipid peroxidation chain reaction. Lipid peroxidation chain reactions initiated by free radicals consists of three major steps (initiation, propagation, and termination), which are marked by green, blue, and red frames, respectively. LH - Lipid molecule, L• - Lipid radical, LOOH - Lipid hydroperoxide, LOO• - Lipid peroxyl radical, LO• - Lipid alhoxyl radical, LOOL - Peroxide bridged dimer, L-L - Fatty acid dimer, OH• - hydroxyl radical, HO2• - perhydroxyl radical. See text for details.
LIPID PEROXIDATION PRODUCTS AND SIGNALING PATHWAYS
Several studies suggest an interesting mechanism protecting tumor cells against superoxide anion-mediated apoptosis by the expression of membrane-associated catalase. Tumor cells, as known, generate extracellular superoxide anions with the participation of NADPH Oxidase 1 (NOX1). NOX1 are multi-subunit enzyme complexes localized in cell membrane and required for the reduction of molecular oxygen. On the one hand, extracellular superoxide anions are needed for the regulation of cancer cell proliferation and for the maintenance of their transformed state. On the other hand, extracellular superoxide anions can induce intercellular apoptosis in transformed cells [125, 126]. As a result, they are selectively eliminated. The ROS signaling by the HOCl and NO/peroxynitrite pathways are involved directly in this apoptosis-derived cell death [127-129]. In the transformed cell, extracellular superoxide anions spontaneously react with hydroxyl ions (H-). They could be also driven by SOD forming an unstable hydrogen peroxide. It is used as a substrate to generate exogenous HOCl by the dual oxidase (DUOX)-coded peroxidase (POD) domain. Then HOCl can interact with superoxide anions, generating a hydroxyl radical, which penetrates intracellular space, induces lipid peroxidation, and consequently promotes the mitochondrial-derived apoptosis. However, tumor cells express the membrane-associated catalase, which can efficiently decompose H2O2 directly after its generation. Moreover, SOD can play a co-modulatory protective role through partial inhibition of HOCl/superoxide anion interactions. SOD-derived generation of H2O2 could be also compensated by interrelation of SOD with a catalase-mediated protective effect. Additionally, in the presence of high concentrations of H2O2, compared with POD, HOCl can react with H2O2 to block the HOCl signaling [130].
NO/peroxynitrite signaling pathway modulating tumor cell death consists of the following: (1) within cells, NO synthase promotes arginine-derived NO synthesis, (2) NO passes through the cell membrane, (3) the formation of peroxynitrite extracellular interaction of NO with superoxide anion and the subsequent reactions lead to the generation of nitrogen dioxide (NO2) and hydroxyl radical, (4) these reactions initiate the lipid peroxidation and apoptosis [131]. Nevertheless, the membrane-associated catalase can protect the tumor cell by preventing peroxynitrite generation and NO oxidation [126]. Moreover, the inhibition of NO/superoxide anion interactions by SOD, as in the previous case, modulates the negative effect on apoptosis pathway. Interestingly, it has been shown that high concentrations of HNE may inactivate membrane-associated catalase, leading to tumor cell death through mitochondria-derived apoptosis and consequently bestowing anti-cancer effects (Figure 3) [126, 132].
In a multitude of studies, it has been shown that HNE and its protein adducts can regulate Nuclear factor-kB (NF-κB) and activator protien 1 transcription factor (AP-1) transcription factors, thereby being responsible for the expression of many genes (Figure 3). It is well known that NF-kB signaling pathway is involved in normal aging, age-related diseases, and cancer [133-138]. NF-κB is a family of proteins, which play a key role in regulating the expression of genes which are responsible for immunity, apoptosis, and cell cycle. NF-κB family consists of five proteins: p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1), and p100/52 (NF-κB2), which associate with each other to form distinct active NF-κB dimers. In the cytosol, NF-κB dimers in inactive form anchored by IκB are found. There are two ways to activate the NF-kB-induced gene transcription. These are triggered by cytokines TNFα and IL-1 (canonical signaling) or antigen receptors CD40 and BAFF (non-canonical/alternative signaling). There are the activation of IKK complex (IKKα, IKKβ, and IKKγ) and the phosphorylation of IκB proteins, which are in interaction with p50-RelA subunit, with cytokines in the course of canonical signaling pathway. This reaction leads to degradation of IkB proteins, and a release of the active p50-RelA NF-κB dimer. NF-kB non-canonical signaling consists of the following: NIK (NF-κB-inducing kinase) induces the activation of IKKα, which phosphorylates p100 NF-κB subunit of p100-RelB NF-κB dimer directly in cytosol. This leads to the generation of an active p52-RelB NF-κB dimer. In both cases, the active NF-κB dimer can translocate to the nucleus and induce the transcription of target genes. NF-κB target genes include regulators of apoptosis (Bcl-XL and IAPs), cytokines (TNFa, IL-1, IL-6, and IL-12), cyclins and growth factors (G-CSF and M-CSF), immunregulatory proteins (VCAM, ICAM, and MHC I), and others [139, 140]. Many different kinases can phosphorylate and activate the IKKα and IKKβ subunits of the IKK complex: glycogen synthase kinase 3β (GSK3β), protein kinases B, R and C (PKB, PKR, and PKC), mitogen-activated type 3-protein kinase 7 (MAP3K7), p38 MAP kinases or c-Jun N-terminal kinases (JNKs) [141]. Thus, it has been shown that HNE could differently regulate PKC isozymes, MAPK (mitogen-activated protein kinase), and JNK in a dose-dependent manner and be involved in NF-kB activation (Figure 3) [142-145].
AP-1 is a heterodimeric/homodimeric complex composed of members of the JUN, FOS, ATF, and MAF families. The combination of protein in the AP-1 complex determines its target genes that play a major role in differentiation, proliferation, and apoptosis. Important examples are Bcl2 family of proteins, EGFR, p53, CD44, and proliferin. AP-1 complex has been shown to be involved in tumorigenesis. Two components of AP-1, c-JUN, and c-FOS, are well known as oncoproteins, but in some cases they can suppress tumor formation [146]. The regulation of AP-1 activity occurs by many ways: differential expression of single AP-1 components, interactions with ancillary proteins, and transcriptional and post-translational regulation. The last way consists in MAPK pathways activation (ERK1/ERK2, JNK/SAPK, and p38) by several external stimuli. This leads to induction of FOS and JUN gene transcription and the formation of further AP-1 complexes. It has been shown that HNE can regulate AP-1 complex generation through induction of FOS and JUN gene expression or activation/inhibition of MAPK pathways [108, 147-150].
Moreover, HNE has been shown to regulate the Fas/Fadd-dependent pathway of apoptosis (Figure 3). Fas is a protein belonging to the TNF-receptor superfamily. The Fas receptor (FasR) contains a death domain, which is a protein interaction module. FADD is an adaptor molecule, which contains a death domain and a death effector domain. FADD can interact with members of the TNF-receptor superfamily and mediates cell apoptotic signals. Binding of FasR with Fas ligand (FasL) induces its trimerization. This leads to interaction with adaptor molecule FADD, which has already been recruited by the receptor-associated death proteases (pro-caspases-8 and -10) through the death effector domain. Thereby, the death-inducing signaling complex I (DISC complex I) is formed. Activated caspase-8 can stimulate the caspase-3 signaling in two ways: (1) caspase-8 cleaves the Bcl-2 interacting protein (Bid), which alters the mitochondrial membrane permeabilization or induces activation of Bak. It triggers cytochrome c release, thereby activating the mitochondrion-dependent pathway of apoptosis; (2) caspase-8 directly activates caspase-3, -7 or -6, leading to the apoptotic DNA fragmentation and cell death [151]. In another case, FasR can recruit death-associated protein (DAXX), which further binds with apoptosis signal-regulating kinase 1 (ASK1) and activates it [152, 153]. Then ASK1 induces JNK/SAPK and p38 MAPK pathways [154-156]. The interaction of FasR with receptor-interacting protein kinase 1 (RIP1) was supposed to be a component of DISC complex II [157]. DISC complex II, depending on the combination of signaling proteins, can promote apoptosis, necrosis, NF-kB, JNK/SAPK and p38 MAPK pathways. A study by Chaudhary and colleagues has demonstrated that HNE could induce a Fas-mediated apoptosis in HepG2 cells. They demonstrate that an exposure of HepG2 cells to sublethal concentrations of HNE promoted the export of DAXX from the nucleus to cytoplasm and facilitated Fas-DAXX binding. In its turn, it activated ASK1, JNK and caspase-3, leading to cell death [158]. The same data were obtained in Jurkat and HLE B-3 cells [159, 160]. Generally, it has been shown that the expression and functions of Fas can be modulated by HNE in a time- and concentration-dependent manner and binding of HNE with Fas was essential for the execution of apoptosis. The involvement of HNE in p53 apoptosis pathway in HepG2 cells was also determined. Treatment of HepG2 cells with HNE resulted in the induction of p53 expression, its phosphorylation, and activation of downstream targets Bax and p21 [158]. Actually, it was shown that HNE treatment increased the expression of p53 family proteins and their targets in the SK-N-BE neuroblastoma cell line [161]. A recent study demonstrated that HNE treatment of SH-SY5Y cell culture induced the abnormal expression of apoptotic markers (p53, Bax and caspase-3) and led to neuronal cell death [162].
As previously mentioned, AD pathogenesis is triggered by the progressive accumulation of the amyloid-β peptide in the form of extracellular amyloid plaques in human brain. Aβ results from a cell surface receptor and transmembrane precursor amyloid protein (APP). Within lipid rafts, APP cleaved by β-secretase leads to a generation of a membrane bound carboxyl (C)-terminal fragment (CTF-β). CTF-β needs to be cleaved by γ-secretase to form Aβ and the amyloid precursor protein intracellular domain (AICD). Then Aβ molecules self-aggregate into soluble oligomers. It was discovered that the levels of this soluble fibrillar oligomers were increased in the brains of AD patients and correlated with the disease [163]. Moreover, Aβ oligomers were found in both intracellular and extracellular species [164-166]. Thus, Aβ oligomers can contribute to AD pathology via different mechanisms, including the induction of neurotoxicity, the formation of insoluble fibrillar amyloid-β aggregates, and the facilitation of tau pathology [167, 168]. Tau is the microtubule-associated protein (MAP), which stabilizes neuronal microtubules and is located, mainly, in the axons of neurons in central nervous system. An abnormal hyperphosphorylation of tau protein in brain leads to the generation of neurofibrillary tangles (NFTs) known as a hallmark of Alzheimer’s disease [169, 170]. It has been shown that HNE bound directly to normal tau and induced the tau Alz50 epitope involved in hyperphosphorylation of tau protein and neurofibrillary tangle formation in Alzheimer’s disease [171, 172]. A study by Liu and co-authors confirmed these findings to show that an antibody against neurofibrillary tangles recognized tau in brains of AD patients more effectively after HNE-treatment, but only when tau was in the phosphorylated state [173]. The immunocytochemical study previously showed that HNE treatment of cultured rat hippocampal neurons caused a moderate increase in the basal levels of tau phosphorylation, and prevented tau dephosphorylation [174].
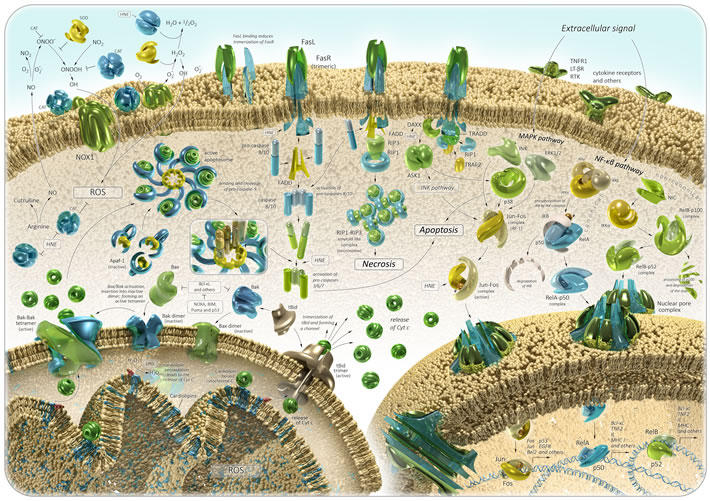
Figure 3: Oxidative stress in aging and cancer: signaling pathways. See text for details.
