
INTRODUCTION
Colorectal cancer (CRC) is one of the most common cancers in the world [1]. Despite the fact that CRC is histologically homogeneous, each tumor has a unique molecular profile, which is characterized by various genetic and epigenetic changes. Several molecular genetic markers, which are currently used for CRC diagnosis, prognosis and treatment assignment, have been identified. Numerous molecular genetic studies of CRC have revealed many genes that are characterized by high frequency of mutations (KRAS, NRAS, BRAF, PIK3CA, APC, TP53, SMAD2, SMAD4, ARID1A, SOX9, FAM123B/WTX, and FBXW7), copy number alterations (ERBB2 and IGF2), methylation status changes (MLH1), impaired expression at the mRNA or protein level, and translocations (NAV2/TCF7L1) [2, 3]. Alteration in many genes have predictive value for assessing prognosis and sensitivity to various drugs. However, the data on their clinical significance are often scattered, ambiguous and even conflicting. In this review, we summarized data on the most important widely used and prospective markers of CRC prognosis, prediction of the response to therapy and evaluated their significance.
Several approaches of CRC classification based on genomic, transcriptomic and proteomic profiling are known. Multiple studies of CRC molecular features suggest the existence of several major molecular subtypes of colorectal cancer, which differ in the mechanisms of their development, course of the disease, and response to various drugs [4-6]. However, exhaustive classification of CRC into molecular subtypes is restricted by a large number of individual tumor features that do not fit the overall picture. The second aim of this review is to describe known approached of CRC classification based on genome-wide features (methylation, expression profiling), alterations in individual genes (driver mutations, deletions) and pathogenic pathways.
CHROMOSOMAL INSTABILITY OVERVIEW OF PATHWAYS INVOLVED IN COLORECTAL CARCINOGENESIS
Tumors with chromosomal instability (CIN) are often thought of as a separate biological entity. CIN is the most common feature (65-85% tumors) of colon adenocarcinomas compared to others (MSI, CIMP) [4, 7]. CIN-positive tumors are characterized with severely increased frequency of duplications or losses of chromosome regions or entire chromosomes. Despite the fact that a number of methods are currently available in clinical laboratories (e.g., FISH, karyotyping, comparative genomic hybridization (CGH), including microarray-based CGH), it is not always possible to carry out the differential diagnosis of CIN phenotype in routine practice. In many ways, this is related to the variety of chromosomal abnormalities following CIN and the difficulty of developing clear quantitative criteria for diagnosis. CIN tumors are conventionally divided into two subgroups depending on the frequency of chromosomal aberrations: CIN-high (CIN-H) and CIN-low (CIN-L) tumors. CIN tumors are characterized with mutations in various tumor suppressor genes: APC (up to 85%), TP53 (40-50%), SMAD2/4 (10-20%), and DCC (5%), and proto-oncogenes: KRAS (30-50%), CTNNB1 (5-15%), and PIK3CA (20%) [4].
The development of most colorectal tumors is caused by dysregulation of several signal transduction pathways - Wnt, TGFβ/BMP, RTK/Ras, PI3K/Akt. Activation of canonical Wnt pathway contributes to the onset and progression of more than 90% colorectal adenocarcinomas and adenomas [3, 8]. In normal cells, Wnt is responsible for a plethora of processes including embryonic development, cell proliferation, cell polarity, specification of cell fate. The main element of Wnt pathway is β-catenin, which interacts to TCF/LEF transcription factors and activates proliferative and pro-survival transcriptional programs (more than 1000 genes including Myc). In cytoplasm, β-catenin forms complex with GSK3β/Axin/APC. This prevents translocation of β-catenin to the nucleus and promotes its proteasomal degradation. Wnt signaling leads to the disassociation of the complex and release of β-catenin.
Absence of normal function of APC tumor suppressor (adenomatous polyposis coli) results to the accumulation of β-catenin, its translocation to nucleus and subsequent induction of proliferative expressional programs. The most of sporadic colorectal adenocarcinomas (90%) harbor driver mutations within the members of Wnt pathway, with APC gene being the most frequent (70-80%) target of inactivating mutations [3, 9, 10]. In many cases, mutations of APC is the first event of the development of colorectal adenomas and subsequent carcinogenesis. In contrast, mutational activation of KRAS cannot initiate cancer in vivo, and only when combined with a mutation in APC mutant KRAS does promote tumor progression [11]. Mutations are not the only cause of APC deficiency. Hypermethylation of the APC promoter (18% primary colorectal carcinomas and adenomas) is alternate mechanism [4, 12].
It is well known that mutations in APC are strongly associated with CIN [3, 4]. However, dysregulation of APC—β-Catenin axis may be not obligate for the development of CIN tumors [13]. Moreover, it is unclear whether loss of APC function is truly the cause underlying CIN and whether CIN occurs before or after APC mutations. Many studies propose APC as initiator of chromosome instability pathway [4, 14-16]. During mitosis, APC clusters at the plus-ends of the spindle microtubules and co-localizes with the kinetochore, the attachment site of the mitotic spindle to the newly duplicated chromosomes [15, 17]. Losses or truncations of APC cause mitotic spindle defects that, upon somatic inactivation of other CIN-associated genes (e.g. spindle and cell cycle checkpoint genes, DNA repair, telomere maintenance, etc.) results in chromosomal abnormalities and aneuploidy as observed in the most of CIN-positive CRCs [17].
The main mechanisms of the development of CIN tumors beyond APC are related to the dysregulation of chromosome segregation, telomere formation and DNA repair. Genes related to the BUB/MAD family are involved in the cell cycle checkpoint system at the stage of separation of two identical chromosome copies during prometaphase. Activation of their protein products is a consequence of improper mitosis and leads to inhibition of the anaphase-promoting complex/cyclosome (APC/C, not to be confused with the APC tumor suppressor) and subsequent cell cycle arrest [18]. Mutations in genes that are involved in chromosome segregation are one of the main causes of CIN. Substitution mutations in BUB1 or BUBR1 impair the G2/M cell cycle checkpoint [19-21]. Furthermore, dominant mutations in the BUBR1 are associated with both disturbances in cell cycle checkpoints and the gradual development of CIN in the cell line that initially did not have the chromosomal instability [22].
Another mechanism for the development of CIN is abnormality of the centrosome system, including the formation of additional microtubule-organizing centers [23]. It has been shown that the formation of extra centrosomes can initiate tumorigenesis of larval brain cells in Drosophila transgenic lines [24]. The amplification and increased expression of Aurora and Polo-like kinase genes AURKA and PLK1 is among major causes of the centrosome abnormalities in colorectal cancer [25-27]. AURKA is involved in centrosome duplication, induction of mitosis, and spindle assembly. AURKA gene amplification is accompanied with the formation of extra centrosomes and inappropriate beginning of anaphase despite defective spindle formation [28]. Moreover, it was shown that AURKA participates in the pro-oncogenic Myc pathway [29].
Polo-like kinases PLK1-4 are involved in centrosome duplication, induction of mitosis, promotion of the metaphase to anaphase transition, and cell division [30]. It was shown that PLKs play a role in the activation of AURKA/B [31]. It has been found that Polo-like kinases are characterized by the increased expression levels in colorectal cancer and participate in the activation of migration and invasion of tumor cells [32, 33]. AURKA up-regulation is associated with chromosomal instability [34]. The suppression of both Aurora (in combination with the MEK) and PLK leads to inhibition of tumor cell proliferation [35, 36]. However, the association between AURKA overexpression and CRC prognosis is ambiguous [29, 37, 38].
The third major mechanism of chromosomal instability in colorectal cancer is associated with telomere dysfunction, which in turn causes impairments like a “breakage-fusion-bridge”, which often results in the amplification of oncogenes localized at telomeric regions [39, 40]. The study with Terc-deficient (telomerase RNA component) mice demonstrated association between telomere shortening and generation of intestinal carcinomas and microadenomas [41]. The importance of telomere dysfunction in the development of CRC in humans has been repeatedly mentioned. Many malignant colorectal tumors have both telomere shortening, which causes chromosomal instability, and their extension, which promotes unlimited cell proliferation [42-45]. Moreover, the positive correlation between telomere length and effectiveness of anti-EGFR therapy has been observed [46]. However, a significant lengthening of telomeres in colorectal cancer is associated with a higher degree of invasion and worse prognosis [47].
Finally, the last major mechanism of CIN development in CRC is related to the impairment of DNA damage response (DDR). It has been shown that haploinsufficiency of the H2AX histone, which is involved in the activation of ATM (ataxia-telangiectasia) during DNA damage, leads to genomic instability, and when combined with TP53 may also initiate tumorigenesis [48-50]. One of the possible causes of association between chromosomal instability and DNA damage is that during mitosis DDR activation selectively stabilizes the interaction between microtubules and kinetochores through AURKA and PLK1 which may result in improper chromosome segregation. In case of inhibition of ATM and CHK2 proteins which are involved in cellular DNA damage response, such an effect was not observed [51].
In general, CIN-H can be treated as a factor of unfavorable prognosis [52]. In CIN tumors, as a rule, there is a loss of 25-30% alleles [7]. Besides, there is a bias in preferential losses/gains of specific regions between microsatellite stable (MSS) and instable (MSI) CIN tumors. In particular, MSS tumors, which comprises most of CIN-positive colorectal adenocarcinomas, tend to have deletions in 18q (about 50% tumors) [53]. This region harbors two crucial tumors suppressors - DCC (deleted in colorectal cancer) and SMAD4. SMADs are key intracellular components of TGF-β/BMP pathway, one of the most frequently inactivated ones in colorectal carcinomas (30-40% tumors) [3, 54].
TGF-β/BMP pathway is responsible for embryonic development, cell differentiation, apoptosis and other cellular processes. After binding of ligands (TGF-β, GDF, BMP or Activin) to the surface receptors, SMAD4 forms complex with active phosphorylated SMAD1/2/3/5/8 (R-SMADs), translocates to the nucleus and engage gene expression. The effect of TGF-β pathway strongly depends on the distinct context; in can either contribute or diminish tumor initiation and progression. Loss of SMAD4 plays an important role in the onset of squamous cell carcinomas of upper digestive tract, skin, adenocarcinomas of gastrointestinal tract [54]. The impact of SMAD4 deficiency on the development of CIN phenotype is not clear; SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway [55, 56]. It is known about Wnt—TGF-β pathways crosstalk: SMAD4-mediated signaling inhibits intestinal neoplasia by decreasing expression of β-catenin [57].
Losses in 18q (including SMAD4 and DCC) and reduced expression of SMAD4 are markers of worse response to fluorouracil-based CRC treatment [58]. Moreover, deletions of 18q, 8p, 4p and 15q, inactivation of SMAD4, DCC is considered as discussible factor of negative prognosis of colorectal cancer and other tumors [59-62].
THE DISTURBANCE IN THE MISMATCH REPAIR SYSTEM, INCREASED FREQUENCY OF MUTATIONS IN VARIOUS GENES AND MICROSATELLITE INSTABILITY
About 15% of colorectal adenocarcinomas are characterized with an increased frequency of mutations (hypermutated, HM) that often occurs against the background of high level of microsatellite instability and disturbances in the DNA repair system (mismatch repair, MMR). According to The Cancer Genome Atlas (TCGA) project data, 60% HM-tumors are characterized by epigenetic inactivation of the MLH1 gene, a high frequency of the BRAF V600E mutation and a low frequency of mutations in APC and KRAS genes. The other 40% HM-tumors, for which methylation of MLH1 is not revealed, contain a significantly greater number of mutations and can be referred to a subtype with an ultra-high number of mutations (ultramutated, UM). UM tumors are characterized by an increased frequency of mutations in APC and KRAS genes and a reduced frequency of activating mutation BRAF V600E [63]. Adenocarcinomas containing mutations in genes of polymerases δ and ε (usually, against the background of microsatellite stability) can be related to UM subtype [64].
Tumors related to HM and UM subtypes develop in fundamentally different ways than most other colorectal adenocarcinomas [63]. HM subtype tumors can be a consequence of germinal mutations in one of several genes (MSH2, MSH3, MSH6, PMS1, and PMS2) in hereditary colorectal cancer. These genes are the tumor suppressors genes, which are inactivated in more than 10% of human cancers. Germinal mutations in these genes are associated with Lynch syndrome [65], which is characterized by a predisposition to the development of a number of malignant tumors, primarily nonpolyposis colorectal cancer and endometrial cancer.
The disturbance in the MMR system is the main cause of the high level of microsatellite instability (MSI-H), since microsatellites are particularly labile and can accumulate errors, which are not corrected [66]. However, MSS colorectal tumors show higher incidences of mutations in the APC, KRAS and TP53 genes. To determine the MSI status, the panel recommended by the National Cancer Institute (NCI) is often used. The panel consists of two mononucleotide repeats (BAT26 and BAT25) and three dinucleotide repeats (D5S346, D2S123, and D17S250). According to the classification based on the results that are obtained with the use of this panel, MSI-H tumors have instability in two or more markers, while tumors with low level of microsatellite instability (MSI-L) and MSS-tumors have instability in no more than one marker [67]. Additionally, MMR status can be determined by immunohistochemical analysis of proteins, which are involved in the repairing of unpaired bases, such as those encoded by MLH1, MSH2, MSH6, and PMS2 genes [68].
Tumors with hypermutated phenotype are characterized by a more favorable prognosis. Microsatellite instability is considered and discussed as a possible marker of overall and disease-free survival, as well as of sensitivity to therapy with 5-fluorouracil (5-FU). Despite the fact that the results are contradictive [69-73], most of the recent studies suggest microsatellite instability as marker of good response to treatment with 5-FU in combination with other drugs, especially in the presence of large deletions in HSP110 [71-73]. The National Comprehensive Cancer Network (NCCN) recommends 5-FU either as monotherapy or in combination with other drugs for patients with stage III and IV colorectal cancer and stage II, if it is associated with negative prognosis. However, several research groups do not recommend using this marker whereas the other groups suggest determining the MSI status for all patients with stage II CRC, because MSI-H patients have a favorable prognosis and 5-FU therapy may be not required [74-76].
Generally, the presence of microsatellite instability in CRC is a positive prognostic factor, especially in the absence of early onset of the disease and mutations in BRAF gene. Against the background of an inactivated MGMT gene, MSI is associated with potential tumor cell resistance to methylating agents (temozolomide, dacarbazine, procarbazine), and potential sensitivity to ethylating agents (nitrosoureas).
CpG ISLAND METHYLATOR PHENOTYPE
The existence of a group of tumors characterized with a large number of simultaneously methylated CpG islands, which results in the inactivation of several key tumor suppressor genes, was first shown for colorectal cancer. This phenomena was called CpG Island Methylator Phenotype (CIMP) [77]. CIMP-positive CRCs have their own precursor lesions, serrated adenomas, distinct from conventional adenomas, which progress and transform into CIMP-negative CRCs [78]. Based on the genome-wide data on the density of CpG island hypermethylation, three groups of tumors are mostly defined: high degree of hypermethylation (CIPM-H), low (CIMP-L) and CIMP-negative (CIMP-N) tumors [79, 80]. It was shown that CIMP-H is highly associated with hypermethylation of the MLH1 gene and MSI-H phenotype [81, 82]. Associations of CIMP-H with proximal tumor localization, older age, female gender, high degree of differentiation and mucinous histological type, mutations in KRAS and BRAF genes and wild-type TP53 were also identified [77, 83-85].
The description of CIMP criteria is not unified among studies. The major challenge is the selection of specific loci, methylation status of which should be used to define CIMP. Most studies use the classic panel: MLH1, p16, MINT1, MINT2, and MINT31 [84, 86, 87]. This panel can be extended to include CACNA1G, CRABP1, IGF2, NEUROG1, RUNX3, SOCS1, HIC1, IGFBP3, and WRN [86, 88]. Most of the CIMP tumors are characterized by microsatellite instability and lack of chromosomal instability [3]. The accurate cause of dense aberrant DNA methylation in CIMP tumors remains incompletely clear. However, several factors that may be involved in this process were found. For example, increased expression of DNA methyltransferase-3B (DNMT3B) was detected in CIMP-H tumors [89]. DNMT3B expression was also increased during tumor progression and correlated with methylation level of CIMP-associated genes (NEUROG1, CACNA1G, and CDKN2A) and SFRP2 gene, suggesting the presence of a certain relationship between these processes [90]. The interconnection of genetic factors, dietary features and bad habits with CIMP-H has been also analyzed in a number of epidemiological studies. Smoking was shown to be associated with the presence of CIMP-H colorectal tumors and BRAF mutations [91, 92]. There is a relationship between the presence of a number of genetic variants in the genes of folate metabolism (MTHFR, MTR, and MTRR). In particular, high probability of CIMP-H occurrence is described for patients with specific alleles of MTHFR gene, low consumption of folate and methionine, and high alcohol consumption [93-95].
The effect of CIMP status on prognosis has been analyzed in a large number of studies, but the results are not self-consistent enough. The results of the systematic review across 36 studies (2003—2015) suggest a poorer prognosis for patients with CIMP-positive/CIMP-H colorectal tumors compared to CIMP-N or CIMP-L CRCs [96]. The prognosis strongly depends on MSI tumor status. CIMP-H seems to be a marker of poor prognosis, but its effect can be compensated in tumors with microsatellite instability: several studies report that shorter cancer-specific survival for CIMP-H group compared to CIMP-negative patients is observed only in the MSS subgroup [97, 98]. However, CIMP+/MSI+ tumors were closely associated with poorer differentiation and worse overall survival of patients compared to CIMP-/MSI+ cancers [99].
Other clinical factors or genetic characteristics can affect the significance of DNA methylation profiles as a prognostic marker. Proximally localized CIMP-H tumors are characterized with a higher rate of recurrence than CIMP-L and CIMP-N, while for the distal tumors such association was not revealed [100]. In another study, it was shown that the CIMP-H phenotype is associated with poor prognosis only in the case of rectal cancer (Asian population) [101]. Several clinical characteristics that correlate with CIMP-H status come from the presence of microsatellite instability, since CIMP-H is most frequently followed with MSI-H [86]. This makes a challenge to extract CIMP features which are not caused by MSI. However, some conclusions can be made here: CIMP is associated with BRAF (and less frequently - KRAS) mutations [3, 102-104], and some authors just ascribe poor clinical outcome of CIMP-H patients to the presence of BRAF/KRAS mutations [102, 105].
The effectiveness of 5-fluorouracil therapy for patients with different CIMP status was also investigated in several studies, but the data are extremely contradictory, and that can be explained by methodological differences, primarily in gene panels that were used for the status determination [70, 106-108].
EXCISION REPAIR SYSTEM
One of the most common drugs used in colorectal cancer chemotherapy is oxaliplatin, which is a third-generation platinum derivative containing 1,2-diaminocyclohexane in its structure. In combination with 5-FU, oxaliplatin has been approved as first-line therapy for metastatic CRC [109, 110]. Like many other platinum-based compounds, oxaliplatin reacts with DNA and forms DNA-platinum macromolecular adducts, inter- and intra-strand crosslinks that block DNA synthesis and further replication. This drug has a wide spectrum of cytotoxicity in vitro and antitumor activity in vivo in different tumor models. Apoptosis of cancer cells can be caused by formation of DNA lesions, block of DNA synthesis, inhibition of RNA synthesis, and triggering of immune reactions [111]. The main role in repairing platinum-induced DNA damage is played by nucleotide excision repair (NER) system. One of the key DNA repair enzymes is ERCC1 (Excision repair cross-complementing group 1) whereas ERCC2 and XRCC1 (X-Ray repair complementing defective repair) are also of paramount importance.
It has been demonstrated that overexpression of ERCC1 truly correlates with low sensitivity of tumors to the platinum-based drugs. Among polymorphic germinal variants of ERCC1, synonymous variants in 118th codon (Asn118Asn) are mostly studied in the context of response to oxaliplatin therapy. This synonymic substitution is associated with the altered gene expression at mRNA and protein levels, and, as a consequence, altered NER efficacy and response to oxaliplatin [112-114]. The frequency of ERCC1 allele, which is associated with reduced gene expression and better response to chemotherapy, is about 35% in the European population [114]. The same observations have been marked for the non-synonymous variants in ERCC2 (Lys751Gln) [115] and XRCC1 (Arg399Gln) genes [116].
However, the results are poorly consistent between studies. Meta-analysis of 22 studies (2004—2013; 2846 patients) revealed no differences of objective response to oxaliplatin-based chemotherapy between patients bearing distinct ERCC1 variants, but did revealed significant differences between progression-free and overall survival. Also the impact of ethnicity factor had been demonstrated [117].
Numerous studies have shown a relationship between ERCC1 overexpression and resistance of various other tumors to platinum-based drugs [118, 119].. In human ovarian and gastric cancers high expression level of ERCC1 is observed in a moderate number of cases, but in colon, lung and breast cancers, the high level of the expression is observed for more than one third of patients. This may suggest the low effect of platinum-based drugs for such type of patients making this kind of therapy not only unreasonable but even dangerous because of the high toxicity of the drugs.
MUTATIONS IN POLYMERASE GENES
Among mutations in genes encoding for different polymerases, mutations in the polymerase ε gene (Pol ε/POLE) are the mostly studied in colorectal cancer. Polymerase ε is one of three polymerases (α, δ, and ε) that mostly responsible for replication of nuclear DNA. Moreover, Pol ε performs the synthesis stage in the DNA repairing process and takes part in the recombination [120]. The significance of POLE mutations in carcinogenesis was confirmed by several studies, mainly in endometrial and colorectal cancers [64, 121, 122]. Although there is only a weak correlation between the presence of mutations in the exonuclease domain of Pol ε and the decrease of overall survival in MSS colorectal tumors [123], it was shown that POLE germinal mutations may be responsible for predisposition to colorectal and other cancers, including Lynch syndrome [122, 124-126]. In endometrial tumors, POLE mutations can play role in the development of microsatellite instability [127].
Polymerase δ also takes part in DNA reparation. It has been shown that germinal mutations in the polymerase δ gene, as well as in the polymerase ε gene, result in a high risk of development of multiple colorectal adenomas and adenocarcinomas [125]. Tumors that are formed as a result of mutations in the polymerase δ and ε genes, are characterized by an extremely high mutation frequency (over 1 million in a genome) against the background of microsatellite stability. Tumors with such characteristics may be isolated into a separate group [64]. In contrast, mutations in the polymerase α gene are rare and thus can be exposed to a negative selection in tumors [128].
SIGNAL TRANSDUCERS (KRAS, NRAS, BRAF, PIK3CA)
KRAS, NRAS, BRAF, and PIK3CA genes belong to proto-oncogenes and encode proteins that are involved in intracellular signal transduction from growth factor receptors. Driver mutations are frequently found in these genes; they are mostly localized in the 2nd exon of KRAS (codons 12 and 13), 3rd exon of NRAS (codon 61), 15th exon of BRAF (codon 600), and 9th, 20th exons of PIK3CA. In normal cells, RAS proteins are activated after binding growth factors to receptor tyrosine kinases. The presence of KRAS (Kirsten Rat Sarcoma Viral Oncogene Homolog) mutations described above results in constitutively active KRAS protein and eliminates the need for EGFR ligand for activation downstream MAPK and PI3K/mTOR pathways. These two pathways are responsible for cell growth, proliferation, differentiation, migration in normal cells and strongly contribute to progression of tumors. BRAF (v-Raf murine sarcoma viral oncogene homolog B) is the direct downstream target of KRAS, the second element of RAS/RAF/MEK/MAPK cascade. Mutations in 600th BRAF codon result in constitutively active MAPK signaling. Unlike KRAS, BRAF does not regulate PI3K/mTOR pathway.