OXIDATION PROTEIN-ADDUCTS
Many studies have reported that HNE- and MDA-protein adducts were associated with Alzheimer’s disease progression. They show that Aβ oligomers can insert into the cell membranes and promote lipid peroxidation. This led to generation of by-products of lipid peroxidation such as MDA and HNE. Moreover, in brain tissue, lipid peroxidation-derived aldehydes can be also formed in many ways mentioned above. Then MDA and HNE can react and covalently modify many critical proteins such as amyloid-β peptide, collapsing response mediator protein 2 (CRMP2), neuronal glucose transporter 3 (GLUT3), neuropolypeptide h3, carbonyl reductase (NADPH), lactate dehydrogenase B (LDHB), heat shock protein 70 (HSP70), elongation factor Tu (EF-Tu), elongation factor 1 alpha (eIF-α), and manganese superoxide dismutase (MnSOD). It promotes neuronal cell impairment and Alzheimer’s disease pathogenesis [175-178]. For example, the immediate reaction of HNE with the Aβ peptide leads to the formation of more toxic diffusible Aβ-oligomers and insoluble aggregates [179]. The HNE-amyloid-β peptide adducts have an increased affinity for lipid membranes and tendency to form amyloid fibrils. Thus, the stimulation of lipid peroxidation by Aβ results in its own modification and accelerates amyloidogenesis [180, 181].
Elevated levels of several HNE-modified proteins of energy metabolism, including alpha-enolase (ENO1), phosphoglycerate kinase 1 (PGK1), pyruvate kinase (PK), mitochondrial ATP synthase α chain (ATP5A), malate dehydrogenase (MDH) and triosephosphate isomerase (TPI), were detected in mild cognitive impairment (MCI) patients associated with a risk for Alzheimer’s disease. Some of these oxidatively modified proteins are enzymes involved in glycolysis. These are ENO1, PGK1, TPI, and PK.
It is well known that brain is one of the greatest consumers of glucose, and glycolysis is required for normal functions of one. Alpha-enolase is a housekeeping enzyme, which catalyzes the hydrolysis dehydration of 2-phospho-D-glycerate (2-PGA) to phosphoenolpyruvate (PEP) in the penultimate step of glycolysis in cytoplasm. Many studies demonstrate that ENO1 is subjected to oxidative modification, which can be accompanied by decreasing its activity in different pathological conditions such as aging [182], Alzheimer’s and Parkinson’s diseases [183-186], Huntington’s disease [187], and cancer [188, 189]. However, ENO1 does not directly affect ATP production in spite of its glycolytic function. Actually, recent studies reported that ENO1 has also a lot of non-glycolytic functions [190-194] and might be involved in more than just metabolic processing of glucose [195].
Phosphoglycerate kinase 1 is an enzyme of the glycolytic pathway regulated by hypoxia-inducible factor-1α (HIF-1α). It catalyzes the conversion of 1,3-biphosphoglycerate (1,3-BPG) to 3-phosphoglycerate (3PG) in glycolysis. HNE can both react directly with PGK1 to form HNE-PGK1-adducts and promote a decrease in PGK1 expression [196]. The altered expression and conformation of PGK1 is correlated with cellular senescence and cancer [197-200]. Furthermore, a recent study has revealed that oxidative damage of PGK1 was markedly increased in aged human frontal cortex in progressive supranuclear palsy [201]. Decreased levels of PGK1 were detected even in the hippocampus of aged rats under beneficial effects of caloric restriction [202].
Pyruvate kinase catalyzes the last step of glycolysis leading to the generation of ATP and pyruvate. PK has a key role for energy homeostasis in brain tissues [203]. It has been revealed that the increasing activity of pyruvate kinase was followed by enhancement of glucose-dependence of brains in aged rats [204]. At the same time, reduction of pyruvate kinase activity, mediated by free radicals, was found in rat cerebrum [205]. Pyruvate kinase could also submit Aβ-induced oxidative modifications in the process of AD pathology [206]. In tumor cells, PK is converted to a less active dimer form PKM2, which is a major regulator of cancer metabolism. Moreover, PKM2 is associated with caspase-independent cell death [207]. This is confirmed by the fact that, in cancer cells, caspases can downregulate primary regulators of pyruvate kinase activity, particularly, phosphoserine [208].
Triosephosphate isomerase is an essential enzyme for glycolysis and gluconeogenesis, which catalyzes the interconvertible isomerization of glyceraldehyde 3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP) [209]. TPI deficiency is a severe glycolytic defect that contributes to progressive neurological dysfunction [210]. Actually, it was demonstrated that the inhibition of TPI might lead to neurodegeneration [211]. Lower TPI activity is detected in aged senescence-accelerated mice brain models, which show early cognitive impairment [212]. Also, modified TPI interacts with tau protein, inducing an intraneuronal aggregation, thereby contributing to the acceleration of AD progression [213]. Conversely, tau can have an effect on TPI, triggering its functional loss and subsequently facilitating neurodegenerative disease development [214]. Many studies elucidated that TPI was also involved in tumorigenesis and anti-drug resistance of cancer cells [215-217].
There are two isoenzymes of malate dehydrogenase: mitochondrial and cytoplasmic ones. Mitochondrial MDH catalyzes the reaction of reversible malate oxidation to oxaloacetate using the reduction of NAD+ to NADH. Conversely, cytoplasmic MDA reduces oxaloacetate to malate, oxidizing NADH to NAD+. Then malate enters into mitochondria from cytoplasm where it can be utilized by mitochondrial MDH. These reactions are components of the TCA cycle and gluconeogenesis from pyruvate. It is known that a large part of glucose molecules utilized by the brain is produced via gluconeogenesis. The altered activity of MDA is associated with Parkinson’s disease [218], Alzheimer’s disease [219], schizophrenia [220], and cancer [221, 222].
Mitochondrial ATP synthase α chain plays a crucial role in the activity of the entire electron transport chain and subsequently in ATP production. Thus, a failure of ATP5A leads to the loss of the whole ATP synthase (complex V) activity. This event, coupled with the changes in complex I, III, and IV, may result in impairment of mitochondrial ATP production, the leakage of electrons from their carrier molecules, and further ROS generation. Schägger and Ohm previously reported ATP-synthase deficiencies in Alzheimer’s disease [223]. In addition, decreased activity of ATP-synthase was detected in brains of patients with late-stage AD [224].
These results confirm the contribution of mitochondrial dysfunction to AD progression [176]. Moreover, the altered activity of energy metabolism enzymes on the whole is one of general stages in the progression of age-related diseases and cancer.
Oxidative DNA damage
Oxidative DNA damage appears to be critical for aging, age-related diseases and cancer [225]. ROS and products of lipid peroxidation can have an effect on both genomic and mitochondrial DNA, leading to various types of DNA damage: double- and single-strand breaks, intra- and interstrand DNA crosslinks, DNA-adduct formation, DNA base and deoxyribose modifications. Subsequently, replication of damaged DNA before repairing results in DNA mutations and genomic instability [226]. The DNA double-strand breaks (DSBs) are the most dangerous impairment. They cause severe genetic mutations leading to various disorders and tumor progression [227-229]. The single-strand breaks (SSBs) are less harmful for cells if they are repaired in time. If they are not repaired rapidly, chromosomal SSBs also result in serious lesions and may contribute to many human diseases [230]. Moreover, DNA replication of SSBs can potentially lead to DSBs formation. It has been shown that a transient increase in DSBs may induce Aβ-derived DNA damage caused by a synaptic dysfunction and being involved in the pathogenesis of Alzheimer’s disease [231-233]. In addition, larger numbers of SSBs and DSBs were observed in the brains of PD patients [234].
8-oxoGuanine (8-OHG) is one from the multiple oxidation products generated in DNA through dG oxidation. 8-OHG can join erroneously to adenine to make G-T and C-A replacements in genome. The nucleoside form of 8-OHG is 8-oxo-2’-deoxyguanosine (8-OHdG), which has been proposed as an indicator of oxidative DNA damage in vivo and in vitro [235, 236]. 8-OHdG further can be subjected to keto-enol tautomerism to favor the oxidized product 8-oxo-7,8-dihydro-2-deoxyguanosine (8-oxodG), which is also commonly used as a marker of oxidative DNA damage [237, 238]. Many studies reported a direct correlation between 8-OHG formation and carcinogenesis [239]. Altered levels of 8-OHG/8-OHdG demonstrated an association with pathogenesis of Alzheimer’s disease, amyotrophic lateral sclerosis, Down’s syndrome, Parkinson’s disease, normal aging, and cancer [237, 240-248]. For example, Ames and colleagues have shown the age-dependent accumulation of 8-OHdG in DNA from various rat organs [249]. Increased levels of 8-OHdG and OH8Gua, one more marker of oxidative DNA damage, in senescent human diploid fibroblast were shown [250]. DNA adducts derived from dC oxidation, 2’-deoxycytidine (dC)-5-hydroxy-2’-deoxycytidine (OH5dC), 5-hydroxy-2’-deoxyuridine (OH5dU), and 5,6-dihydroxy-5,6-dihydro-2’-deoxyuridine (dUg), were also detected in organs of different aged rats at levels similar to those of 8-OHdG [251]. It may contribute to spontaneous mutagenesis, leading to cancer and aging. A study of 5,6-hydroxy-5,6-dihydrothymine/dihydrothymidine (dTg), a product of dT oxidation, in mouse and monkey urine revealed a correlation between the specific metabolic rate of a species and the urinary outputs of dTg. The urinary dTg levels from mice were higher than those from monkey [252]. These data demonstrated that increased metabolic rate and oxidative DNA damage were associated with the shorter life span typical for smaller mammals. Thus, the presence of DNA oxidation products may be related to a higher oxidative stress and lower level of some antioxidants. In this case, there is a higher level of DNA lesions, which are not completely repaired.
Mitochondria generate large amounts of ROS directly exposing mtDNA to oxidative stress. The levels of oxidative mtDNA damage are more than a half higher and more extensive compared to nuclear DNA [235]. mtDNA lacks “protective” histones and has a limited repertoire of available DNA repair pathways, therefore it is very sensitive to oxidative damage. There is a “vicious cycle theory of mitochondrial ROS production”. It consists in the following: mtDNA damage results in mitochondrial dysfunction leading to an increase in ROS production, which subsequently elevates the accumulation rate of mtDNA mutations, which will further impair respiratory chain function [253]. However, this theory is still being discussed in the field of research on aging, cancer, and oxidative stress [254-259].
There is an evidence that the accumulation of oxidative mitochondrial DNA damage during normal aging is a risk factor for the development of age-associated neurodegenerative disorders [260]. It has been shown that the frequency of point mtDNA mutations increased approximately 5-fold during an 80-year lifespan [261]. The accumulation of somatic mtDNA mutations was demonstrated to be a feature of accelerated aging in knock-in mice, expressing a proofreading-deficient version of the mitochondrial DNA polymerase G (POLG) and to promote apoptosis [262]. Another similar study confirmed these data and showed a causative link between mtDNA mutations and aging phenotypes [263]. The mtDNA damage, correlated with elevated mitochondrial ROS formation, was demonstrated to significantly contribute to age-dependent endothelial dysfunction in vessels [264]. Aliev and co-authors showed that mtDNA deletions were accompanied by increased levels of APP, 8-OHG and cytochrome c oxidase (COX), and correlated with endothelial lesions in vessels [265]. These findings explain the fact that aging-related impairment of cerebral perfusion results in brain hypoperfusion, which contributes to the development of AD and consequently to neurodegeneration [266]. Increased of mtDNA damage and 8-OHG levels, associated with reduced mtDNA content, were observed in AD brains [267-270]. The age-related increase in mtDNA damage was shown in patients with sporadic Parkinson’s disease [271]. Greater accumulation of mtDNA deletions was detected in the dopaminergic neurons of substantia nigra in old rats compared to young ones [272].
In recemt years, several mutations and depletions of mtDNA were identified in different types of cancers. Mutations associated with the development of tumors were found to be present in both the non-coding and coding regions of mtDNA in patients with leukemia and various types of carcinoma [273, 274]. However, it is unclear whether the mitochondrial dysfunction is a cause or a consequence of cancer. A recent study of a cohort including 311 individuals with mitochondrial dysfunction (90% maternally inherited mtDNA mutation) has not shown an increased risk of cancer compared with the general population. However, these results do not contradict the hypothesis that secondary mtDNA alterations are formed during tumorigenesis, which can play an essential role in the further malignant transformation [275-277].
MITOCHONDRIAL HORMESIS
The MFRTA suggests that oxidative damage is accumulated with age and drives the aging process. There is a linear dose-response relationship between the increasing amounts of ROS and the oxidative stress. However, several lines of research demonstrated the potential beneficial role of ROS as redox signaling molecules. Mitochondria are fully integrated into the cell, and any significant deficiency in mitochondrial function may trigger an adaptive nuclear response, thereby altering nuclear gene expression [278]. Thus, it has been reported that low doses of ROS exposure decreased mortality and increased stress resistance, while higher doses exerted opposite effects [279]. Moreover, the effect of antioxidants on the mitochondrial ROS signal impairs the general health and prevents the extension of lifespan [280, 281]. ROS is termed mitochondrial hormesis or mitohormesis [282]. It has been detected that mitohormesis extended the lifespan in many model organisms, including Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans and mice [278, 280]. The concept of mitohormesis bases on the physiological effects of calorie and glucose restriction, reduction of specific macronutrients, and physical exercises, which are required to promote health and longevity, and the role of ROS as essential signaling molecules in these processes.
It was found that ROS influenced stress resistance and lifespan through several transcription factors, such as FOXO/DAF-16, NRF2/SKN1, and HSF-1 [279]. For instance, NF-E2-Related Factor 2 (NRF2), which is activated by ROS, can bind with the antioxidants responsive elements (AREs) and mediate mitohormesis [283, 284]. Other transcription factors, such as Forkhead transcription factors (FOXOs) and heat shock factor 1 (HSF-1), activate many genes involved in cellular stress response. HSF-1 is a major repressor of heat shock genes, which encode proteins rapidly induced after temperature stress. FOXOs regulate the transcription of superoxide dismutase and catalase genes, which encode the enzymes involved in detoxification of ROS [285, 286]. Several studies show that the mechanisms underlying extended lifespan are dependent on the AMP-activated protein kinase (AMPK) [281, 287-290]. AMPK is a cellular energy sensor, which is activated by metabolic stress. AMPK upregulates the activities of many key metabolic enzymes, thereby, compensates for the energy deficit and increases the oxidative stress resistance and survival rates [287]. It is suggested that the extension of lifespan requires activation of p38 MAP kinase, which can induce ROS formation [291, 292]. The impairment of the mTOR pathway is shown to extend the lifespan in various organisms [293-295]. The impairment of insulin/IGF-1 signaling prolongs the lifespan of mice [296]. HIF-1 responds to hypoxia by activating the transcription of many genes. It is shown that HIF-1 can down-regulate the mitochondrial activity and be responsible for lifespan extending through RNAi-mediated knockdown of several mitochondrial proteins [279, 297, 298].
It should be noted that the longevity is closely related to an increased risk of cancer and neurodegenerative disorder incidences. Thus, it is not surprising that the molecular mechanism underlying mitohormesis can be associated with age-related diseases and cancer. In support of this, many studies showed the critical role of Nrf2, HIF-1, and p38 MAP kinase in progression of neurodegenerative disease and cancer, in which oxidative stress is closely implicated [299-312].
Cellular senescence and tumor suppression
The cellular senescence has been first described by Hayflick and colleagues more than 40 years ago [313, 314]. They demonstrated that normal diploid cells had a limited replicative potential. In contrast to cancer cells, normal cells at the end of their replicative life span are in a process known as cellular senescence which is characterized by irreversible cell cycle arrest, morphological changes, epigenetic modifications, lack of response to growth factors, telomere shortening and dysfunction, sustained metabolic activity and elevated DNA damage [315]. Further, cellular senescence has been supposed to be a tumor-suppressor mechanism [316]. Many studies showed the activation of the negative growth regulatory genes and proteins, which inhibit the initiation of DNA synthesis, during senescence. For example, p53 and Rb, the tumor suppressors, were shown to play a role in the regulation of cellular senescence [317]. Inactivation of p53 and Rb genes contributed to extend the proliferative lifespan of normal fibroblasts [318-320]. However, it is not sufficient for malignant transformation. Studies by Cairns revealed that at least four or five mutations were required for the transformation from normal to cancer cells [321, 322]. Using a genetic model of colorectal tumorigenesis, Vogelstein and collaborators also proposed at least four genetic alterations that could underlie tumor development [323]. The other growth inhibitory genes such as p21, p16, p33, p19, and p27 were also regarded as key effectors of cellular senescence [317, 324].
Ras and Myc oncogenes have been shown to be involved in the regulation of senescence-inducing pathways. Serrano and colleagues in 1997 demonstrated that oncogenic transformation of human diploid and mouse embryo fibroblasts by RAS resulted in a permanent cell-cycle arrest, simultaneous induction of the p53 and p16 tumor suppressor proteins, and cellular senescence. They found that oncogenic transformation of the cells by RAS required either a cooperating oncogene or the inactivation of tumor suppressors. However, escape from RAS-induced arrest by disruption of p53 or p16/Rb pathways may lead to cell transformation [325]. In contrast to RAS, Myc overexpression resulted in apoptosis in primary cells. This effect could be mediated by activation of p19ARF/p53 and p14ARF/E2F-1 pathways [326, 327]. The expression of other oncoproteins, such as Raf and MEK, also induces cell cycle arrest and activates mediators of senescence (p53, p21Cip1, and p16Ink4a) in human fibroblasts [328-330]. Jacobs and co-authors showed that overexpression of Bmi-1 oncogene induced primary mouse fibroblast immortalization and downregulated expression of the tumor suppressors p16 and p19Arf. Bmi-1-deficient mouse fibroblasts, in turn, showed an increased expression of p16 and p19Arf and premature senescence phenotype [331].
Telomere shortening is one of the major mechanisms inducing cellular senescence and inhibiting tumorigenesis [332, 333]. However, it can trigger not only senescence response but telomere crisis as well. Progressive telomere shortening and a loss of tumor suppressor function result in a massive chromosomal instability, secondary genetic alterations and facilitate carcinogenesis [334, 335]. For example, a study of a telomerase-knockout mouse, heterozygous for mutant p53, revealed that a loss of telomere function and the consequent genomic instability could cooperate with p53 deficiency and promote tumorigenic initiation [336]. In addition, a series of studies revealed the relative roles of senescence and apoptosis induced by telomere dysfunction and p53 activation in tumor suppression [337].
mTOR pathway is involved in both senescent phenotype and cancer, which have been extensively studied by Blagosklonny and colleagues. mTOR pathway drives the process of conversion from proliferative arrest to irreversible senescence and is involved in longevity [338, 339]. On the other hand, it is activated by mutations in oncogenes such as Raf, Ras, and PI3K and inhibition of many tumor suppressor genes, including p53. However, cells with TOR-activating oncogenes are required in deactivation of cell cycle checkpoints to proliferate [340].
Senescing cells are characterized by persistent DNA-damage response (DDR) signaling, which could be induced by mitochondrial dysfunction and oxidative stress. It detects DNA lesions, signals the presence of genomic DNA damage, and promotes their repair [341]. Indeed, DDR was recognized as an anticancer mechanism leading to cell cycle arrest followed by cellular senescence or apoptosis [342, 343]. Markers of DDR, such as overexpression of p21, the activation of checkpoint kinases ATM and Chk2, the phosphorylation of histone H2AX, p53 accumulation or phosphorylation, commonly occur at the early stages of human tumors and precursor lesions [344-346]. These data indicate that, at an early stage of tumorigenesis, cells activate DDR to delay or prevent cancer. Thus, DDR can be considered yet another mechanism of tumor suppression that could be triggered by oxidative stress and in which senescence is involved.
Data from several sources show that the progression of cancer is also slower in aged individuals, and metastases are often less frequent [347, 348]. The loss in genome “plasticity” during the development and aging is a possible explanation for this decline [349]. The deregulation of chromosome recombination potential predisposes to malignant transformation, but there is a decline in it with aging. Thus, during senescence, the loss in “plasticity” protects from deviations in cell proliferation, slowing down tumor growth and metastasis in the old. Moreover, cellular senescence is also mentioned as a mechanism of reversion of tumor cells to normality [350]. In tumor cell population, there are nondividing cells with growth arrest also termed senescence [351].
Normal cells have a genetically programmed time limit for cell replication and aim to avoid excessive proliferation. There is no doubt that senescence is one of the major defense mechanisms against malignant transformation. However, it was shown that under some conditions, normal cells can “escape senescence”. The treatment of primary cells with viral oncogenes results in either death of the cells or apoptosis. At a certain frequency, cells can become immortal [352]. In vivo, during senescence, a lot of alterations, including oncogenic ones, can be accumulated. Moreover, senescent cells secrete factors that can promote other cell growth and tumorigenesis [353]. Thus, despite senescence is recognized as a potent tumor-suppressor mechanism it remains a risk factor for cancer.
ConclusionS
There is much evidence that the normal aging and carcinogenesis are multistep processes, which can be induced by ROS (Figure 4). Mitochondria generate ROS during normal metabolism. ROS, in turn, may have an effect on many important intracellular components. For example, they attack the mitochondrial membranes and mtDNA directly near the place of their formation. This leads to mitochondrial dysfunctions and more ROS production. Furthermore, ROS interacts extensively with nuclear DNA and proteins, leading to DNA damage and protein-adduct formation. ROS also interacts with lipids of cell membranes to disturb their functions. Actually, large amounts of DNA mutations or arrangements, genomic instability, impairment of protein functions and altered metabolic and signal pathways, induced by oxidative stress, have been found in cells subjected to pathological conditions, such as aging or malignant transformation. It has been demonstrated that age-related neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases and different types of cancers, have similar disturbances. The energy metabolism is altered in both neurodegenerative disorders and tumors. However, in the former case, this leads to nerve cell death. In the latter case, such alteration results in tumorigenesis.
Thus, we suggest that mitochondrial dysfunction and oxidative damage inevitably occur in normal aging and can lead to age-related neurodegenerative disorders. Under various conditions, these processes may be a risk factor for cancer.
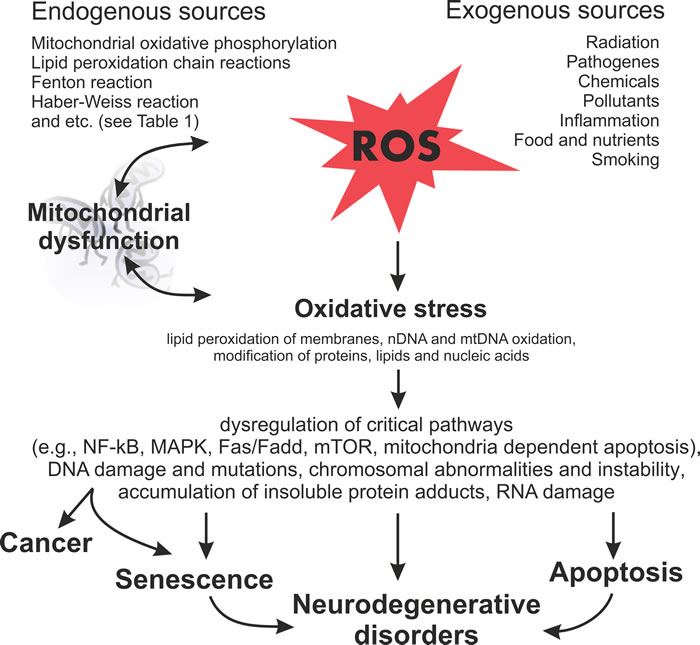
Figure 4: Schematic diagram illustrating the harmful effects of ROS on the cellular processes and subsequent outcomes.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was financially supported by the Russian Foundation for Basic Research (projects 16-04-01521a and 15-34-70035 mol_a_mos), grant МК-8047.2016.4 of the President of the Russian Federation, the grant from RAS Presidium Program “Molecular and Cellular Biology” (for AVK), and ICGEB project CRP/RUS15-01.
References
1. Ernster L and Schatz G. Mitochondria: a historical review. The Journal of cell biology. 1981; 91:227s-255s.
2. Wanders RJ, Ruiter JP, L IJ, Waterham HR and Houten SM. The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. Journal of inherited metabolic disease. 2010; 33:479-494.
3. Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J, Liu J, Pan X and Murphy E. The ins and outs of mitochondrial calcium. Circulation research. 2015; 116:1810-1819.
4. Guda P, Guda C and Subramaniam S. Reconstruction of pathways associated with amino acid metabolism in human mitochondria. Genomics, proteomics & bioinformatics. 2007; 5:166-176.
5. Atamna H. Heme, iron, and the mitochondrial decay of ageing. Ageing research reviews. 2004; 3:303-318.
6. Paul VD and Lill R. Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochimica et biophysica acta. 2015; 1853:1528-1539.
7. Kroemer G, Galluzzi L and Brenner C. Mitochondrial membrane permeabilization in cell death. Physiological reviews. 2007; 87:99-163.
8. Tait SW and Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature reviews Molecular cell biology. 2010; 11:621-632.
9. Jiang X and Wang X. Cytochrome C-mediated apoptosis. Annual review of biochemistry. 2004; 73:87-106.
10. Rossier MF. T channels and steroid biosynthesis: in search of a link with mitochondria. Cell calcium. 2006; 40:155-164.
11. Yager JD and Chen JQ. Mitochondrial estrogen receptors—new insights into specific functions. Trends in endocrinology and metabolism: TEM. 2007; 18:89-91.
12. Alvarez-Delgado C, Mendoza-Rodriguez CA, Picazo O and Cerbon M. Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V. Experimental gerontology. 2010; 45:580-585.
13. Gellerich FN, Trumbeckaite S, Opalka JR, Seppet E, Rasmussen HN, Neuhoff C and Zierz S. Function of the mitochondrial outer membrane as a diffusion barrier in health and diseases. Biochemical Society transactions. 2000; 28:164-169.
14. Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochimica et biophysica acta. 2006; 1763:542-548.
15. Vogel F, Bornhovd C, Neupert W and Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. The Journal of cell biology. 2006; 175:237-247.
16. Zick M, Rabl R and Reichert AS. Cristae formation-linking ultrastructure and function of mitochondria. Biochimica et biophysica acta. 2009; 1793:5-19.
17. Bogenhagen D and Clayton DA. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell. 1977; 11:719-727.
18. Park CB and Larsson NG. Mitochondrial DNA mutations in disease and aging. The Journal of cell biology. 2011; 193:809-818.
19. Chinnery PF and Hudson G. Mitochondrial genetics. British medical bulletin. 2013; 106:135-159.
20. Muller-Hocker J, Horvath R, Schafer S, Hessel H, Muller-Felber W, Kuhr J, Copeland WC and Seibel P. Mitochondrial DNA depletion and fatal infantile hepatic failure due to mutations in the mitochondrial polymerase gamma (POLG) gene: a combined morphological/enzyme histochemical and immunocytochemical/biochemical and molecular genetic study. Journal of cellular and molecular medicine. 2011; 15:445-456.
21. Zhang H, Meng LH and Pommier Y. Mitochondrial topoisomerases and alternative splicing of the human TOP1mt gene. Biochimie. 2007; 89:474-481.
22. Youle RJ and van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012; 337:1062-1065.
23. Arakaki N, Nishihama T, Kohda A, Owaki H, Kuramoto Y, Abe R, Kita T, Suenaga M, Himeda T, Kuwajima M, Shibata H and Higuti T. Regulation of mitochondrial morphology and cell survival by Mitogenin I and mitochondrial single-stranded DNA binding protein. Biochimica et biophysica acta. 2006; 1760:1364-1372.
24. Suenaga M. [Study on the novel factors regulating mitochondrial dynamics]. Yakugaku zasshi : Journal of the Pharmaceutical Society of Japan. 2010; 130:1543-1547.
25. Huang CY, Chiang SF, Lin TY, Chiou SH and Chow KC. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PloS one. 2012; 7:e33657.
26. Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of gerontology. 1956; 11:298-300.
27. Mills GC. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. The Journal of biological chemistry. 1957; 229:189-197.
28. Mills GC. The purification and properties of glutathione peroxidase of erythrocytes. The Journal of biological chemistry. 1959; 234:502-506.
29. Mills GC. Glutathione peroxidase and the destruction of hydrogen peroxide in animal tissues. Archives of biochemistry and biophysics. 1960; 86:1-5.
30. McCord JM and Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). The Journal of biological chemistry. 1969; 244:6049-6055.
31. Jensen PK. Antimycin-insensitive oxidation of succinate and reduced nicotinamide-adenine dinucleotide in electron-transport particles. I. pH dependency and hydrogen peroxide formation. Biochimica et biophysica acta. 1966; 122:157-166.
32. Boveris A and Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. The Biochemical journal. 1973; 134:707-716.
33. Loschen G and Azzi A. On the formation of hydrogen peroxide and oxygen radicals in heart mitochondria. Recent advances in studies on cardiac structure and metabolism. 1975; 7:3-12.
34. Boveris A, Oshino N and Chance B. The cellular production of hydrogen peroxide. The Biochemical journal. 1972; 128:617-630.
35. Loschen G and Azzi A. Dibromothymoquinone: a new inhibitor of mitochondrial electron transport at the level of ubiquinone. FEBS letters. 1974; 41:115-117.
36. Loschen G, Azzi A and Flohe L. Mitochondrial H2O2 formation: relationship with energy conservation. FEBS letters. 1973; 33:84-87.
37. Sorgato MC, Sartorelli L, Loschen G and Azzi A. Oxygen radicals and hydrogen peroxide in rat brain mitochondria. FEBS letters. 1974; 45:92-95.
38. Chance B, Sies H and Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological reviews. 1979; 59:527-605.
39. Boveris A. Mitochondrial production of hydrogen peroxide in Saccharomyces cerevisiae. Acta physiologica latino americana. 1976; 26:303-309.
40. Harman D. The biologic clock: the mitochondria? Journal of the American Geriatrics Society. 1972; 20:145-147.
41. Ray PD, Huang BW and Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling. 2012; 24:981-990.
42. Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. Journal of bioenergetics and biomembranes. 1999; 31:347-366.
43. St-Pierre J, Buckingham JA, Roebuck SJ and Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. The Journal of biological chemistry. 2002; 277:44784-44790.
44. Raha S and Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends in biochemical sciences. 2000; 25:502-508.
45. Rus DA, Sastre J, Vina J and Pallardo FV. Induction of mitochondrial xanthine oxidase activity during apoptosis in the rat mammary gland. Frontiers in bioscience : a journal and virtual library. 2007; 12:1184-1189.
46. Li X, Fang P, Mai J, Choi ET, Wang H and Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. Journal of hematology & oncology. 2013; 6:19.
47. Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009; 417:1-13.
48. Forstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nature clinical practice Cardiovascular medicine. 2008; 5:338-349.
49. Loesch A, Belai A and Burnstock G. An ultrastructural study of NADPH-diaphorase and nitric oxide synthase in the perivascular nerves and vascular endothelium of the rat basilar artery. Journal of neurocytology. 1994; 23:49-59.
50. Tatoyan A and Giulivi C. Purification and characterization of a nitric-oxide synthase from rat liver mitochondria. The Journal of biological chemistry. 1998; 273:11044-11048.
51. Giulivi C, Poderoso JJ and Boveris A. Production of nitric oxide by mitochondria. The Journal of biological chemistry. 1998; 273:11038-11043.
52. Lacza Z, Snipes JA, Zhang J, Horvath EM, Figueroa JP, Szabo C and Busija DW. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free radical biology & medicine. 2003; 35:1217-1228.
53. Lacza Z, Horn TF, Snipes JA, Zhang J, Roychowdhury S, Horvath EM, Figueroa JP, Kollai M, Szabo C and Busija DW. Lack of mitochondrial nitric oxide production in the mouse brain. Journal of neurochemistry. 2004; 90:942-951.
54. Lacza Z, Pankotai E, Csordas A, Gero D, Kiss L, Horvath EM, Kollai M, Busija DW and Szabo C. Mitochondrial NO and reactive nitrogen species production: does mtNOS exist? Nitric oxide. 2006; 14:162-168.
55. Girotti AW. Mechanisms of lipid peroxidation. Journal of free radicals in biology & medicine. 1985; 1:87-95.
56. Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. Journal of lipid research. 1998; 39:1529-1542.
57. Esterbauer H, Schaur RJ and Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free radical biology & medicine. 1991; 11:81-128.
58. Michiels C and Remacle J. Cytotoxicity of linoleic acid peroxide, malondialdehyde and 4-hydroxynonenal towards human fibroblasts. Toxicology. 1991; 66:225-234.
59. Esterbauer H, Benedetti A, Lang J, Fulceri R, Fauler G and Comporti M. Studies on the mechanism of formation of 4-hydroxynonenal during microsomal lipid peroxidation. Biochimica et biophysica acta. 1986; 876:154-166.
60. Spiteller P, Kern W, Reiner J and Spiteller G. Aldehydic lipid peroxidation products derived from linoleic acid. Biochimica et biophysica acta. 2001; 1531:188-208.
61. Griesser M, Boeglin WE, Suzuki T and Schneider C. Convergence of the 5-LOX and COX-2 pathways: heme-catalyzed cleavage of the 5S-HETE-derived di-endoperoxide into aldehyde fragments. Journal of lipid research. 2009; 50:2455-2462.
62. Negre-Salvayre A, Coatrieux C, Ingueneau C and Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. British journal of pharmacology. 2008; 153:6-20.
63. Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, Chapple S, Cohen G, Feher J, Grune T, Lengyel G, Mann GE, Pamplona R, et al. Pathological aspects of lipid peroxidation. Free radical research. 2010; 44:1125-1171.
64. Esterbauer H, Eckl P and Ortner A. Possible mutagens derived from lipids and lipid precursors. Mutation research. 1990; 238:223-233.
65. Shoeb M, Ansari NH, Srivastava SK and Ramana KV. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Current medicinal chemistry. 2014; 21:230-237.
66. Barrera G, Pizzimenti S, Ciamporcero ES, Daga M, Ullio C, Arcaro A, Cetrangolo GP, Ferretti C, Dianzani C, Lepore A and Gentile F. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxidants & redox signaling. 2015; 22:1681-1702.
67. Raghavan S, Subramaniyam G and Shanmugam N. Proinflammatory effects of malondialdehyde in lymphocytes. Journal of leukocyte biology. 2012; 92:1055-1067.
68. Jomova K, Vondrakova D, Lawson M and Valko M. Metals, oxidative stress and neurodegenerative disorders. Molecular and cellular biochemistry. 2010; 345:91-104.
69. Zarkovic N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Molecular aspects of medicine. 2003; 24:281-291.
70. Jorgensen P, Milkovic L, Zarkovic N, Waeg G and Rattan SI. Lipid peroxidation-derived 4-hydroxynonenal-modified proteins accumulate in human facial skin fibroblasts during ageing in vitro. Biogerontology. 2014; 15:105-110.
71. Testa G, Gamba P, Di Scipio F, Sprio AE, Salamone P, Gargiulo S, Sottero B, Biasi F, Berta GN, Poli G and Leonarduzzi G. Potentiation of amyloid-beta peptide neurotoxicity in human dental-pulp neuron-like cells by the membrane lipid peroxidation product 4-hydroxynonenal. Free radical biology & medicine. 2012; 53:1708-1717.
72. Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G and Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. Journal of neurochemistry. 1997; 69:273-284.
73. Zhu X, Castellani RJ, Moreira PI, Aliev G, Shenk JC, Siedlak SL, Harris PL, Fujioka H, Sayre LM, Szweda PA, Szweda LI, Smith MA and Perry G. Hydroxynonenal-generated crosslinking fluorophore accumulation in Alzheimer disease reveals a dichotomy of protein turnover. Free radical biology & medicine. 2012; 52:699-704.
74. Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG and Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. Journal of neurochemistry. 1997; 68:2092-2097.
75. Dei R, Takeda A, Niwa H, Li M, Nakagomi Y, Watanabe M, Inagaki T, Washimi Y, Yasuda Y, Horie K, Miyata T and Sobue G. Lipid peroxidation and advanced glycation end products in the brain in normal aging and in Alzheimer’s disease. Acta neuropathologica. 2002; 104:113-122.
76. Molina JA, Jimenez-Jimenez FJ, Fernandez-Calle P, Lalinde L, Tenias JM, Pondal M, Vazquez A and Codoceo R. Serum lipid peroxides in patients with Parkinson’s disease. Neuroscience letters. 1992; 136:137-140.