Mutation status of KRAS, NRAS, BRAF and PIK3CA genes can be used as a prognostic and especially as predictive marker: the presence of a mutation in the hotspots of any of these genes is associated with a potential resistance to inhibitors of receptor tyrosine kinases, as well as to monotherapy with mTOR inhibitors [129-131]. Moreover, detection and quantification of mutated alleles in circulating tumor cells (CTC) enables non-invasive monitoring of response to therapy [132].
KRAS and NRAS mutations
The RAS subfamily consists of about 30 structurally related proteins, small GTPases, mainly regulating cell proliferation [133]. However, only three members, KRAS, NRAS and HRAS, are of paramount importance in the context of cancer development and clinical significance. Mutations of these gene have been identified in many tumor types. In solid tumors, such as colorectal, lung and pancreatic cancers, mutations in KRAS gene occur much more frequently than in NRAS gene. The inverse picture has been observed in some hematological diseases such as acute lymphoblastic and chronic myelomonocytic leukemia and Hodgkin’s lymphoma [134]. Approximately 90% of KRAS activating mutations are located in codons 12 and 13, which belong to the second exon. Another driver mutations are located in codons 59, 61 (third exon), 117, 146 (fourth exon) [135-137]. 30%-50% of colorectal cancers have KRAS mutations [3, 138]. HRAS mutations are not significant in CRC.
In a massive study involving patients with stage II and III colon cancer, it was shown that KRAS mutations do not have predictive value in assessing the overall and relapse-free survivals [139]. However, a number of studies have suggested clinical significance of KRAS mutations as prognostic markers [131, 140]. The presence of activating mutations in KRAS and NRAS (as well as inactivating mutations in PTEN) is marker of insensitivity to anti-EGFR therapy, particularly, using antibodies (cetuximab, panitumumab) [141, 142]. Absence of mutations in KRAS and NRAS does not imply the sensitivity to the therapy, but their presence can exactly predict the lack of response. In this case, combining mTOR and EGFR inhibition as well as simultaneous inhibition of mTOR and Bcl-2/Bcl-xL may improve therapeutic outcome in patients with KRAS- or BRAF-mutant CRC [143, 144].
It appears that there are other mutations associated with the resistance to cetuximab and panitumumab. For example, it has been reported that mutations in the PIK3CA and BRAF genes may are also predictive markers of absence of objective response to the anti-EGFR therapy, but conclusive demonstration on a large set of patients is currently absent. Treatment with cetuximab and panitumumab should be carried out only after obtaining data on the mutation status of the three exons of KRAS and NRAS genes, as this allows to prevent undesirable toxic effects of the drugs in the absence of objective response to the therapy.
According to the results of preclinical studies, mutations in NRAS, KRAS, BRAF genes are associated with sensitivity of tumors to inhibitors of Hsp90 and combinations of 1) sorafenib and irinotecan (in heavily pretreated mCRC); 2) MEK and PI3K/mTOR inhibitors; 3) sorafenib or MEK inhibitors; 4) Bcl-2/Bcl-xL and mTOR inhibitors; 5) anti-EGFR drugs and mTOR inhibitors [143-147]. Mutations in the KRAS and NRAS genes are actively studied as markers of response to anti-VEGF therapy (bevacizumab) [148-151].
BRAF mutations
BRAF is one of three members of the RAF (Rapidly Accelerated Fibrosarcoma) serine/threonine protein kinase family. BRAF is a downstream target of KRAS. In normal cells, RAF activation occurs as a result of many complex processes, which needs binding of proteins and ligands, conformational changes, and numerous (de)phosphorylation events having regulatory character [152, 153]. BRAF mutations in 600th codon lead to the constitutive activation of the BRAF protein and downstream elements of MAPK cascade. BRAF mutations are found in different types of cancer, especially in those with poor prognosis, for example, in more than 60% of metastatic melanoma, 40-70% of papillary thyroid cancer, and up to 18% of CRC [154]. Various studies have demonstrated different frequency of BRAF mutation occurrence in CRC - from 4% to 18% [155-157]. To date, more than 50 different BRAF mutations have been documented for CRC. However, in 80% of cases, mutations are represented by V600E substitution [156]. BRAF mutations are found highly enriched in right-sided proximal tumors [158].
BRAF mutations are considered to be oncogenic drivers, since they occur at early stages of carcinogenesis, causing transformation of epithelia into serrated adenomas [159]. BRAF plays an important role in CRC progression and metastasis. Particularly, BRAF constitutive activation was shown to induce disturbance of the polarity of epithelial cells via activating the expression of Myc [160].
BRAF mutations frequently occur in microsatellite instable tumors, which are usually associated with a failure of a mismatch repair system. According to the results of several studies, this group of patients have a favorable prognosis, and presence of BRAF mutations in MSI-H tumors do not correlate with reduced overall or disease-free survival [139]. BRAF mutations are rarely found in MSS or MSI-L tumors, which are characterized by poor prognosis [139, 161-163]. Large-scale study involving 1404 patients with stage II and III CRC demonstrated the association of BRAF mutations with the female gender, localization of tumor in the right side, older age (60 years or more), a high grade of anaplasia, and microsatellite instability. At the same time, BRAF mutations do not have prognostic potential for the assessment of time to disease progression, but can represent a marker of overall survival of MSI-L and MSS patients [139]. However, some authors assess prognostic significance of BRAF mutations not taking into account MSI status. The results of these studies suggest that BRAF mutations may be considered as an independent prognostic factor for disease-free and overall survivals in locally advanced and recurrent CRC: presence of BRAF mutations is associated with metastasis and represents a marker of unfavorable prognosis [157, 161, 164, 165].
In contract to KRAS, the predictive value of mutated BRAF in the context of response to anti-EGFR therapy is still discussible [159]. However, the results of two recent meta-analyses support the necessity of BRAF mutation study before initiation of treatment with anti-EGFR therapy. The first study by Pietrantonio et al included 463 RAS-wt/BRAF-mut patients from 10 trials and demonstrated that the addition of cetuximab and panitumumab treatment for the BRAF-mut patients did not improve progression free, overall survival, overall response rates [166]. The second study by Roland et al covered 8 trials with 3168 RAS-wt participants including 351 RAS-wt/BRAF-mut patients [167]. The results of this meta-analysis demonstrated increased both progression-free and overall survival in RAS-wt/BRAF-wt patients compared to RAS-wt/BRAF-mut (20-40% decreased hazard ratio). However, the results did not meet statistical significance criteria [167].
“Classical” BRAF mutations (substitutions in codons 600 and 601) are associated with tumor cell sensitivity to BRAF inhibitors: most commonly used of them are vemurafenib and dabrafenib, whereas other are also known: encorafenib, and experimental XL281 (Exelixis), CEP-32496. However, BRAF-targeted therapy may eventually fail because of the developing resistance. The vast majority of these escape mechanisms are driven by feedback reactivation of EGFR that activates in turn MAPK via other RAFs (CRAF) and RAS [159, 168, 169]. This suggest usage of anti-BRAF therapy in combination with inhibitors of EGFR and MEK, but here we also expect pitfalls as an acquired resistance driven by alterations in MAPK participants, which may be overcome via ERK inhibition [170]. “Non-classical” BRAF mutations may be responsible for tumor cell resistance to vemurafenib and dabrafenib, but increased sensitivity to sorafenib (multi-kinase inhibitor) and MEK inhibitors. Most likely, “non-classical” BRAF mutations are markers of favorable prognosis [171].
PIK3CA mutations
PIK3CA is a catalytic subunit of phosphatidylinositol 3 kinase (PIK3). Mutations of PIK3CA gene are frequently found in various solid tumors, particularly in CRC. According to the results of several studies, PIK3CA mutations are present in 10-20% of CRC cases [172-175]. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers [176]. Presence of PIK3CA mutations is associated with the mucinous CRC phenotype [174, 177]. About 80% of PIK3CA mutations are located in hotspots of 9th exon (codons 542 and 545) and 20th exon (codon 1047) [178]. Mutations in the 9th exon are found more frequently, whereas simultaneous mutations in both 9th and 20th exons are very rare [177, 179, 180]. There is a gradual decrease in frequency of PIK3CA mutations as it moves from the proximal (cecum, 21-25%) to distal (sigmoid colon, rectum, 8-9%) sites of the intestine [177, 179]. Against the background of absence of RAS mutations, PIK3CA substitutions in exon 20 seem to be marker of poor prognosis and potential inefficacy of anti-EGFR therapy [181-183].
Surprisingly, PIK3CA mutations as well as overexpression of cyclooxygenase COX-2 represent a marker of good response to therapy with Aspirin [172, 184-186]. The results of meta-analysis by Li et al across 7 studies (35 thousand patients) suggest that post-diagnosis aspirin therapy improves CRC overall survival, especially for patients with tumors positive for PTGS2 (COX-2) expression and PIK3CA mutations [187]. Another meta-analysis by Paleari et al included 4589 patients and revealed 29%-reduced total mortality with Aspirin treatment of PIK3CA-mut tumors, whereas no significant improvements were found for PIK3CA-wt tumors [185].
The major targets of Aspirin are constitutively expressed isoform of cyclooxygenase (COX-1/PTGS1) and its inducible isoform (COX-2/PTGS2), which is expressed under cytokine, inflammatory stimuli, and some growth factors. Aspirin has approximately 150—200-fold more inhibition potency for COX-1 rather than COX-2 [188]. Aspirin catalyzes acetylation of COX-1 at serine-529 in the substrate binding channel and inactivates the enzyme irreversibly, whereas most of other nonsteroidal anti-inflammatory drugs do it reversibly [189]. The exact mechanisms of interplay between mutations in PIK3CA and aspirin are still elusive [183, 190].
Anti-cancer activity of Aspirin may include several mechanisms. First, Aspirin contributes to the downregulating MAPK, β-catenin, Akt/mTOR, PKA, NF-kB pathways, as it was shown in different models [191-195]. Inhibition of COX-2 by Aspirin prevents synthesis of prostaglandins E2 and subsequent activation of EP1-4 prostaglandin receptors, some of which are participating colonic tumorigenesis and invasion [194, 196]. In colon cancer cells, EP2 is capable of activating β-catenin, the central element of Wnt/β-catenin pathway, which is the major player in colorectal cancer induction, growth and invasion. EP2 stimulate dissociation of β-catenin/GSK3β/Axin/APC complex and subsequent release of β-catenin by two ways: 1) α-subunit of EP2-coupling trimeric G-protein directly interacts with Axin, and 2) β/γ-subunits of G-protein activate PI3K and Akt, whereas the last one phosphorylates GSK3β [196] (Figure 1). Another prostaglandin receptor, EP4 is capable of activating MAPK pathway also by two ways: 1) EP4 induces Src—β-arrestin-mediated transactivation of EGFR, and 2) activating MAPK signaling by means of PI3K [192, 197, 198]. Pro-angiogenic activity of EP4 mediated via protein kinase A pathways is also known [199].