77. Baillet A, Chanteperdrix V, Trocme C, Casez P, Garrel C and Besson G. The role of oxidative stress in amyotrophic lateral sclerosis and Parkinson’s disease. Neurochemical research. 2010; 35:1530-1537.
78. Sanyal J, Bandyopadhyay SK, Banerjee TK, Mukherjee SC, Chakraborty DP, Ray BC and Rao VR. Plasma levels of lipid peroxides in patients with Parkinson’s disease. European review for medical and pharmacological sciences. 2009; 13:129-132.
79. Gustaw-Rothenberg K, Kowalczuk K and Stryjecka-Zimmer M. Lipids’ peroxidation markers in Alzheimer’s disease and vascular dementia. Geriatrics & gerontology international. 2010; 10:161-166.
80. Padurariu M, Ciobica A, Hritcu L, Stoica B, Bild W and Stefanescu C. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neuroscience letters. 2010; 469:6-10.
81. Morel P, Tallineau C, Pontcharraud R, Piriou A and Huguet F. Effects of 4-hydroxynonenal, a lipid peroxidation product, on dopamine transport and Na+/K+ ATPase in rat striatal synaptosomes. Neurochemistry international. 1998; 33:531-540.
82. Shin Y, White BH, Uh M and Sidhu A. Modulation of D1-like dopamine receptor function by aldehydic products of lipid peroxidation. Brain research. 2003; 968:102-113.
83. Monroy CA, Doorn JA and Roman DL. Modification and functional inhibition of regulator of G-protein signaling 4 (RGS4) by 4-hydroxy-2-nonenal. Chemical research in toxicology. 2013; 26:1832-1839.
84. Selley ML. (E)-4-hydroxy-2-nonenal may be involved in the pathogenesis of Parkinson’s disease. Free radical biology & medicine. 1998; 25:169-174.
85. Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER and Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:2696-2701.
86. Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Agid Y, Lees A, Jenner P and Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. Journal of neurochemistry. 1989; 52:381-389.
87. Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova, II, Zhao Q, Zou M, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature chemical biology. 2005; 1:223-232.
88. Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C and Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell death and differentiation. 2006; 13:1423-1433.
89. Orrenius S and Zhivotovsky B. Cardiolipin oxidation sets cytochrome c free. Nature chemical biology. 2005; 1:188-189.
90. Petrosillo G, Ruggiero FM and Paradies G. Role of reactive oxygen species and cardiolipin in the release of cytochrome c from mitochondria. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003; 17:2202-2208.
91. Houtkooper RH and Vaz FM. Cardiolipin, the heart of mitochondrial metabolism. Cellular and molecular life sciences : CMLS. 2008; 65:2493-2506.
92. Ma ZA. The role of peroxidation of mitochondrial membrane phospholipids in pancreatic beta -cell failure. Current diabetes reviews. 2012; 8:69-75.
93. Reed JC, Jurgensmeier JM and Matsuyama S. Bcl-2 family proteins and mitochondria. Biochimica et biophysica acta. 1998; 1366:127-137.
94. Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM and Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007; 315:856-859.
95. Pagliari LJ, Kuwana T, Bonzon C, Newmeyer DD, Tu S, Beere HM and Green DR. The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:17975-17980.
96. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M and Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004; 303:1010-1014.
97. Lomonosova E and Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008; 27 Suppl 1:S2-19.
98. Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB and Korsmeyer SJ. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes & development. 2000; 14:2060-2071.
99. Brunelle JK and Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. Journal of cell science. 2009; 122:437-441.
100. Ji C, Amarnath V, Pietenpol JA and Marnett LJ. 4-hydroxynonenal induces apoptosis via caspase-3 activation and cytochrome c release. Chemical research in toxicology. 2001; 14:1090-1096.
101. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997; 91:479-489.
102. Liu X, Kim CN, Yang J, Jemmerson R and Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996; 86:147-157.
103. Ferraro E, Pulicati A, Cencioni MT, Cozzolino M, Navoni F, di Martino S, Nardacci R, Carri MT and Cecconi F. Apoptosome-deficient cells lose cytochrome c through proteasomal degradation but survive by autophagy-dependent glycolysis. Molecular biology of the cell. 2008; 19:3576-3588.
104. Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicology letters. 2004; 149:19-23.
105. Zhong H and Yin H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox biology. 2015; 4:193-199.
106. Zhong H, Lu J, Xia L, Zhu M and Yin H. Formation of electrophilic oxidation products from mitochondrial cardiolipin in vitro and in vivo in the context of apoptosis and atherosclerosis. Redox biology. 2014; 2:878-883.
107. Oberley TD, Toyokuni S and Szweda LI. Localization of hydroxynonenal protein adducts in normal human kidney and selected human kidney cancers. Free radical biology & medicine. 1999; 27:695-703.
108. Siddiqui MA, Kumar V, Kashyap MP, Agarwal M, Singh AK, Khanna VK, Al-Khedhairy AA, Musarrat J, Pant AB and Jahan S. Short-term exposure of 4-hydroxynonenal induces mitochondria-mediated apoptosis in PC12 cells. Human & experimental toxicology. 2012; 31:336-345.
109. Skrzydlewska E, Sulkowski S, Koda M, Zalewski B, Kanczuga-Koda L and Sulkowska M. Lipid peroxidation and antioxidant status in colorectal cancer. World journal of gastroenterology : WJG. 2005; 11:403-406.
110. Chang B, Nishikawa M, Nishiguchi S and Inoue M. L-carnitine inhibits hepatocarcinogenesis via protection of mitochondria. International journal of cancer Journal international du cancer. 2005; 113:719-729.
111. Ji GR, Yu NC, Xue X and Li ZG. 4-Hydroxy-2-nonenal induces apoptosis by inhibiting AKT signaling in human osteosarcoma cells. TheScientificWorldJournal. 2014; 2014:873525.
112. Li YP, Tian FG, Shi PC, Guo LY, Wu HM, Chen RQ and Xue JM. 4-Hydroxynonenal promotes growth and angiogenesis of breast cancer cells through HIF-1alpha stabilization. Asian Pacific journal of cancer prevention : APJCP. 2014; 15:10151-10156.
113. Karki K, Pande D, Negi R, Khanna S, Khanna RS and Khanna HD. Correlation of serum toll like receptor 9 and trace elements with lipid peroxidation in the patients of breast diseases. Journal of trace elements in medicine and biology : organ of the Society for Minerals and Trace Elements. 2015; 30:11-16.
114. Szychta P, Zadrozny M, Lewinski A and Karbownik-Lewinska M. Increased oxidative damage to membrane lipids following surgery for breast cancer. Neuro endocrinology letters. 2014; 35:602-607.
115. Kilic N, Yavuz Taslipinar M, Guney Y, Tekin E and Onuk E. An investigation into the serum thioredoxin, superoxide dismutase, malondialdehyde, and advanced oxidation protein products in patients with breast cancer. Annals of surgical oncology. 2014; 21:4139-4143.
116. Maia FM, Santos EB and Reis GE. Oxidative stress and plasma lipoproteins in cancer patients. Einstein. 2014; 12:480-484.
117. Herrera AC, Victorino VJ, Campos FC, Verenitach BD, Lemos LT, Aranome AM, Oliveira SR, Cecchini AL, Simao AN, Abdelhay E, Panis C and Cecchini R. Impact of tumor removal on the systemic oxidative profile of patients with breast cancer discloses lipid peroxidation at diagnosis as a putative marker of disease recurrence. Clinical breast cancer. 2014; 14:451-459.
118. do Val Carneiro JL, Nixdorf SL, Mantovani MS, da Silva do Amaral Herrera AC, Aoki MN, Amarante MK, Fabris BA, Pelegrinelli Fungaro MH and Ehara Watanabe MA. Plasma malondialdehyde levels and CXCR4 expression in peripheral blood cells of breast cancer patients. Journal of cancer research and clinical oncology. 2009; 135:997-1004.
119. Taysi S, Uslu C, Akcay F and Sutbeyaz MY. Malondialdehyde and nitric oxide levels in the plasma of patients with advanced laryngeal cancer. Surgery today. 2003; 33:651-654.
120. Didziapetriene J, Bublevic J, Smailyte G, Kazbariene B and Stukas R. Significance of blood serum catalase activity and malondialdehyde level for survival prognosis of ovarian cancer patients. Medicina. 2014; 50:204-208.
121. Chole RH, Patil RN, Basak A, Palandurkar K and Bhowate R. Estimation of serum malondialdehyde in oral cancer and precancer and its association with healthy individuals, gender, alcohol, and tobacco abuse. Journal of cancer research and therapeutics. 2010; 6:487-491.
122. Kosova F, Cetin B, Akinci M, Aslan S, Ari Z, Sepici A, Altan N and cetin A. Advanced oxidation protein products, ferrous oxidation in xylenol orange, and malondialdehyde levels in thyroid cancer. Annals of surgical oncology. 2007; 14:2616-2620.
123. Kaynar H, Meral M, Turhan H, Keles M, Celik G and Akcay F. Glutathione peroxidase, glutathione-S-transferase, catalase, xanthine oxidase, Cu-Zn superoxide dismutase activities, total glutathione, nitric oxide, and malondialdehyde levels in erythrocytes of patients with small cell and non-small cell lung cancer. Cancer letters. 2005; 227:133-139.
124. Kaur J, Politis C and Jacobs R. Salivary 8-hydroxy-2-deoxyguanosine, malondialdehyde, vitamin C, and vitamin E in oral pre-cancer and cancer: diagnostic value and free radical mechanism of action. Clinical oral investigations. 2015.
125. Bauer G. Targeting extracellular ROS signaling of tumor cells. Anticancer research. 2014; 34:1467-1482.
126. Bauer G and Zarkovic N. Revealing mechanisms of selective, concentration-dependent potentials of 4-hydroxy-2-nonenal to induce apoptosis in cancer cells through inactivation of membrane-associated catalase. Free radical biology & medicine. 2015; 81:128-144.
127. Herdener M, Heigold S, Saran M and Bauer G. Target cell-derived superoxide anions cause efficiency and selectivity of intercellular induction of apoptosis. Free radical biology & medicine. 2000; 29:1260-1271.
128. Heigold S, Sers C, Bechtel W, Ivanovas B, Schafer R and Bauer G. Nitric oxide mediates apoptosis induction selectively in transformed fibroblasts compared to nontransformed fibroblasts. Carcinogenesis. 2002; 23:929-941.
129. Bauer G. Tumor cell-protective catalase as a novel target for rational therapeutic approaches based on specific intercellular ROS signaling. Anticancer research. 2012; 32:2599-2624.
130. Bauer G. HOCl-dependent singlet oxygen and hydroxyl radical generation modulate and induce apoptosis of malignant cells. Anticancer research. 2013; 33:3589-3602.
131. Rahman K. Studies on free radicals, antioxidants, and co-factors. Clinical interventions in aging. 2007; 2:219-236.
132. West JD, Ji C, Duncan ST, Amarnath V, Schneider C, Rizzo CJ, Brash AR and Marnett LJ. Induction of apoptosis in colorectal carcinoma cells treated with 4-hydroxy-2-nonenal and structurally related aldehydic products of lipid peroxidation. Chemical research in toxicology. 2004; 17:453-462.
133. Salminen A, Kauppinen A and Kaarniranta K. Emerging role of NF-kappaB signaling in the induction of senescence-associated secretory phenotype (SASP). Cellular signalling. 2012; 24:835-845.
134. Salminen A and Kaarniranta K. NF-kappaB signaling in the aging process. Journal of clinical immunology. 2009; 29:397-405.
135. Xia Y, Shen S and Verma IM. NF-kappaB, an active player in human cancers. Cancer immunology research. 2014; 2:823-830.
136. Tilstra JS, Clauson CL, Niedernhofer LJ and Robbins PD. NF-kappaB in Aging and Disease. Aging and disease. 2011; 2:449-465.
137. Moskalev A and Shaposhnikov M. Pharmacological inhibition of NF-kappaB prolongs lifespan of Drosophila melanogaster. Aging. 2011; 3:391-394.
138. Shaposhnikov MV, Moskalev AA and Plyusnina EN. Effect of PARP-1 overexpression and pharmacological inhibition of NF-kB on the lifespan of Drosophila melanogaster. Advances in gerontology = Uspekhi gerontologii / Rossiiskaia akademiia nauk, Gerontologicheskoe obshchestvo. 2011; 24:405-419.