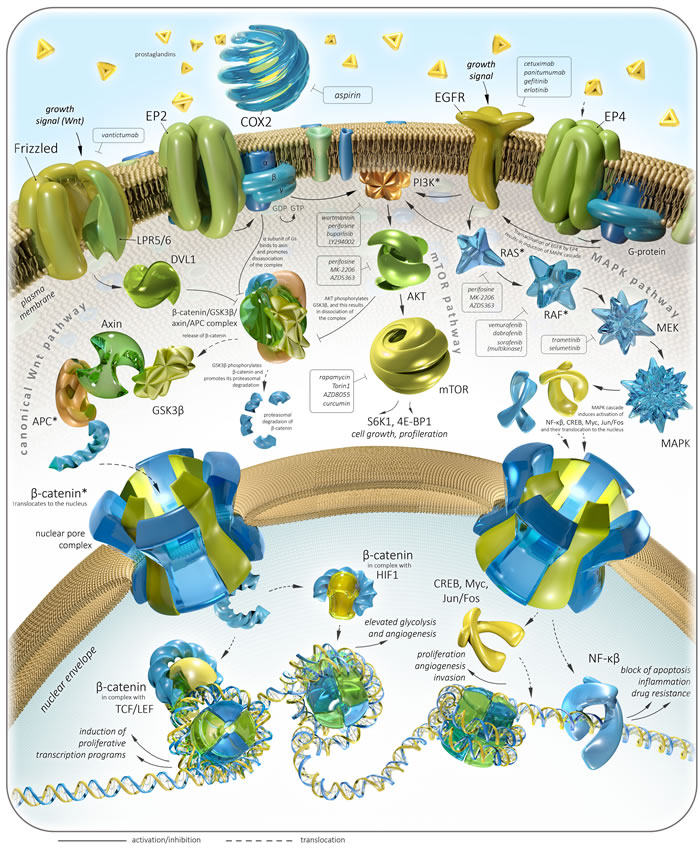
Figure 1: Colorectal cancer-related pathways and therapeutic targets with focus on Aspirin. Asterisks (*) indicate genes harboring the most frequent and clinically significant driver mutations in CRC.
Second, inhibition of COX-2, shifts balance between pro- and anti-inflammatory cytokines, including these secreted with peripheral blood mononuclear cells in response to tumor cells [200]. In turn, inflammation is well known contributor to tumor initiation, progression, invasion and metastasis [201, 202].
The mechanisms of Aspirin anti-metastatic activity include disrupting interaction between platelets and tumor cells. In platelets, COX-1 is involved in the generation of thromboxane A2 (TXA2), which promotes platelet activation and aggregation. In the recent study, Paloma Guillem-Llobat et al found that co-cultivation of HT29 CRC cells with platelets resulted in the formation of mesenchymal-like tumor cells with enhanced cell mobility, aggregative properties regarding to platelets and significantly increased metastatic potential. Aspirin prevented these changes [203]. Another mechanism of platelet-mediated metastasis is related to platelet aggregation and thrombi formation around circulating tumor cells. Such thrombi protect CTCs from natural killer cells activity and other factors, enable adhesion to macrophages, which promote the invasion [204-208]. The anti-metastatic mechanisms of Aspirin are not limited to interactions of tumor cells and platelets: recently it was shown that Aspirin suppresses both the growth and metastasis of osteosarcoma through the NF-κB pathway [209]. In addition, combination of tyrosine kinase inhibitor sorafenib with Aspirin may be preferred to sorafenib alone with respect to preventing metastasis [210].
Surprisingly, aspirin with combination of isoform-specific COX2 inhibitor celecoxib and lipid-lowering statin atorvastatin demonstrated more efficient inhibition of azoxymethane-induced colon carcinogenesis in rats rather than when these agents were given individually at higher doses [211].
TP53 MUTATIONS
The TP53 (p53) tumor suppressor plays a crucial role in the response to stress [212]. It is a component of the cell cycle checkpoint system, it sustains genome integrity, induces cell cycle arrest and apoptosis. Active TP53 is maintained at low levels in the most of cells: once activated, TP53 upregulates E3 ubiquitin-ligase MDM2, which promote TP53 proteasomal degradation [213, 214]. Activated TP53 can either induce cell cycle arrest, inhibit cell growth or promote cell apoptosis depending on different type of stress (including DNA damage) and the other context [213]. TP53 can trigger both intrinsic (mitochondrial) and extrinsic (death receptor-induced) apoptotic pathways: TP53 activates the expression of pro-apoptotic proteins Bax, Noxa and PUMA and downregulates anti-apoptotic Bcl-2. This leads to the formation of mitochondrial permeability pore and subsequent release of cytochrome c which is responsible for the induction of caspase cascade [215-217]. On the other hand, active TP53 upregulates the expression of some death receptor genes, such as Fas, DR5 and PIDD [214]. Among TP53 targets there are well known tumor suppressors GADD45, WAF1 (p21), Rb, 14-3-3-σ. The role of TP53 has been shown to go beyond its effects on cell cycle and apoptosis [218]. TP53 is involved in the metabolic reprogramming, regulation of intracellular reactive oxygen species levels, once more highlighting its role in cell death or cell survival [219-221].
Failure of TP53 pathways is one of the hallmarks of cancer cells [222, 223]. Up to 50% of human malignancies contain TP53 mutations. In the rest of tumors, TP53 pathways have other deficiencies to ensure uncontrolled growth and proliferation. These alterations play an important role in CRC onset and progression and may represent a marker of the response to chemotherapy, radiotherapy or their combination. Driver mutations of most of tumor suppressor genes are associated with almost complete loss of protein function or expression. In contrast, not all of TP53 driver mutations result in complete loss of TP53 functionality. Tumor-associated mutant proteoforms of TP53 often gain new tumorigenic activities. In particular, not only completely losses of TP53 activity but ‘gain-of-function’ TP53-mutants mediate tumor metabolic reprogramming which promotes tumor progression and invasion [220, 221, 224, 225]. TP53 mutant proteoforms may inhibit apoptosis, which normally occurs in the response to ionizing radiation and anti-cancer drugs (doxorubicin, platinum-containing agents, etc.) [226-228]. The presence of TP53 Pro72Arg germlinne variant (rs1042522) may be associated with increased sensitivity to 5-fluorouracil [229].
GERMINAL VARIATIONS IN GENES enCODING DRUG METABOLISING ENZYMES
Mutations of dihydropyrimidine dehydrogenase (DPYD)
The DPYD gene encodes a cytoplasmic enzyme involved in pyrimidine catabolism. Its expression level and enzymatic activity negatively correlates with both efficiency and toxicity of 5-fluorouracil and related antitumor drugs [230]. Germline inactivating DPYD mutations cause the so-called DPYD syndrome involving life-threatening complications of 5-FU or capecitabine treatment [231]. In order to prevent these potentially fatal outcomes, DPYD testing before administration of pyrimidine antimetabolites is advocated by several groups [232]. For the carriers of a non-functional DPYD allele, current guidelines recommend complete avoidance of 5-FU and related drugs in case of homozygous mutation and at least 50% dose reduction in case of heterozygous one [233]. DPYD mutation analysis may be rather cumbersome due to absence of mutation hotspots (except 3 very high risk alleles) [234], unknown significance of many allelic variants and possible involvement of epigenetic mechanisms (including but not limited to the levels of DPYD promoter methylation and miR-494 expression [235]). Therefore, ELISA-based testing for DPYD enzymatic activity seems to be a reasonable alternative.
However, wide clinical acceptance of DPYD testing in general is hampered by relatively low incidence of DPYD mutations (minor allele frequency for each substitution is below 0.01%). In CRC (as well as in gastric cancer and several other solid tumors), high levels of DPYD expression and activity in tumor cells correlate with relative resistance to 5-FU and capecitabine [236, 237] but not to S-1 or raltitrexed [238], whereas tumors with low DPYD, low TYMS and high TYMP expression levels were found to be exquisitely sensitive to capecitabine [239]. These trends were seen in many retrospective analyses, but no definitive prospective studies on the subject were ever conducted, so validated algorithms for antimetabolite therapy personalization in CRC are still lacking.
Variations in UDP glucuronosyltransferase (UGT1A1)
The UGT1A1 gene encodes a cytoplasmic enzyme involved in detoxification of a wide range of metabolites and xenobiotics including bilirubin and SN-38, the most active metabolite of irinotecan. Several allelic variants in the TATA-box of the UGT1A1 promoter (UGT1A1*28 is the most common alternative allele) are associated with a decreased level of its expression and activity, which in turn leads to rapid accumulation of its unmodified substrates in several tissues and organs, mainly in the liver and blood. Individuals harboring UGT1A1*28 allele are at high risk for transitory neonatal hyperbilirubinemia (Gilbert and Kriggler-Nayyaar syndromes) and for life-threatening toxicities of irinotecan, including deep neutropenia and fatal diarrhea. Therefore, irinotecan dose adjustment is highly required in CRC patients homozygous for this allele and may also be discussed in heterozygous cases having any other risk factors for increased sensitivity to irinotecan [240, 241].
CLASSIFICATION OF COLORECTAL CANCER
A set of molecular markers is usually used for CRC classification, which should accelerate understanding of the causation and facilitate clinical management in the areas of both prevention and treatment. Traditionally, colorectal cancer has been classified according to three molecular features: chromosomal instability, microsatellite instability, and the CpG island methylator phenotype. Unfortunately, such a classification is very conditional, and sets of markers of each group do not uniquely represent their unequivocal signs. In 2007, with the development of molecular genetics, Jeremy Jass proposed to classify CRC into five molecular subtypes (Figure 2):
1. CIMP-H, MSI-H and BRAF mutation;
2. CIMP-H, MSI-L or MSS/BRAF mutation;
3. CIMP-L/MSS or MSI-L/KRAS mutation;
4. CIMP-negative/MSS;
5. CIMP-negative/MSI-H or Lynch syndrome.
The molecular pathways are determined at an early stage of tumor formation. Serrated polyps seem to be the precursors of CRC types 1 and 2, whereas CRC types 4 and 5 evolve through the adenoma-carcinoma transformation. CRC types 1 and 4 represent two “reference points” with minimal overlap, Each type has its own histological features and clinical picture [242].
The evolution of CRC classification tended to shift from descriptive to integrative approaches and to link of traditional CRC classification (CIMP, MSI/MSS, CIN) to the underlying alterations of signaling pathways (Wnt, TGF-β, Ras) and driver mutations. In 1990, Eric Fearon and Bert Vogelstein suggested a multistep model of colorectal carcinogenesis which became a paradigm for the next years [243]. This model describes APC inactivating mutations as the first event of CRC development. Then, KRAS activating mutation are taking place. Further, other mutations in elements of TP53, PI3K, TGF-β and other pathways occur [243, 244]. This model became a basis for understanding colorectal cancer development. During next years, the principle of the model remained unchanged: several hits are needed for CRC onset. However, the model was subject to many refinements and additions [245, 246].
In 2010, Barbara Leggett and Vicki Whitehall summarized accumulated data on CRC development and suggested new classification system of sporadic CRC based on molecular pathways of its onset and progression [5]:
- Serrated pathway. The initial event is BRAF mutation, which leads to the formation of microvesicular hyperplastic polyp. Subsequent promoter methylation of p16, IGFBP7 results in the formation of sessile serrated adenomas. Finally, methylation of either MLH1 and TGFβ receptor II genes occurs (and this results to MSI-H, CIMP-H cancer) or other genes along with possible mutations in TP53, losses of 18q, deregulation of Wnt pathway (this results to MSS, CIMP-H cancer).
- Alternative pathway. This pathway has two alternate starting points. The first one is KRAS mutations followed with possible methylation of p16, IGFBP7, deregulation of Wnt pathway. This leads to the development of traditional serrated adenomas. The second starting point is mutation in APC leading to the formation of small tubular adenomas. Then, MGMT methylation, KRAS mutations along with chromosomal instability occurs. This results in the onset of tubulovillous adenoma. Both tubular and tubulovillous adenomas develop into MSS, CIMP-L colorectal cancer.
- Traditional pathway. Like alternate pathway, starting point here is APC mutation with subsequent formation of small tubular adenoma. Then, TP53 mutations, losses of 18q and chromosomal instability take place. This results in the growth of tubular adenomas with the development of severe dysplasia. Finally, it can result in MSS, CIN-H, CIMP-negative colorectal cancer with no mutations in BRAF and KRAS.
With the spread of high-throughput methods of genome and transcriptome sequencing, several novel approaches for CRC classification have evolved. The Cancer Genome Atlas (TCGA), the greatest project in the field of molecular oncology, comprises genome, transcriptome and methylome data for several hundred CRC samples. In 2012, analysis of these multidimensional datasets (RNA-Seq, mutations, methylation) allowed for a new look at the classification of colorectal cancer [3]. Two major classes of CRC were found: hypermutated (HM, > 10 non-silent substitutions per 1 Mb) and non-hypermutated (non-HM) tumors (Figure 2). These groups demonstrated the most dramatic difference in gene expression profiles between each other. Intra-group variation was significantly lower than inter-group. In turn, each group could be subdivided into several subgroups according to alterations in the signaling pathways (Wnt, TGF-β, RTK/RAS, PI3K, TP53), driver mutations and classical subtyping (CIMP, CIN, MSI). HM and non-HM tumors revealed the following differences in Wnt, TGF-β and RAS - three pathways which are the mostly altered in CRC:
- TGF-β signaling was repressed in 84% and only 27% of HM and non-HM tumors, accordingly;
- 80% and 59% of HM and non-HM tumors demonstrated activation of RTK/RAS signaling;
- 97% HM tumors demonstrated activation of Wnt pathway as opposed to only 92% of non-HM tumors;
Non-HM tumors have more dramatic changes in gene expression profiles. The following regularities can be remarked here:
- down-regulation of FOXA1 targets and genes involved in inflammatory response (only non-HM tumors);
- up-regulation of integrins and genes responsible for angiogenesis (only non-HM);
- increased expression of MYB targets in HM tumors; increased expression of p63 targets and non-homologous end joining repair system genes in HM tumors.
The further subdivision of CRC into groups smaller than HM and non-HM is strongly complicated because of the presence of various combinations of pathway alterations. However, some observations can be done here. For example, non-HM tumors with repressed TGF-β pathway have higher frequency of RTK/RAS activation than non-HM tumors with quasi-intact TGF-β pathway. Among the 30 HM tumor samples, 23 (77%) were MSI-H but none of non-HM tumors had MSI-H phenotype. Moreover, 19 of these MSI-H 23 tumors had MLH1 methylation, 17 of 23 had CIMP-H phenotype. Remarkably, excluding hypermutated samples, colon and rectum cancers demonstrated similar patterns of genomic alterations [3].
One year earlier, in 2011, Anita Sveen et al evaluated frequencies of exon skipping or inclusion in 160 colorectal cancer samples using microarrays and identifies transcriptome instability (TIN) as one of the CRC characteristics [247]. TIN tumors show skewed exon usage profiles, which strongly correlated with the expression of almost half of 280 splicing factors. Tumors of TIN phenotype account for 30-55% colorectal cancers. There were significant associations between transcriptome instability and reduced patient survival. However, no association between MSI and TIN was found. In 2014, Sveen extended this conception to other cancer types and identified TIN as pan-cancer characteristics [248].

Figure 2: Four approaches of colorectal cancer classification. Size of boxes indicates the frequency of the corresponding group or feature (except Leggett’s approach). Dotted boxes indicate that the frequency of feature/group is evaluative
Another approach for CRC classification has emerged from the results of CRC proteomic study in 2014 [2]. Protein abundances in 79 TCGA colorectal cancer samples were analyzed using panoramic mass-spectrometry approaches (LC-MS/MS). Cluster analysis of these data revealed five groups of samples with the distinct patterns of protein expression. These subtypes (A-E) contained 15, 9, 25, 11, and 19 samples, respectively. It was also shown that the mRNA level does not always reliably predict protein abundance. 32% of genes did not have statistically significant correlation between mRNA and protein levels. According to the results of gene set enrichment analysis based on Gene Ontology data, biological functions of proteins that have consistent mRNA and protein levels were different from those without the consistency.
The authors studied the associations between protein expression patterns and genomic and epigenomic characteristics of the tumors. Below we provide a summary for each subtype:
A. MSI-L or MSS, 18q losses, KRAS mutations, absence of POLE and BRAF mutations, CIMP-N;
B. MSI-H, hypermutation, POLE and BRAF mutations, absence of TP53 mutations and 18q losses, CIMP-H;
C. medium rate of TP53 mutations, several samples with MSI-H, BRAF and POLE mutations, rare CIMP; this is the largest and heterogeneous group;
D. MSI-L or MSS, 18q losses, TP53 mutations, rare CIMP;
E. “invasive” TCGA subtype; MSI-L or MSS, ubiquitous TP53 mutations and 18q losses, rare CIMP, HNF4α amplification and overexpression.
Finally, in 2013 Enric Domingo et al analyzed somatic mutations in 906 specimens (stages II and III) taken from patients participating VICTOR clinical trial and found many associations, both novel and well-known (e.g. co-incidence of CIN and mutations of TP53; MSI and BRAF mutation). Based on this data, Domingo et al suggested alternate system for CRC classification which assumes division into seven groups:
- MSI and/or BRAF mutations;
- CIN and/or TP53 mutations, wild-type KRAS and PIK3CA;
- KRAS and/or PIK3CA mutations, CIN, wild-type TP53;
- KRAS and/or PIK3CA mutations, CIN-negative, wild-type TP53;
- NRAS mutations;
- no mutations;
- others.
Such complexity of CRC classification reflects the diversity of possible scenarios of CRC development. These pathways not only have different starting points but also a number of bifurcation points during the disease progression from polyps, adenomas to adenocarcinomas. Future investigations are needed to construct the unified and exhaustive CRC molecular classification that can finally be translated into clinical practice. In conclusion, below we summarize the data regarding markers of prognosis, efficacy of treatment and side effects of chemotherapy.:
- Markers of unfavorable prognosis: CIN; deletions in 18q (discussable), 8p, 4p, 15q regions and inactivation of SMAD4, DCC, lengthening of telomeres in CIN tumors; CIMP-H in combination with MSS; BRAF mutations in MSS or MSI-L tumors; mutations in PIK3CA (20th exon), KRAS, NRAS.
- Markers of favorable prognosis: hypermutated tumor phenotype; MSI-H, especially in the absence of early onset of the disease and mutations in BRAF gene; CIMP-H in combination with MSI; proximal localization of CIMP-H tumors.
- Mutations in KRAS, NRAS, BRAF, PIK3CA (20th exon) are associated with resistance to targeted anti-EGFR therapy (cetuximab, panitumumab): .
- Mutations in KRAS, NRAS, BRAF are potential markers of sensitivity to inhibitors of Hsp90, combinations of 1) sorafenib and irinotecan (in heavily pretreated mCRC), 2) MEK and PI3K/mTOR inhibitors, 3) sorafenib or MEK inhibitors, 4) Bcl-2/Bcl-xL and mTOR inhibitors, 5) anti-EGFR drugs and mTOR inhibitors.
- MSI-H is associated with sensitivity to 5-FU and other fluoropyrimidines.
- Loss of 18q (including SMAD4 and DCC), reduced expression of SMAD4 is associated with lower response to 5-FU.
- Low expression level and specific allele variants of ERCC1, ERCC2, XRCC1 are associated with sensitivity of tumors to the platinum-based drugs.
- PIK3CA mutations and COX-2 overexpression are associated with longer PFS and OS in patients receiving adjuvant therapy with COX inhibitors including aspirin.
- TP53 Pro72Arg germlinne variant (rs1042522) may be associated with increased sensitivity to 5-FU.
- Expression level and specific allele variants of DPYD may seriously influence both efficacy and toxicity of 5-FU, capecitabine.
- Several germline variants in UGT1A1 are associated with severe toxicities of irinotecan.
ACKNOWLEDGMENTS
Authors thank National Medical Research Center of Radiology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, and Orekhovich Institute of Biomedical Chemistry for scientific advice and useful suggestions.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was financially supported by the Russian Science Foundation (RSCF), grant # 14-15-01083.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015; 65:87-108.
2. Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, Davies SR, Wang S, Wang P, Kinsinger CR, Rivers RC, Rodriguez H, et al. Proteogenomic characterization of human colon and rectal cancer. Nature. 2014; 513:382-387.
3. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330-337.
4. Pino MS and Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010; 138:2059-2072.
5. Leggett B and Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology. 2010; 138:2088-2100.
6. Boland CR and Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010; 138:2073-2087 e2073.
7. Lengauer C, Kinzler KW and Vogelstein B. Genetic instabilities in human cancers. Nature. 1998; 396:643-649.
8. Schneikert J and Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007; 56:417-425.
9. Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M, Maeda Y, Iwama T, Mishima Y, Mori T, Koike M. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer research. 1994; 54:3011-3020.
10. Najdi R, Holcombe RF and Waterman ML. Wnt signaling and colon carcinogenesis: beyond APC. Journal of carcinogenesis. 2011; 10:5.
11. Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M and Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nature genetics. 2008; 40:600-608.
12. Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, Baylin SB and Herman JG. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer research. 2000; 60:4366-4371.
13. Abdel-Rahman WM, Lotsari-Salomaa JE, Kaur S, Niskakoski A, Knuutila S, Jarvinen H, Mecklin JP and Peltomaki P. The Role of Chromosomal Instability and Epigenetics in Colorectal Cancers Lacking beta-Catenin/TCF Regulated Transcription. Gastroenterology research and practice. 2016; 2016:6089658.
14. Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH and Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nature cell biology. 2001; 3:433-438.
15. Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK and Nathke IS. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nature cell biology. 2001; 3:429-432.
16. Aoki K, Aoki M, Sugai M, Harada N, Miyoshi H, Tsukamoto T, Mizoshita T, Tatematsu M, Seno H, Chiba T, Oshima M, Hsieh CL and Taketo MM. Chromosomal instability by beta-catenin/TCF transcription in APC or beta-catenin mutant cells. Oncogene. 2007; 26:3511-3520.
17. Alberici P and Fodde R. The role of the APC tumor suppressor in chromosomal instability. Genome dynamics. 2006; 1:149-170.
18. Herzog F, Primorac I, Dube P, Lenart P, Sander B, Mechtler K, Stark H and Peters JM. Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science. 2009; 323:1477-1481.
19. Zhao Y, Ando K, Oki E, Ikawa-Yoshida A, Ida S, Kimura Y, Saeki H, Kitao H, Morita M and Maehara Y. Aberrations of BUBR1 and TP53 gene mutually associated with chromosomal instability in human colorectal cancer. Anticancer research. 2014; 34:5421-5427.
20. Overlack K, Primorac I, Vleugel M, Krenn V, Maffini S, Hoffmann I, Kops GJ and Musacchio A. A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. eLife. 2015; 4:e05269.
21. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW and Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998; 392:300-303.
22. Bardelli A, Cahill DP, Lederer G, Speicher MR, Kinzler KW, Vogelstein B and Lengauer C. Carcinogen-specific induction of genetic instability. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:5770-5775.
23. Ganem NJ, Godinho SA and Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009; 460:278-282.
24. Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A and Raff JW. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008; 133:1032-1042.
25. Lukasiewicz KB and Lingle WL. Aurora A, centrosome structure, and the centrosome cycle. Environmental and molecular mutagenesis. 2009; 50:602-619.