139. Oeckinghaus A and Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor perspectives in biology. 2009; 1:a000034.
140. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harbor perspectives in biology. 2009; 1:a001651.
141. Hoesel B and Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Molecular cancer. 2013; 12:86.
142. Chiarpotto E, Domenicotti C, Paola D, Vitali A, Nitti M, Pronzato MA, Biasi F, Cottalasso D, Marinari UM, Dragonetti A, Cesaro P, Isidoro C and Poli G. Regulation of rat hepatocyte protein kinase C beta isoenzymes by the lipid peroxidation product 4-hydroxy-2,3-nonenal: A signaling pathway to modulate vesicular transport of glycoproteins. Hepatology. 1999; 29:1565-1572.
143. Parola M, Robino G, Marra F, Pinzani M, Bellomo G, Leonarduzzi G, Chiarugi P, Camandola S, Poli G, Waeg G, Gentilini P and Dianzani MU. HNE interacts directly with JNK isoforms in human hepatic stellate cells. The Journal of clinical investigation. 1998; 102:1942-1950.
144. Song BJ, Soh Y, Bae M, Pie J, Wan J and Jeong K. Apoptosis of PC12 cells by 4-hydroxy-2-nonenal is mediated through selective activation of the c-Jun N-terminal protein kinase pathway. Chemico-biological interactions. 2001; 130-132:943-954.
145. Yadav UC and Ramana KV. Regulation of NF-kappaB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxidative medicine and cellular longevity. 2013; 2013:690545.
146. Eferl R and Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nature reviews Cancer. 2003; 3:859-868.
147. Awasthi YC, Sharma R, Cheng JZ, Yang Y, Sharma A, Singhal SS and Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Molecular aspects of medicine. 2003; 24:219-230.
148. Kutuk O, Poli G and Basaga H. Resveratrol protects against 4-hydroxynonenal-induced apoptosis by blocking JNK and c-JUN/AP-1 signaling. Toxicological sciences. 2006; 90:120-132.
149. Kakishita H and Hattori Y. Vascular smooth muscle cell activation and growth by 4-hydroxynonenal. Life sciences. 2001; 69:689-697.
150. Kutuk O and Basaga H. Apoptosis signalling by 4-hydroxynonenal: a role for JNK-c-Jun/AP-1 pathway. Redox report : communications in free radical research. 2007; 12:30-34.
151. Ko YG, Kang YS, Park H, Seol W, Kim J, Kim T, Park HS, Choi EJ and Kim S. Apoptosis signal-regulating kinase 1 controls the proapoptotic function of death-associated protein (Daxx) in the cytoplasm. The Journal of biological chemistry. 2001; 276:39103-39106.
152. Chang HY, Nishitoh H, Yang X, Ichijo H and Baltimore D. Activation of apoptosis signal-regulating kinase 1 (ASK1) by the adapter protein Daxx. Science. 1998; 281:1860-1863.
153. Curtin JF and Cotter TG. Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cellular signalling. 2003; 15:983-992.
154. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K and Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997; 275:90-94.
155. Yang X, Khosravi-Far R, Chang HY and Baltimore D. Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell. 1997; 89:1067-1076.
156. Dhanasekaran DN and Reddy EP. JNK signaling in apoptosis. Oncogene. 2008; 27:6245-6251.
157. Morgan MJ, Kim YS and Liu ZG. Membrane-bound Fas ligand requires RIP1 for efficient activation of caspase-8 within the death-inducing signaling complex. Journal of immunology. 2009; 183:3278-3284.
158. Chaudhary P, Sharma R, Sharma A, Vatsyayan R, Yadav S, Singhal SS, Rauniyar N, Prokai L, Awasthi S and Awasthi YC. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry. 2010; 49:6263-6275.
159. Sharma R, Sharma A, Dwivedi S, Zimniak P, Awasthi S and Awasthi YC. 4-Hydroxynonenal self-limits fas-mediated DISC-independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochemistry. 2008; 47:143-156.
160. Li J, Sharma R, Patrick B, Sharma A, Jeyabal PV, Reddy PM, Saini MK, Dwivedi S, Dhanani S, Ansari NH, Zimniak P, Awasthi S and Awasthi YC. Regulation of CD95 (Fas) expression and Fas-mediated apoptotic signaling in HLE B-3 cells by 4-hydroxynonenal. Biochemistry. 2006; 45:12253-12264.
161. Laurora S, Tamagno E, Briatore F, Bardini P, Pizzimenti S, Toaldo C, Reffo P, Costelli P, Dianzani MU, Danni O and Barrera G. 4-Hydroxynonenal modulation of p53 family gene expression in the SK-N-BE neuroblastoma cell line. Free radical biology & medicine. 2005; 38:215-225.
162. Abarikwu SO, Pant AB and Farombi EO. 4-Hydroxynonenal induces mitochondrial-mediated apoptosis and oxidative stress in SH-SY5Y human neuronal cells. Basic & clinical pharmacology & toxicology. 2012; 110:441-448.
163. Tomic JL, Pensalfini A, Head E and Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiology of disease. 2009; 35:352-358.
164. Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA and Gouras GK. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. The Journal of neuroscience. 2004; 24:3592-3599.
165. Gaspar RC, Villarreal SA, Bowles N, Hepler RW, Joyce JG and Shughrue PJ. Oligomers of beta-amyloid are sequestered into and seed new plaques in the brains of an AD mouse model. Experimental neurology. 2010; 223:394-400.
166. Kayed R and Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. Journal of Alzheimer’s disease : JAD. 2013; 33 Suppl 1:S67-78.
167. Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA and Ashe KH. Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain : a journal of neurology. 2013; 136:1383-1398.
168. Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL and LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. The Journal of biological chemistry. 2006; 281:1599-1604.
169. Mandelkow EM and Mandelkow E. Tau in Alzheimer’s disease. Trends in cell biology. 1998; 8:425-427.
170. Gong CX and Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Current medicinal chemistry. 2008; 15:2321-2328.
171. Takeda A, Smith MA, Avila J, Nunomura A, Siedlak SL, Zhu X, Perry G and Sayre LM. In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. Journal of neurochemistry. 2000; 75:1234-1241.
172. Ueda K, Masliah E, Saitoh T, Bakalis SL, Scoble H and Kosik KS. Alz-50 recognizes a phosphorylated epitope of tau protein. The Journal of neuroscience. 1990; 10:3295-3304.
173. Liu Q, Smith MA, Avila J, DeBernardis J, Kansal M, Takeda A, Zhu X, Nunomura A, Honda K, Moreira PI, Oliveira CR, Santos MS, Shimohama S, et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free radical biology & medicine. 2005; 38:746-754.
174. Mattson MP, Fu W, Waeg G and Uchida K. 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. Neuroreport. 1997; 8:2275-2281.
175. Pizzimenti S, Ciamporcero E, Daga M, Pettazzoni P, Arcaro A, Cetrangolo G, Minelli R, Dianzani C, Lepore A, Gentile F and Barrera G. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Frontiers in physiology. 2013; 4:242.
176. Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR and Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiology of disease. 2008; 30:107-120.
177. Reed TT, Pierce WM, Markesbery WR and Butterfield DA. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain research. 2009; 1274:66-76.
178. Butterfield DA, Poon HF, St Clair D, Keller JN, Pierce WM, Klein JB and Markesbery WR. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiology of disease. 2006; 22:223-232.
179. Siegel SJ, Bieschke J, Powers ET and Kelly JW. The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry. 2007; 46:1503-1510.
180. Murray IV, Liu L, Komatsu H, Uryu K, Xiao G, Lawson JA and Axelsen PH. Membrane-mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. The Journal of biological chemistry. 2007; 282:9335-9345.
181. Liu L, Komatsu H, Murray IV and Axelsen PH. Promotion of amyloid beta protein misfolding and fibrillogenesis by a lipid oxidation product. Journal of molecular biology. 2008; 377:1236-1250.
182. Perluigi M, Di Domenico F, Giorgi A, Schinina ME, Coccia R, Cini C, Bellia F, Cambria MT, Cornelius C, Butterfield DA and Calabrese V. Redox proteomics in aging rat brain: involvement of mitochondrial reduced glutathione status and mitochondrial protein oxidation in the aging process. Journal of neuroscience research. 2010; 88:3498-3507.
183. Newman SF, Sultana R, Perluigi M, Coccia R, Cai J, Pierce WM, Klein JB, Turner DM and Butterfield DA. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. Journal of neuroscience research. 2007; 85:1506-1514.
184. Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR and Butterfield DA. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD. Neurobiology of aging. 2006; 27:1564-1576.
185. Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR and Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. Journal of neurochemistry. 2002; 82:1524-1532.
186. Poon HF, Frasier M, Shreve N, Calabrese V, Wolozin B and Butterfield DA. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice—a model of familial Parkinson’s disease. Neurobiology of disease. 2005; 18:492-498.
187. Perluigi M, Poon HF, Maragos W, Pierce WM, Klein JB, Calabrese V, Cini C, De Marco C and Butterfield DA. Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Molecular & cellular proteomics : MCP. 2005; 4:1849-1861.
188. Perluigi M, Giorgi A, Blarzino C, De Marco F, Foppoli C, Di Domenico F, Butterfield DA, Schinina ME, Cini C and Coccia R. Proteomics analysis of protein expression and specific protein oxidation in human papillomavirus transformed keratinocytes upon UVB irradiation. Journal of cellular and molecular medicine. 2009; 13:1809-1822.
189. Gentile F, Pizzimenti S, Arcaro A, Pettazzoni P, Minelli R, D’Angelo D, Mamone G, Ferranti P, Toaldo C, Cetrangolo G, Formisano S, Dianzani MU, Uchida K, Dianzani C and Barrera G. Exposure of HL-60 human leukaemic cells to 4-hydroxynonenal promotes the formation of adduct(s) with alpha-enolase devoid of plasminogen binding activity. The Biochemical journal. 2009; 422:285-294.
190. Hattori T, Takei N, Mizuno Y, Kato K and Kohsaka S. Neurotrophic and neuroprotective effects of neuron-specific enolase on cultured neurons from embryonic rat brain. Neuroscience research. 1995; 21:191-198.
191. Aaronson RM, Graven KK, Tucci M, McDonald RJ and Farber HW. Non-neuronal enolase is an endothelial hypoxic stress protein. The Journal of biological chemistry. 1995; 270:27752-27757.
192. Subramanian A and Miller DM. Structural analysis of alpha-enolase. Mapping the functional domains involved in down-regulation of the c-myc protooncogene. The Journal of biological chemistry. 2000; 275:5958-5965.
193. Pancholi V and Fischetti VA. alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. The Journal of biological chemistry. 1998; 273:14503-14515.
194. Bottermann K, Reinartz M, Barsoum M, Kotter S and Godecke A. Systematic Analysis Reveals Elongation Factor 2 and alpha-Enolase as Novel Interaction Partners of AKT2. PloS one. 2013; 8:e66045.
195. Butterfield DA and Lange ML. Multifunctional roles of enolase in Alzheimer’s disease brain: beyond altered glucose metabolism. Journal of neurochemistry. 2009; 111:915-933.
196. Li D and Ellis EM. 4-Hydroxynonenal induces an increase in expression of Receptor for Activating C Kinase 1 (RACK1) in Chinese hamster V79-4 lung cells. Chemico-biological interactions. 2014; 213:13-20.
197. O’Connell K, Gannon J, Doran P and Ohlendieck K. Proteomic profiling reveals a severely perturbed protein expression pattern in aged skeletal muscle. International journal of molecular medicine. 2007; 20:145-153.
198. Sharma HK, Prasanna HR and Rothstein M. Altered phosphoglycerate kinase in aging rats. The Journal of biological chemistry. 1980; 255:5043-5050.
199. Ahmad SS, Glatzle J, Bajaeifer K, Buhler S, Lehmann T, Konigsrainer I, Vollmer JP, Sipos B, Ahmad SS, Northoff H, Konigsrainer A and Zieker D. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. International journal of oncology. 2013; 43:586-590.
200. Chen XL, Zhou L, Yang J, Shen FK, Zhao SP and Wang YL. Hepatocellular carcinoma-associated protein markers investigated by MALDI-TOF MS. Molecular medicine reports. 2010; 3:589-596.