26. Zou J, Zhang D, Qin G, Chen X, Wang H and Zhang D. BRCA1 and FancJ cooperatively promote interstrand crosslinker induced centrosome amplification through the activation of polo-like kinase 1. Cell cycle. 2014; 13:3685-3697.
27. Ewart-Toland A, Briassouli P, de Koning JP, Mao JH, Yuan J, Chan F, MacCarthy-Morrogh L, Ponder BA, Nagase H, Burn J, Ball S, Almeida M, Linardopoulos S and Balmain A. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nature genetics. 2003; 34:403-412.
28. Anand S, Penrhyn-Lowe S and Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer cell. 2003; 3:51-62.
29. Takahashi Y, Sheridan P, Niida A, Sawada G, Uchi R, Mizuno H, Kurashige J, Sugimachi K, Sasaki S, Shimada Y, Hase K, Kusunoki M, Kudo S, Watanabe M, Yamada K, Sugihara K, et al. The AURKA/TPX2 axis drives colon tumorigenesis cooperatively with MYC. Annals of oncology. 2015; 26:935-942.
30. Baran V, Solc P, Kovarikova V, Rehak P and Sutovsky P. Polo-like kinase 1 is essential for the first mitotic division in the mouse embryo. Molecular reproduction and development. 2013; 80:522-534.
31. Archambault V and Carmena M. Polo-like kinase-activating kinases: Aurora A, Aurora B and what else? Cell cycle. 2012; 11:1490-1495.
32. Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y and Saji S. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer science. 2003; 94:148-152.
33. Han DP, Zhu QL, Cui JT, Wang PX, Qu S, Cao QF, Zong YP, Feng B, Zheng MH and Lu AG. Polo-like kinase 1 is overexpressed in colorectal cancer and participates in the migration and invasion of colorectal cancer cells. Medical science monitor. 2012; 18:BR237-246.
34. Baba Y, Nosho K, Shima K, Irahara N, Kure S, Toyoda S, Kirkner GJ, Goel A, Fuchs CS and Ogino S. Aurora-A expression is independently associated with chromosomal instability in colorectal cancer. Neoplasia. 2009; 11:418-425.
35. Davis SL, Robertson KM, Pitts TM, Tentler JJ, Bradshaw-Pierce EL, Klauck PJ, Bagby SM, Hyatt SL, Selby HM, Spreafico A, Ecsedy JA, Arcaroli JJ, Messersmith WA, Tan AC and Eckhardt SG. Combined inhibition of MEK and Aurora A kinase in KRAS/PIK3CA double-mutant colorectal cancer models. Frontiers in pharmacology. 2015; 6:120.
36. Xu WJ, Zhang S, Yang Y, Zhang N, Wang W, Liu SY, Tian HW, Dai L, Xie Q, Zhao X, Wei YQ and Deng HX. Efficient inhibition of human colorectal carcinoma growth by RNA interference targeting polo-like kinase 1 in vitro and in vivo. Cancer biotherapy & radiopharmaceuticals. 2011; 26:427-436.
37. Goktas S, Yildirim M, Suren D, Alikanoglu AS, Dilli UD, Bulbuller N, Sezer C and Yildiz M. Prognostic role of Aurora-A expression in metastatic colorectal cancer patients. Journal of BUON. 2014; 19:686-691.
38. Goos JA, Coupe VM, Diosdado B, Delis-Van Diemen PM, Karga C, Belien JA, Carvalho B, van den Tol MP, Verheul HM, Geldof AA, Meijer GA, Hoekstra OS, Fijneman RJ and DeCoDe PETg. Aurora kinase A (AURKA) expression in colorectal cancer liver metastasis is associated with poor prognosis. British journal of cancer. 2013; 109:2445-2452.
39. Bunting SF and Nussenzweig A. End-joining, translocations and cancer. Nature reviews Cancer. 2013; 13:443-454.
40. Matsui A, Ihara T, Suda H, Mikami H and Semba K. Gene amplification: mechanisms and involvement in cancer. Biomolecular concepts. 2013; 4:567-582.
41. Rudolph KL, Millard M, Bosenberg MW and DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nature genetics. 2001; 28:155-159.
42. Baichoo E and Boardman LA. Toward a molecular classification of colorectal cancer: the role of telomere length. Frontiers in oncology. 2014; 4:158.
43. Nakamura K, Furugori E, Esaki Y, Arai T, Sawabe M, Okayasu I, Fujiwara M, Kammori M, Mafune K, Kato M, Oshimura M, Sasajima K and Takubo K. Correlation of telomere lengths in normal and cancers tissue in the large bowel. Cancer letters. 2000; 158:179-184.
44. Engelhardt M, Drullinsky P, Guillem J and Moore MA. Telomerase and telomere length in the development and progression of premalignant lesions to colorectal cancer. Clinical cancer research. 1997; 3:1931-1941.
45. Chadeneau C, Hay K, Hirte HW, Gallinger S and Bacchetti S. Telomerase activity associated with acquisition of malignancy in human colorectal cancer. Cancer research. 1995; 55:2533-2536.
46. Augustine TA, Baig M, Sood A, Budagov T, Atzmon G, Mariadason JM, Aparo S, Maitra R and Goel S. Telomere length is a novel predictive biomarker of sensitivity to anti-EGFR therapy in metastatic colorectal cancer. British journal of cancer. 2015; 112:313-318.
47. Gertler R, Rosenberg R, Stricker D, Friederichs J, Hoos A, Werner M, Ulm K, Holzmann B, Nekarda H and Siewert JR. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. Journal of clinical oncology. 2004; 22:1807-1814.
48. Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C and Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003; 114:359-370.
49. Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM and Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003; 114:371-383.
50. Kobayashi J, Tauchi H, Chen B, Burma S, Tashiro S, Matsuura S, Tanimoto K, Chen DJ and Komatsu K. Histone H2AX participates the DNA damage-induced ATM activation through interaction with NBS1. Biochemical and biophysical research communications. 2009; 380:752-757.
51. Bakhoum SF, Kabeche L, Murnane JP, Zaki BI and Compton DA. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer discovery. 2014; 4:1281-1289.
52. Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Yamada H, Hayama T, Inoue E, Tamura J, Iinuma H, Akiyoshi T and Muto T. Chromosomal instability (CIN) phenotype, CIN high or CIN low, predicts survival for colorectal cancer. Journal of clinical oncology. 2012; 30:2256-2264.
53. Li LS, Kim NG, Kim SH, Park C, Kim H, Kang HJ, Koh KH, Kim SN, Kim WH, Kim NK and Kim H. Chromosomal imbalances in the colorectal carcinomas with microsatellite instability. The American journal of pathology. 2003; 163:1429-1436.
54. Yang G and Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. International journal of biological sciences. 2010; 6:1-8.
55. Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF and Tomlinson IP. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:9719-9723.
56. Tariq K and Ghias K. Colorectal cancer carcinogenesis: a review of mechanisms. Cancer biology & medicine. 2016; 13:120-135.
57. Freeman TJ, Smith JJ, Chen X, Washington MK, Roland JT, Means AL, Eschrich SA, Yeatman TJ, Deane NG and Beauchamp RD. Smad4-mediated signaling inhibits intestinal neoplasia by inhibiting expression of beta-catenin. Gastroenterology. 2012; 142:562-571 e562.
58. Kozak MM, von Eyben R, Pai J, Vossler SR, Limaye M, Jayachandran P, Anderson EM, Shaffer JL, Longacre T, Pai RK, Koong AC and Chang DT. Smad4 inactivation predicts for worse prognosis and response to fluorouracil-based treatment in colorectal cancer. Journal of clinical pathology. 2015; 68:341-345.
59. Popat S, Zhao D, Chen Z, Pan H, Shao Y, Chandler I and Houlston RS. Relationship between chromosome 18q status and colorectal cancer prognosis: a prospective, blinded analysis of 280 patients. Anticancer research. 2007; 27:627-633.
60. Lebok P, Mittenzwei A, Kluth M, Ozden C, Taskin B, Hussein K, Moller K, Hartmann A, Lebeau A, Witzel I, Mahner S, Wolber L, Janicke F, Geist S, Paluchowski P, Wilke C, et al. 8p deletion is strongly linked to poor prognosis in breast cancer. Cancer biology & therapy. 2015; 16:1080-1087.
61. El Gammal AT, Bruchmann M, Zustin J, Isbarn H, Hellwinkel OJ, Kollermann J, Sauter G, Simon R, Wilczak W, Schwarz J, Bokemeyer C, Brummendorf TH, Izbicki JR, Yekebas E, Fisch M, Huland H, et al. Chromosome 8p deletions and 8q gains are associated with tumor progression and poor prognosis in prostate cancer. Clinical cancer research. 2010; 16:56-64.
62. Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA and Domany E. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:7131-7136.
63. Donehower LA, Creighton CJ, Schultz N, Shinbrot E, Chang K, Gunaratne PH, Muzny D, Sander C, Hamilton SR, Gibbs RA and Wheeler D. MLH1-silenced and non-silenced subgroups of hypermutated colorectal carcinomas have distinct mutational landscapes. The Journal of pathology. 2013; 229:99-110.
64. Briggs S and Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. The Journal of pathology. 2013; 230:148-153.
65. Silva FC, Valentin MD, Ferreira Fde O, Carraro DM and Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo medical journal = Revista paulista de medicina. 2009; 127:46-51.
66. Ionov Y, Peinado MA, Malkhosyan S, Shibata D and Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993; 363:558-561.
67. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN and Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer research. 1998; 58:5248-5257.
68. Lindor NM, Burgart LJ, Leontovich O, Goldberg RM, Cunningham JM, Sargent DJ, Walsh-Vockley C, Petersen GM, Walsh MD, Leggett BA, Young JP, Barker MA, Jass JR, Hopper J, Gallinger S, Bapat B, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. Journal of clinical oncology. 2002; 20:1043-1048.
69. Thomas ML, Hewett PJ, Ruszkiewicz AR and Moore JW. Clinicopathological predictors of benefit from adjuvant chemotherapy for stage C colorectal cancer: Microsatellite unstable cases benefit. Asia-Pacific journal of clinical oncology. 2015; 11:343-351.
70. Carethers JM, Smith EJ, Behling CA, Nguyen L, Tajima A, Doctolero RT, Cabrera BL, Goel A, Arnold CA, Miyai K and Boland CR. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004; 126:394-401.
71. Klingbiel D, Saridaki Z, Roth AD, Bosman FT, Delorenzi M and Tejpar S. Prognosis of stage II and III colon cancer treated with adjuvant 5-fluorouracil or FOLFIRI in relation to microsatellite status: results of the PETACC-3 trial. Annals of oncology. 2015; 26:126-132.
72. Collura A, Lagrange A, Svrcek M, Marisa L, Buhard O, Guilloux A, Wanherdrick K, Dorard C, Taieb A, Saget A, Loh M, Soong R, Zeps N, Platell C, Mews A, Iacopetta B, et al. Patients with colorectal tumors with microsatellite instability and large deletions in HSP110 T17 have improved response to 5-fluorouracil-based chemotherapy. Gastroenterology. 2014; 146:401-411 e401.
73. Tougeron D, Mouillet G, Trouilloud I, Lecomte T, Coriat R, Aparicio T, Des Guetz G, Lecaille C, Artru P, Sickersen G, Cauchin E, Sefrioui D, Boussaha T, Ferru A, Matysiak-Budnik T, Silvain C, et al. Efficacy of Adjuvant Chemotherapy in Colon Cancer With Microsatellite Instability: A Large Multicenter AGEO Study. Journal of the National Cancer Institute. 2016; 108.