201. Martinez A, Dalfo E, Muntane G and Ferrer I. Glycolitic enzymes are targets of oxidation in aged human frontal cortex and oxidative damage of these proteins is increased in progressive supranuclear palsy. Journal of neural transmission. 2008; 115:59-66.
202. Poon HF, Shepherd HM, Reed TT, Calabrese V, Stella AM, Pennisi G, Cai J, Pierce WM, Klein JB and Butterfield DA. Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: Mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiology of aging. 2006; 27:1020-1034.
203. Lepper TW, Oliveira E, Koch GD, Berlese DB and Feksa LR. Lead inhibits in vitro creatine kinase and pyruvate kinase activity in brain cortex of rats. Toxicology in vitro : an international journal published in association with BIBRA. 2010; 24:1045-1051.
204. Vitorica J, Andres A, Satrustegui J and Machado A. Age-related quantitative changes in enzyme activities of rat brain. Neurochemical research. 1981; 6:127-136.
205. Delwing D, Delwing de Lima D, Scolaro B, Kuss GG, Cruz JG and Wyse AT. Protective effect of antioxidants on cerebrum oxidative damage caused by arginine on pyruvate kinase activity. Metabolic brain disease. 2009; 24:469-479.
206. Sultana R, Newman SF, Abdul HM, Cai J, Pierce WM, Klein JB, Merchant M and Butterfield DA. Protective effect of D609 against amyloid-beta1-42-induced oxidative modification of neuronal proteins: redox proteomics study. Journal of neuroscience research. 2006; 84:409-417.
207. Stetak A, Veress R, Ovadi J, Csermely P, Keri G and Ullrich A. Nuclear translocation of the tumor marker pyruvate kinase M2 induces programmed cell death. Cancer research. 2007; 67:1602-1608.
208. Pradelli LA, Villa E, Zunino B, Marchetti S and Ricci JE. Glucose metabolism is inhibited by caspases upon the induction of apoptosis. Cell death & disease. 2014; 5:e1406.
209. Orosz F, Olah J and Ovadi J. Triosephosphate isomerase deficiency: new insights into an enigmatic disease. Biochimica et biophysica acta. 2009; 1792:1168-1174.
210. Orosz F, Olah J and Ovadi J. Triosephosphate isomerase deficiency: facts and doubts. IUBMB life. 2006; 58:703-715.
211. Bonnet R, Pavlovic S, Lehmann J and Rommelspacher H. The strong inhibition of triosephosphate isomerase by the natural beta-carbolines may explain their neurotoxic actions. Neuroscience. 2004; 127:443-453.
212. Zhao L, Jia Y, Yan D, Zhou C, Han J and Yu J. Aging-related changes of triose phosphate isomerase in hippocampus of senescence accelerated mouse and the intervention of acupuncture. Neuroscience letters. 2013; 542:59-64.
213. Tajes M, Guivernau B, Ramos-Fernandez E, Bosch-Morato M, Palomer E, Guix FX and Munoz FJ. The pathophysiology of triose phosphate isomerase dysfunction in Alzheimer’s disease. Histology and histopathology. 2013; 28:43-51.
214. Park SA, Park HW, Kim NH, Kim YH, Kwak MJ, Shin JS and Kim CW. Effects of Tau on the activity of triose phosphate isomerase (TPI) in brain cells. Neurochemistry international. 2010; 56:886-892.
215. Qi YJ, He QY, Ma YF, Du YW, Liu GC, Li YJ, Tsao GS, Ngai SM and Chiu JF. Proteomic identification of malignant transformation-related proteins in esophageal squamous cell carcinoma. Journal of cellular biochemistry. 2008; 104:1625-1635.
216. Zhang D, Tai LK, Wong LL, Chiu LL, Sethi SK and Koay ES. Proteomic study reveals that proteins involved in metabolic and detoxification pathways are highly expressed in HER-2/neu-positive breast cancer. Molecular & cellular proteomics : MCP. 2005; 4:1686-1696.
217. Wang X, Lu Y, Yang J, Shi Y, Lan M, Liu Z, Zhai H and Fan D. Identification of triosephosphate isomerase as an anti-drug resistance agent in human gastric cancer cells using functional proteomic analysis. Journal of cancer research and clinical oncology. 2008; 134:995-1003.
218. Yu G, Liu H, Zhou W, Zhu X, Yu C, Wang N, Zhang Y, Ma J, Zhao Y, Xu Y, Liao L, Ji H, Yuan C and Ma J. In vivo protein targets for increased quinoprotein adduct formation in aged substantia nigra. Experimental neurology. 2015; 271:13-24.
219. Shi Q and Gibson GE. Up-regulation of the mitochondrial malate dehydrogenase by oxidative stress is mediated by miR-743a. Journal of neurochemistry. 2011; 118:440-448.
220. Bubber P, Hartounian V, Gibson GE and Blass JP. Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2011; 21:254-260.
221. Rajendran P, Ekambaram G and Sakthisekaran D. Effect of mangiferin on benzo(a)pyrene induced lung carcinogenesis in experimental Swiss albino mice. Natural product research. 2008; 22:672-680.
222. Kudriavtsev Iu V. [Histocytospectrophotometric characteristics of lung cancer]. Arkhiv patologii. 1980; 42:20-27.
223. Schagger H and Ohm TG. Human diseases with defects in oxidative phosphorylation. 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. European journal of biochemistry / FEBS. 1995; 227:916-921.
224. Perluigi M, Sultana R, Cenini G, Di Domenico F, Memo M, Pierce WM, Coccia R and Butterfield DA. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics Clinical applications. 2009; 3:682-693.
225. Ames BN. Endogenous oxidative DNA damage, aging, and cancer. Free radical research communications. 1989; 7:121-128.
226. Dehennaut V, Loison I, Dubuissez M, Nassour J, Abbadie C and Leprince D. DNA double-strand breaks lead to activation of hypermethylated in cancer 1 (HIC1) by SUMOylation to regulate DNA repair. The Journal of biological chemistry. 2013; 288:10254-10264.
227. Zhou BB and Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000; 408:433-439.
228. Bassing CH and Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA repair. 2004; 3:781-796.
229. Tchurikov NA, Fedoseeva DM, Sosin DV, Snezhkina AV, Melnikova NV, Kudryavtseva AV, Kravatsky YV and Kretova OV. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenetic regulation. Journal of molecular cell biology. 2015; 7:366-382.
230. Caldecott KW. Single-strand break repair and genetic disease. Nature reviews Genetics. 2008; 9:619-631.
231. Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC and Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nature neuroscience. 2013; 16:613-621.
232. Li JC and Kaminskas E. Deficient repair of DNA lesions in Alzheimer’s disease fibroblasts. Biochemical and biophysical research communications. 1985; 129:733-738.
233. Shackelford DA. DNA end joining activity is reduced in Alzheimer’s disease. Neurobiology of aging. 2006; 27:596-605.
234. Hegde ML, Gupta VB, Anitha M, Harikrishna T, Shankar SK, Muthane U, Subba Rao K and Jagannatha Rao KS. Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Archives of biochemistry and biophysics. 2006; 449:143-156.
235. Richter C, Park JW and Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proceedings of the National Academy of Sciences of the United States of America. 1988; 85:6465-6467.
236. Kasai H, Crain PF, Kuchino Y, Nishimura S, Ootsuyama A and Tanooka H. Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis. 1986; 7:1849-1851.
237. Valavanidis A, Vlachogianni T and Fiotakis C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis. Journal of environmental science and health Part C, Environmental carcinogenesis & ecotoxicology reviews. 2009; 27:120-139.
238. Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H and Fraifeld VE. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing research reviews. 2013; 12:661-684.
239. Olinski R, Gackowski D, Rozalski R, Foksinski M and Bialkowski K. Oxidative DNA damage in cancer patients: a cause or a consequence of the disease development? Mutation research. 2003; 531:177-190.
240. Castellani RJ, Gupta Y, Sheng B, Siedlak SL, Harris PL, Coller JM, Perry G, Lee HG, Tabaton M, Smith MA, Wang X and Zhu X. A novel origin for granulovacuolar degeneration in aging and Alzheimer’s disease: parallels to stress granules. Laboratory investigation; a journal of technical methods and pathology. 2011; 91:1777-1786.
241. Aguirre N, Beal MF, Matson WR and Bogdanov MB. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free radical research. 2005; 39:383-388.
242. Nunomura A, Perry G, Pappolla MA, Friedland RP, Hirai K, Chiba S and Smith MA. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. Journal of neuropathology and experimental neurology. 2000; 59:1011-1017.
243. Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG and Montine TJ. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. The American journal of pathology. 1999; 154:1423-1429.
244. Hagen TM, Huang S, Curnutte J, Fowler P, Martinez V, Wehr CM, Ames BN and Chisari FV. Extensive oxidative DNA damage in hepatocytes of transgenic mice with chronic active hepatitis destined to develop hepatocellular carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:12808-12812.
245. Wu LL, Chiou CC, Chang PY and Wu JT. Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clinica chimica acta; international journal of clinical chemistry. 2004; 339:1-9.
246. Yano T, Shoji F, Baba H, Koga T, Shiraishi T, Orita H and Kohno H. Significance of the urinary 8-OHdG level as an oxidative stress marker in lung cancer patients. Lung cancer. 2009; 63:111-114.
247. Gmitterova K, Heinemann U, Gawinecka J, Varges D, Ciesielczyk B, Valkovic P, Benetin J and Zerr I. 8-OHdG in cerebrospinal fluid as a marker of oxidative stress in various neurodegenerative diseases. Neuro-degenerative diseases. 2009; 6:263-269.
248. Rall LC, Roubenoff R, Meydani SN, Han SN and Meydani M. Urinary 8-hydroxy-2’-deoxyguanosine (8-OHdG) as a marker of oxidative stress in rheumatoid arthritis and aging: effect of progressive resistance training. The Journal of nutritional biochemistry. 2000; 11:581-584.
249. Fraga CG, Shigenaga MK, Park JW, Degan P and Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2’-deoxyguanosine in rat organ DNA and urine. Proceedings of the National Academy of Sciences of the United States of America. 1990; 87:4533-4537.
250. Chen Q, Fischer A, Reagan JD, Yan LJ and Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92:4337-4341.
251. Wagner JR, Hu CC and Ames BN. Endogenous oxidative damage of deoxycytidine in DNA. Proceedings of the National Academy of Sciences of the United States of America. 1992; 89:3380-3384.
252. Adelman R, Saul RL and Ames BN. Oxidative damage to DNA: relation to species metabolic rate and life span. Proceedings of the National Academy of Sciences of the United States of America. 1988; 85:2706-2708.
253. Bandy B and Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free radical biology & medicine. 1990; 8:523-539.
254. Sanz A, Caro P, Gomez J and Barja G. Testing the vicious cycle theory of mitochondrial ROS production: effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. Journal of bioenergetics and biomembranes. 2006; 38:121-127.
255. de Grey AD. Reactive oxygen species production in the mitochondrial matrix: implications for the mechanism of mitochondrial mutation accumulation. Rejuvenation research. 2005; 8:13-17.
256. Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT and Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102:17993-17998.
257. Cheng Z and Ristow M. Mitochondria and metabolic homeostasis. Antioxidants & redox signaling. 2013; 19:240-242.
258. Shokolenko IN, Wilson GL and Alexeyev MF. Aging: A mitochondrial DNA perspective, critical analysis and an update. World journal of experimental medicine. 2014; 4:46-57.
259. Hiona A and Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Experimental gerontology. 2008; 43:24-33.
260. Jeppesen DK, Bohr VA and Stevnsner T. DNA repair deficiency in neurodegeneration. Progress in neurobiology. 2011; 94:166-200.
261. Kennedy SR, Salk JJ, Schmitt MW and Loeb LA. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS genetics. 2013; 9:e1003794.
262. Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005; 309:481-484.
263. Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT and Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004; 429:417-423.
264. Wenzel P, Schuhmacher S, Kienhofer J, Muller J, Hortmann M, Oelze M, Schulz E, Treiber N, Kawamoto T, Scharffetter-Kochanek K, Munzel T, Burkle A, Bachschmid MM and Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovascular research. 2008; 80:280-289.