74. Sturgeon CM, Duffy MJ, Stenman UH, Lilja H, Brunner N, Chan DW, Babaian R, Bast RC, Jr., Dowell B, Esteva FJ, Haglund C, Harbeck N, Hayes DF, Holten-Andersen M, Klee GG, Lamerz R, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Clinical chemistry. 2008; 54:e11-79.
75. Duffy MJ, van Dalen A, Haglund C, Hansson L, Holinski-Feder E, Klapdor R, Lamerz R, Peltomaki P, Sturgeon C and Topolcan O. Tumour markers in colorectal cancer: European Group on Tumour Markers (EGTM) guidelines for clinical use. European journal of cancer. 2007; 43:1348-1360.
76. Locker GY, Hamilton S, Harris J, Jessup JM, Kemeny N, Macdonald JS, Somerfield MR, Hayes DF, Bast RC, Jr. and Asco. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. Journal of clinical oncology. 2006; 24:5313-5327.
77. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB and Issa JP. CpG island methylator phenotype in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:8681-8686.
78. Bae JM, Kim JH and Kang GH. Epigenetic alterations in colorectal cancer: the CpG island methylator phenotype. Histology and histopathology. 2013; 28:585-595.
79. Ang PW, Loh M, Liem N, Lim PL, Grieu F, Vaithilingam A, Platell C, Yong WP, Iacopetta B and Soong R. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC cancer. 2010; 10:227.
80. Sideris M and Papagrigoriadis S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer research. 2014; 34:2061-2068.
81. Ahuja N, Mohan AL, Li Q, Stolker JM, Herman JG, Hamilton SR, Baylin SB and Issa JP. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer research. 1997; 57:3370-3374.
82. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics. 2006; 38:787-793.
83. Toyota M, Ohe-Toyota M, Ahuja N and Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97:710-715.
84. Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, O’Connor T and Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002; 122:1376-1387.
85. Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK and Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005; 129:837-845.
86. Nazemalhosseini Mojarad E, Kuppen PJ, Aghdaei HA and Zali MR. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterology and hepatology from bed to bench. 2013; 6:120-128.
87. Barault L, Charon-Barra C, Jooste V, de la Vega MF, Martin L, Roignot P, Rat P, Bouvier AM, Laurent-Puig P, Faivre J, Chapusot C and Piard F. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer research. 2008; 68:8541-8546.
88. Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M and Fuchs CS. Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. The Journal of molecular diagnostics. 2007; 9:305-314.
89. Nosho K, Shima K, Irahara N, Kure S, Baba Y, Kirkner GJ, Chen L, Gokhale S, Hazra A, Spiegelman D, Giovannucci EL, Jaenisch R, Fuchs CS and Ogino S. DNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancer. Clinical cancer research. 2009; 15:3663-3671.
90. Ibrahim AE, Arends MJ, Silva AL, Wyllie AH, Greger L, Ito Y, Vowler SL, Huang TH, Tavare S, Murrell A and Brenton JD. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut. 2011; 60:499-508.
91. Samowitz WS, Albertsen H, Sweeney C, Herrick J, Caan BJ, Anderson KE, Wolff RK and Slattery ML. Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. Journal of the National Cancer Institute. 2006; 98:1731-1738.
92. Limsui D, Vierkant RA, Tillmans LS, Wang AH, Weisenberger DJ, Laird PW, Lynch CF, Anderson KE, French AJ, Haile RW, Harnack LJ, Potter JD, Slager SL, Smyrk TC, Thibodeau SN, Cerhan JR, et al. Cigarette smoking and colorectal cancer risk by molecularly defined subtypes. Journal of the National Cancer Institute. 2010; 102:1012-1022.
93. van Engeland M, Weijenberg MP, Roemen GM, Brink M, de Bruine AP, Goldbohm RA, van den Brandt PA, Baylin SB, de Goeij AF and Herman JG. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer research. 2003; 63:3133-3137.
94. de Vogel S, Wouters KA, Gottschalk RW, van Schooten FJ, de Goeij AF, de Bruine AP, Goldbohm RA, van den Brandt PA, Weijenberg MP and van Engeland M. Genetic variants of methyl metabolizing enzymes and epigenetic regulators: associations with promoter CpG island hypermethylation in colorectal cancer. Cancer epidemiology, biomarkers & prevention. 2009; 18:3086-3096.
95. Hazra A, Fuchs CS, Kawasaki T, Kirkner GJ, Hunter DJ and Ogino S. Germline polymorphisms in the one-carbon metabolism pathway and DNA methylation in colorectal cancer. Cancer causes & control. 2010; 21:331-345.
96. Jia M, Gao X, Zhang Y, Hoffmeister M and Brenner H. Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: a systematic review. Clinical epigenetics. 2016; 8:25.
97. Dahlin AM, Palmqvist R, Henriksson ML, Jacobsson M, Eklof V, Rutegard J, Oberg A and Van Guelpen BR. The role of the CpG island methylator phenotype in colorectal cancer prognosis depends on microsatellite instability screening status. Clinical cancer research. 2010; 16:1845-1855.
98. Ward RL, Cheong K, Ku SL, Meagher A, O’Connor T and Hawkins NJ. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. Journal of clinical oncology. 2003; 21:3729-3736.
99. Bae JM, Kim MJ, Kim JH, Koh JM, Cho NY, Kim TY and Kang GH. Differential clinicopathological features in microsatellite instability-positive colorectal cancers depending on CIMP status. Virchows Archiv. 2011; 459:55-63.
100. Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC, Kim NK and Issa JP. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2011; 117:1847-1854.
101. Bae JM, Kim JH, Cho NY, Kim TY and Kang GH. Prognostic implication of the CpG island methylator phenotype in colorectal cancers depends on tumour location. British journal of cancer. 2013; 109:1004-1012.
102. Lee S, Cho NY, Choi M, Yoo EJ, Kim JH and Kang GH. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathology international. 2008; 58:104-113.
103. Issa JP. CpG island methylator phenotype in cancer. Nature reviews Cancer. 2004; 4:988-993.
104. Hughes LA, Khalid-de Bakker CA, Smits KM, van den Brandt PA, Jonkers D, Ahuja N, Herman JG, Weijenberg MP and van Engeland M. The CpG island methylator phenotype in colorectal cancer: progress and problems. Biochimica et biophysica acta. 2012; 1825:77-85.
105. Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, Giovannucci EL and Fuchs CS. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009; 58:90-96.
106. Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, Tu D, Redston M and Gallinger S. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. The New England journal of medicine. 2003; 349:247-257.
107. Jover R, Zapater P, Castells A, Llor X, Andreu M, Cubiella J, Pinol V, Xicola RM, Bujanda L, Rene JM, Clofent J, Bessa X, Morillas JD, Nicolas-Perez D, Paya A, Alenda C, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006; 55:848-855.
108. Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K and Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clinical cancer research. 2003; 9:2898-2903.
109. Alberts SR, Horvath WL, Sternfeld WC, Goldberg RM, Mahoney MR, Dakhil SR, Levitt R, Rowland K, Nair S, Sargent DJ and Donohue JH. Oxaliplatin, fluorouracil, and leucovorin for patients with unresectable liver-only metastases from colorectal cancer: a North Central Cancer Treatment Group phase II study. Journal of clinical oncology. 2005; 23:9243-9249.
110. Cassidy J, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F and Saltz L. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. Journal of clinical oncology. 2008; 26:2006-2012.
111. Alcindor T and Beauger N. Oxaliplatin: a review in the era of molecularly targeted therapy. Current oncology. 2011; 18:18-25.
112. Chang PM, Tzeng CH, Chen PM, Lin JK, Lin TC, Chen WS, Jiang JK, Wang HS and Wang WS. ERCC1 codon 118 C—>T polymorphism associated with ERCC1 expression and outcome of FOLFOX-4 treatment in Asian patients with metastatic colorectal carcinoma. Cancer science. 2009; 100:278-283.
113. Woelfelschneider A, Popanda O, Lilla C, Linseisen J, Mayer C, Celebi O, Debus J, Bartsch H, Chang-Claude J and Schmezer P. A distinct ERCC1 haplotype is associated with mRNA expression levels in prostate cancer patients. Carcinogenesis. 2008; 29:1758-1764.
114. Park DJ, Zhang W, Stoehlmacher J, Tsao-Wei D, Groshen S, Gil J, Yun J, Sones E, Mallik N and Lenz HJ. ERCC1 gene polymorphism as a predictor for clinical outcome in advanced colorectal cancer patients treated with platinum-based chemotherapy. Clinical advances in hematology & oncology : H&O. 2003; 1:162-166.
115. Ruzzo A, Graziano F, Loupakis F, Rulli E, Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P, Bisonni R, Masi G, Schiavon G, Giordani P, Giustini L, Falcone A, Tonini G, et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. Journal of clinical oncology. 2007; 25:1247-1254.
116. Ye F, Liu Z, Tan A, Liao M, Mo Z and Yang X. XRCC1 and GSTP1 polymorphisms and prognosis of oxaliplatin-based chemotherapy in colorectal cancer: a meta-analysis. Cancer chemotherapy and pharmacology. 2013; 71:733-740.
117. Qian YY, Liu XY, Wu Q, Song X, Chen XF, Liu YQ, Pei D, Shen LZ and Shu YQ. The ERCC1 C118T polymorphism predicts clinical outcomes of colorectal cancer patients receiving oxaliplatin-based chemotherapy: a meta-analysis based on 22 studies. Asian Pacific journal of cancer prevention. 2014; 15:8383-8390.
118. Milovic-Kovacevic M, Srdic-Rajic T, Radulovic S, Bjelogrlic S and Gavrilovic D. Expression of ERCC1 protein in biopsy specimen predicts survival in advanced ovarian cancer patients treated with platinum-based chemotherapy. Journal of BUON. 2011; 16:708-714.
119. Leng XF, Chen MW, Xian L, Dai L, Ma GY and Li MH. Combined analysis of mRNA expression of ERCC1, BAG-1, BRCA1, RRM1 and TUBB3 to predict prognosis in patients with non-small cell lung cancer who received adjuvant chemotherapy. Journal of experimental & clinical cancer research. 2012; 31:25.
120. Pospiech H and Syvaoja JE. DNA polymerase epsilon - more than a polymerase. TheScientificWorldJournal. 2003; 3:87-104.
121. Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, ter Haar N, Noske A, Amant F, Tomlinson IP, Wild PJ, Lambrechts D, Jurgenliemk-Schulz IM, Jobsen JJ, Smit VT, Creutzberg CL and Bosse T. Prognostic significance of POLE proofreading mutations in endometrial cancer. Journal of the National Cancer Institute. 2015; 107:402.
122. Billingsley CC, Cohn DE, Mutch DG, Stephens JA, Suarez AA and Goodfellow PJ. Polymerase varepsilon (POLE) mutations in endometrial cancer: clinical outcomes and implications for Lynch syndrome testing. Cancer. 2015; 121:386-394.
123. Stenzinger A, Pfarr N, Endris V, Penzel R, Jansen L, Wolf T, Herpel E, Warth A, Klauschen F, Kloor M, Roth W, Blaker H, Chang-Claude J, Brenner H, Hoffmeister M and Weichert W. Mutations in POLE and survival of colorectal cancer patients—link to disease stage and treatment. Cancer medicine. 2014; 3:1527-1538.
124. Valle L, Hernandez-Illan E, Bellido F, Aiza G, Castillejo A, Castillejo MI, Navarro M, Segui N, Vargas G, Guarinos C, Juarez M, Sanjuan X, Iglesias S, Alenda C, Egoavil C, Segura A, et al. New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis. Human molecular genetics. 2014; 23:3506-3512.
125. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, Ma Y, Kaur K, Dobbins S, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature genetics. 2013; 45:136-144.