265. Aliev G, Seyidova D, Lamb BT, Obrenovich ME, Siedlak SL, Vinters HV, Friedland RP, LaManna JC, Smith MA and Perry G. Mitochondria and vascular lesions as a central target for the development of Alzheimer’s disease and Alzheimer disease-like pathology in transgenic mice. Neurological research. 2003; 25:665-674.
266. Mancuso M, Calsolaro V, Orsucci D, Siciliano G and Murri L. Is there a primary role of the mitochondrial genome in Alzheimer’s disease? Journal of bioenergetics and biomembranes. 2009; 41:411-416.
267. de la Monte SM, Luong T, Neely TR, Robinson D and Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Laboratory investigation; a journal of technical methods and pathology. 2000; 80:1323-1335.
268. Mecocci P, MacGarvey U and Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Annals of neurology. 1994; 36:747-751.
269. Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S and Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. The Journal of neuroscience. 1999; 19:1959-1964.
270. Mancuso M, Calsolaro V, Orsucci D, Carlesi C, Choub A, Piazza S and Siciliano G. Mitochondria, cognitive impairment, and Alzheimer’s disease. International journal of Alzheimer’s disease. 2009; 2009.
271. Arthur CR, Morton SL, Dunham LD, Keeney PM and Bennett JP, Jr. Parkinson’s disease brain mitochondria have impaired respirasome assembly, age-related increases in distribution of oxidative damage to mtDNA and no differences in heteroplasmic mtDNA mutation abundance. Molecular neurodegeneration. 2009; 4:37.
272. Parkinson GM, Dayas CV and Smith DW. Increased mitochondrial DNA deletions in substantia nigra dopamine neurons of the aged rat. Current aging science. 2014; 7:155-160.
273. Chatterjee A, Mambo E and Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2006; 25:4663-4674.
274. Minocherhomji S, Tollefsbol TO and Singh KK. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics. 2012; 7:326-334.
275. Lund M, Melbye M, Diaz LJ, Duno M, Wohlfahrt J and Vissing J. Mitochondrial dysfunction and risk of cancer. British journal of cancer. 2015; 112:1134-1140.
276. Gogvadze V, Orrenius S and Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends in cell biology. 2008; 18:165-173.
277. Woo DK, Green PD, Santos JH, D’Souza AD, Walther Z, Martin WD, Christian BE, Chandel NS and Shadel GS. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. The American journal of pathology. 2012; 180:24-31.
278. Yun J and Finkel T. Mitohormesis. Cell metabolism. 2014; 19:757-766.
279. Ristow M and Schmeisser K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-response : a publication of International Hormesis Society. 2014; 12:288-341.
280. Ristow M and Zarse K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Experimental gerontology. 2010; 45:410-418.
281. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M and Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell metabolism. 2007; 6:280-293.
282. Tapia PC. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Medical hypotheses. 2006; 66:832-843.
283. Kim YC, Masutani H, Yamaguchi Y, Itoh K, Yamamoto M and Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. The Journal of biological chemistry. 2001; 276:18399-18406.
284. Rushmore TH, Morton MR and Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. The Journal of biological chemistry. 1991; 266:11632-11639.
285. Klotz LO, Sanchez-Ramos C, Prieto-Arroyo I, Urbanek P, Steinbrenner H and Monsalve M. Redox regulation of FoxO transcription factors. Redox biology. 2015; 6:51-72.
286. Akasaki Y, Alvarez-Garcia O, Saito M, Carames B, Iwamoto Y and Lotz MK. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis & rheumatology. 2014; 66:3349-3358.
287. Hardie DG, Hawley SA and Scott JW. AMP-activated protein kinase—development of the energy sensor concept. The Journal of physiology. 2006; 574:7-15.
288. Apfeld J, O’Connor G, McDonagh T, DiStefano PS and Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes & development. 2004; 18:3004-3009.
289. Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP and Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Current biology : CB. 2007; 17:1646-1656.
290. Pan DA and Hardie DG. A homologue of AMP-activated protein kinase in Drosophila melanogaster is sensitive to AMP and is activated by ATP depletion. The Biochemical journal. 2002; 367:179-186.
291. Schmeisser S, Priebe S, Groth M, Monajembashi S, Hemmerich P, Guthke R, Platzer M and Ristow M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Molecular metabolism. 2013; 2:92-102.
292. Schmeisser S, Zarse K and Ristow M. Lonidamine extends lifespan of adult Caenorhabditis elegans by increasing the formation of mitochondrial reactive oxygen species. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2011; 43:687-692.
293. Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S and Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310:1193-1196.
294. Jia K, Chen D and Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004; 131:3897-3906.
295. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V and Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Current biology : CB. 2004; 14:885-890.
296. Quarrie JK and Riabowol KT. Murine models of life span extension. Science of aging knowledge environment : SAGE KE. 2004; 2004:re5.
297. Tormos KV and Chandel NS. Inter-connection between mitochondria and HIFs. Journal of cellular and molecular medicine. 2010; 14:795-804.
298. Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J and Kenyon C. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002; 298:2398-2401.
299. Martin-Montalvo A, Villalba JM, Navas P and de Cabo R. NRF2, cancer and calorie restriction. Oncogene. 2011; 30:505-520.
300. Carbajo-Pescador S, Mauriz JL, Garcia-Palomo A and Gonzalez-Gallego J. FoxO proteins: regulation and molecular targets in liver cancer. Current medicinal chemistry. 2014; 21:1231-1246.
301. Neckers L. Heat shock protein 90: the cancer chaperone. Journal of biosciences. 2007; 32:517-530.
302. Monteverde T, Muthalagu N, Port J and Murphy DJ. Evidence of cancer-promoting roles for AMPK and related kinases. The FEBS journal. 2015.
303. Perl A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Annals of the New York Academy of Sciences. 2015; 1346:33-44.
304. Smith EJ, Shay KP, Thomas NO, Butler JA, Finlay LF and Hagen TM. Age-related loss of hepatic Nrf2 protein homeostasis: Potential role for heightened expression of miR-146a. Free radical biology & medicine. 2015.
305. Johnson DA and Johnson JA. Nrf2-a therapeutic target for the treatment of neurodegenerative diseases. Free radical biology & medicine. 2015; 88:253-267.
306. Maiese K. FoxO proteins in the nervous system. Analytical cellular pathology. 2015; 2015:569392.
307. Maiese K. Targeting molecules to medicine with mTOR, autophagy, and neurodegenerative disorders. British journal of clinical pharmacology. 2015.
308. Ou JR, Tan MS, Xie AM, Yu JT and Tan L. Heat shock protein 90 in Alzheimer’s disease. BioMed research international. 2014; 2014:796869.
309. Zhang Z, Yan J, Chang Y, ShiDu Yan S and Shi H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Current medicinal chemistry. 2011; 18:4335-4343.
310. Zemva J and Schubert M. The role of neuronal insulin/insulin-like growth factor-1 signaling for the pathogenesis of Alzheimer’s disease: possible therapeutic implications. CNS & neurological disorders drug targets. 2014; 13:322-337.
311. Kim EK and Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochimica et biophysica acta. 2010; 1802:396-405.
312. Lee JM and Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. Journal of biochemistry and molecular biology. 2004; 37:139-143.
313. Hayflick L and Moorhead PS. The serial cultivation of human diploid cell strains. Experimental cell research. 1961; 25:585-621.
314. Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Experimental cell research. 1965; 37:614-636.
315. Collado M and Serrano M. Senescence in tumours: evidence from mice and humans. Nature reviews Cancer. 2010; 10:51-57.
316. Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends in cell biology. 2001; 11:S27-31.
317. Garkavtsev I, Hull C and Riabowol K. Molecular aspects of the relationship between cancer and aging: tumor suppressor activity during cellular senescence. Experimental gerontology. 1998; 33:81-94.
318. Shay JW, Pereira-Smith OM and Wright WE. A role for both RB and p53 in the regulation of human cellular senescence. Experimental cell research. 1991; 196:33-39.
319. Hara E, Tsurui H, Shinozaki A, Nakada S and Oda K. Cooperative effect of antisense-Rb and antisense-p53 oligomers on the extension of life span in human diploid fibroblasts, TIG-1. Biochemical and biophysical research communications. 1991; 179:528-534.
320. Bond JA, Wyllie FS and Wynford-Thomas D. Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene. 1994; 9:1885-1889.
321. Cairns J. Mutation selection and the natural history of cancer. Nature. 1975; 255:197-200.
322. Cairns J. The cancer problem. Scientific American. 1975; 233:64-72, 77-68.
323. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759-767.
324. Bringold F and Serrano M. Tumor suppressors and oncogenes in cellular senescence. Experimental gerontology. 2000; 35:317-329.
325. Serrano M, Lin AW, McCurrach ME, Beach D and Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88:593-602.
326. Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ and Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & development. 1998; 12:2424-2433.
327. Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL and Vousden KH. p14ARF links the tumour suppressors RB and p53. Nature. 1998; 395:124-125.
328. Lundberg AS, Hahn WC, Gupta P and Weinberg RA. Genes involved in senescence and immortalization. Current opinion in cell biology. 2000; 12:705-709.
329. Zhu J, Woods D, McMahon M and Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes & development. 1998; 12:2997-3007.
330. Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M and Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & development. 1998; 12:3008-3019.
331. Jacobs JJ, Kieboom K, Marino S, DePinho RA and van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999; 397:164-168.
332. Kipling D, Wynford-Thomas D, Jones CJ, Akbar A, Aspinall R, Bacchetti S, Blasco MA, Broccoli D, DePinho RA, Edwards DR, Effros RB, Harley CB, Lansdorp PM, et al. Telomere-dependent senescence. Nature biotechnology. 1999; 17:313-314.
333. Shay JW. Aging and cancer: are telomeres and telomerase the connection? Molecular medicine today. 1995; 1:378-384.
334. Artandi SE and DePinho RA. A critical role for telomeres in suppressing and facilitating carcinogenesis. Current opinion in genetics & development. 2000; 10:39-46.
335. Artandi SE and DePinho RA. Mice without telomerase: what can they teach us about human cancer? Nature medicine. 2000; 6:852-855.
336. Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW and DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999; 97:527-538.
337. Artandi SE and DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010; 31:9-18.
338. Blagosklonny MV. Cell cycle arrest is not senescence. Aging. 2011; 3:94-101.
339. Moskalev AA and Shaposhnikov MV. Pharmacological inhibition of phosphoinositide 3 and TOR kinases improves survival of Drosophila melanogaster. Rejuvenation research. 2010; 13:246-247.
340. Blagosklonny MV. Molecular damage in cancer: an argument for mTOR-driven aging. Aging. 2011; 3:1130-1141.
341. Jackson SP and Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071-1078.
342. Reimann M, Loddenkemper C, Rudolph C, Schildhauer I, Teichmann B, Stein H, Schlegelberger B, Dorken B and Schmitt CA. The Myc-evoked DNA damage response accounts for treatment resistance in primary lymphomas in vivo. Blood. 2007; 110:2996-3004.
343. Rai P. Oxidation in the nucleotide pool, the DNA damage response and cellular senescence: Defective bricks build a defective house. Mutation research. 2010; 703:71-81.
344. Morelli MB, Amantini C, Santoni M, Soriani A, Nabissi M, Cardinali C, Santoni A and Santoni G. Axitinib induces DNA damage response leading to senescence, mitotic catastrophe, and increased NK cell recognition in human renal carcinoma cells. Oncotarget. 2015; 6:36245-36259. doi: 10.18632/oncotarget.5768.
345. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J and Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434:864-870.
346. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C and Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005; 434:907-913.
347. Macieira-Coelho A. Putative mechanisms responsible for the decline in cancer prevalence during organism senescence. Biogerontology. 2015; 16:559-565.
348. Carreca I, Balducci L and Extermann M. Cancer in the older person. Cancer treatment reviews. 2005; 31:380-402.
349. Zainullin VG and Moskalev AA. [The role of genetic instability in cell aging]. Genetika. 2000; 36:1013-1016.
350. Macieira-Coelho A. Control of cell replication during aging. Interdisciplinary topics in gerontology. 2014; 39:24-44.
351. Martinez AO, Norwood TH, Prothero JW and Martin GM. Evidence for clonal attenuation of growth potential in HeLa cells. In vitro. 1978; 14:996-1002.
352. Bryan TM and Reddel RR. SV40-induced immortalization of human cells. Critical reviews in oncogenesis. 1994; 5:331-357.
353. Krtolica A, Parrinello S, Lockett S, Desprez PY and Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:12072-12077.