126. Yoshida R, Miyashita K, Inoue M, Shimamoto A, Yan Z, Egashira A, Oki E, Kakeji Y, Oda S and Maehara Y. Concurrent genetic alterations in DNA polymerase proofreading and mismatch repair in human colorectal cancer. European journal of human genetics. 2011; 19:320-325.
127. Konstantinopoulos PA and Matulonis UA. POLE mutations as an alternative pathway for microsatellite instability in endometrial cancer: implications for Lynch syndrome testing. Cancer. 2015; 121:331-334.
128. Popanda O, Zheng C, Magdeburg JR, Buttner J, Flohr T, Hagmuller E and Thielmann HW. Mutation analysis of replicative genes encoding the large subunits of DNA polymerase alpha and replication factors A and C in human sporadic colorectal cancers. International journal of cancer Journal international du cancer. 2000; 86:318-324.
129. Deng Y, Wang L, Tan S, Kim GP, Dou R, Chen D, Cai Y, Fu X, Wang L, Zhu J and Wang J. KRAS as a predictor of poor prognosis and benefit from postoperative FOLFOX chemotherapy in patients with stage II and III colorectal cancer. Molecular oncology. 2015; 9:1341-1347.
130. Brudvik KW, Kopetz SE, Li L, Conrad C, Aloia TA and Vauthey JN. Meta-analysis of KRAS mutations and survival after resection of colorectal liver metastases. The British journal of surgery. 2015; 102:1175-1183.
131. Arrington AK, Heinrich EL, Lee W, Duldulao M, Patel S, Sanchez J, Garcia-Aguilar J and Kim J. Prognostic and predictive roles of KRAS mutation in colorectal cancer. International journal of molecular sciences. 2012; 13:12153-12168.
132. Kalikaki A, Politaki H, Souglakos J, Apostolaki S, Papadimitraki E, Georgoulia N, Tzardi M, Mavroudis D, Georgoulias V and Voutsina A. KRAS genotypic changes of circulating tumor cells during treatment of patients with metastatic colorectal cancer. PloS one. 2014; 9:e104902.
133. Wennerberg K, Rossman KL and Der CJ. The Ras superfamily at a glance. Journal of cell science. 2005; 118:843-846.
134. Fernandez-Medarde A and Santos E. Ras in cancer and developmental diseases. Genes & cancer. 2011; 2:344-358. doi: 10.1177/1947601911411084.
135. Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. British journal of cancer. 2009; 101:715-721.
136. Al-Shamsi HO, Alhazzani W and Wolff RA. Extended RAS testing in metastatic colorectal cancer-Refining the predictive molecular biomarkers. Journal of gastrointestinal oncology. 2015; 6:314-321.
137. Boeckx N, Peeters M, Van Camp G, Pauwels P, Op de Beeck K and Deschoolmeester V. Prognostic and Predictive Value of RAS Gene Mutations in Colorectal Cancer: Moving Beyond KRAS Exon 2. Drugs. 2015; 75:1739-1756.
138. Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, Jullian E, Laurent-Puig P, Olschwang S, et al. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. British journal of cancer. 2001; 85:692-696.
139. Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, Aranda E, Nordlinger B, Cisar L, Labianca R, Cunningham D, Van Cutsem E, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. Journal of clinical oncology. 2010; 28:466-474.
140. Yokota T. Are KRAS/BRAF mutations potent prognostic and/or predictive biomarkers in colorectal cancers? Anti-cancer agents in medicinal chemistry. 2012; 12:163-171.
141. Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S and Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta oncologica. 2014; 53:852-864.
142. Rizzo S, Bronte G, Fanale D, Corsini L, Silvestris N, Santini D, Gulotta G, Bazan V, Gebbia N, Fulfaro F and Russo A. Prognostic vs predictive molecular biomarkers in colorectal cancer: is KRAS and BRAF wild type status required for anti-EGFR therapy? Cancer treatment reviews. 2010; 36 Suppl 3:S56-61.
143. Belmont PJ, Jiang P, McKee TD, Xie T, Isaacson J, Baryla NE, Roper J, Sinnamon MJ, Lee NV, Kan JL, Guicherit O, Wouters BG, O’Brien CA, Shields D, Olson P, VanArsdale T, et al. Resistance to dual blockade of the kinases PI3K and mTOR in KRAS-mutant colorectal cancer models results in combined sensitivity to inhibition of the receptor tyrosine kinase EGFR. Science signaling. 2014; 7:ra107.
144. Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT, Boisvert JL, Milano RJ, Roper J, Kodack DP, Jain RK, Corcoran RB, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer discovery. 2014; 4:42-52.
145. Samalin E, Bouche O, Thezenas S, Francois E, Adenis A, Bennouna J, Taieb J, Desseigne F, Seitz JF, Conroy T, Galais MP, Assenat E, Crapez E, Poujol S, Bibeau F, Boissiere F, et al. Sorafenib and irinotecan (NEXIRI) as second- or later-line treatment for patients with metastatic colorectal cancer and KRAS-mutated tumours: a multicentre Phase I/II trial. British journal of cancer. 2014; 110:1148-1154.
146. Cercek A, Shia J, Gollub M, Chou JF, Capanu M, Raasch P, Reidy-Lagunes D, Proia DA, Vakiani E, Solit DB and Saltz LB. Ganetespib, a novel Hsp90 inhibitor in patients with KRAS mutated and wild type, refractory metastatic colorectal cancer. Clinical colorectal cancer. 2014; 13:207-212.
147. E J, Xing J, Gong H, He J and Zhang W. Combine MEK inhibition with PI3K/mTOR inhibition exert inhibitory tumor growth effect on KRAS and PIK3CA mutation CRC xenografts due to reduced expression of VEGF and matrix metallopeptidase-9. Tumour biology. 2015; 36:1091-1097.
148. Petrelli F, Coinu A, Cabiddu M, Ghilardi M and Barni S. KRAS as prognostic biomarker in metastatic colorectal cancer patients treated with bevacizumab: a pooled analysis of 12 published trials. Medical oncology. 2013; 30:650.
149. Bruera G, Cannita K, Tessitore A, Russo A, Alesse E, Ficorella C and Ricevuto E. The prevalent KRAS exon 2 c.35 G>A mutation in metastatic colorectal cancer patients: A biomarker of worse prognosis and potential benefit of bevacizumab-containing intensive regimens? Critical reviews in oncology/hematology. 2015; 93:190-202.
150. Janku F, Wheler JJ, Hong DS and Kurzrock R. Bevacizumab-based treatment in colorectal cancer with a NRAS Q61K mutation. Targeted oncology. 2013; 8:183-188.
151. Kubackova K, Bortlicek Z, Pikus T, Linke Z, Pokorna P, Vyzula R and Prausova J. Bevacizumab with chemotherapy in patients with KRAS wild-type metastatic colorectal cancer: Czech registry data. Future oncology. 2015; 11:225-232.
152. Yarden Y and Sliwkowski MX. Untangling the ErbB signalling network. Nature reviews Molecular cell biology. 2001; 2:127-137.
153. Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D and Zhang XF. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent progress in hormone research. 2001; 56:127-155.
154. Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, Beller U, Westra WH, Ladenson PW and Sidransky D. BRAF mutation in papillary thyroid carcinoma. Journal of the National Cancer Institute. 2003; 95:625-627.
155. Bishehsari F, Mahdavinia M, Malekzadeh R, Verginelli F, Catalano T, Sotoudeh M, Bazan V, Agnese V, Esposito DL, De Lellis L, Semeraro D, Colucci G, Hormazdi M, Rakhshani N, Cama A, Piantelli M, et al. Patterns of K-ras mutation in colorectal carcinomas from Iran and Italy (a Gruppo Oncologico dell’Italia Meridionale study): influence of microsatellite instability status and country of origin. Annals of oncology. 2006; 17 Suppl 7:vii91-96.
156. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949-954.
157. Barault L, Veyrie N, Jooste V, Lecorre D, Chapusot C, Ferraz JM, Lievre A, Cortet M, Bouvier AM, Rat P, Roignot P, Faivre J, Laurent-Puig P and Piard F. Mutations in the RAS-MAPK, PI(3)K (phosphatidylinositol-3-OH kinase) signaling network correlate with poor survival in a population-based series of colon cancers. International journal of cancer Journal international du cancer. 2008; 122:2255-2259.
158. Missiaglia E, Jacobs B, D’Ario G, Di Narzo AF, Soneson C, Budinska E, Popovici V, Vecchione L, Gerster S, Yan P, Roth AD, Klingbiel D, Bosman FT, Delorenzi M and Tejpar S. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Annals of oncology. 2014; 25:1995-2001.
159. Barras D. BRAF Mutation in Colorectal Cancer: An Update. Biomarkers in cancer. 2015; 7:9-12.
160. Magudia K, Lahoz A and Hall A. K-Ras and B-Raf oncogenes inhibit colon epithelial polarity establishment through up-regulation of c-myc. The Journal of cell biology. 2012; 198:185-194.
161. Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK and Slattery ML. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer research. 2005; 65:6063-6069.
162. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B and Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002; 418:934.
163. Oikonomou E, Makrodouli E, Evagelidou M, Joyce T, Probert L and Pintzas A. BRAF(V600E) efficient transformation and induction of microsatellite instability versus KRAS(G12V) induction of senescence markers in human colon cancer cells. Neoplasia. 2009; 11:1116-1131.
164. Oikonomou E, Koustas E, Goulielmaki M and Pintzas A. BRAF vs RAS oncogenes: are mutations of the same pathway equal? Differential signalling and therapeutic implications. Oncotarget. 2014; 5:11752-11777. doi: 10.18632/oncotarget.2555.
165. Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A and Li G. The prognostic value of BRAF mutation in colorectal cancer and melanoma: a systematic review and meta-analysis. PloS one. 2012; 7:e47054.
166. Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi I, Lonati V, Ghilardi M, de Braud F and Barni S. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. European journal of cancer. 2015; 51:587-594.
167. Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS and Sorich MJ. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. British journal of cancer. 2015; 112:1888-1894.
168. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A and Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012; 483:100-103.
169. Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, Flaherty KT, Piris A, Wargo JA, Settleman J, Mino-Kenudson M and Engelman JA. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer discovery. 2012; 2:227-235.
170. Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, Godfrey JT, Nishimura K, Lynch KD, Mermel CH, Lockerman EL, Kalsy A, Gurski JM, Jr., Bahl S, Anderka K, Green LM, et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer discovery. 2015; 5:358-367.
171. Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, Sartore-Bianchi A, Fontanini G, Milione M, Lauricella C, Siena S, Falcone A, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Annals of oncology. 2015.
172. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, Giovannucci E, Fuchs CS, Chan AT, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. The New England journal of medicine. 2012; 367:1596-1606.
173. Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, Chan AT, Engelman JA, Kraft P, Cantley LC, Giovannucci EL and Fuchs CS. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. Journal of clinical oncology. 2009; 27:1477-1484.
174. Nosho K, Kawasaki T, Ohnishi M, Suemoto Y, Kirkner GJ, Zepf D, Yan L, Longtine JA, Fuchs CS and Ogino S. PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia. 2008; 10:534-541.
175. Hsieh LL, Er TK, Chen CC, Hsieh JS, Chang JG and Liu TC. Characteristics and prevalence of KRAS, BRAF, and PIK3CA mutations in colorectal cancer by high-resolution melting analysis in Taiwanese population. Clinica chimica acta. 2012; 413:1605-1611.
176. Janku F, Lee JJ, Tsimberidou AM, Hong DS, Naing A, Falchook GS, Fu S, Luthra R, Garrido-Laguna I and Kurzrock R. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PloS one. 2011; 6:e22769.
177. Rosty C, Young JP, Walsh MD, Clendenning M, Sanderson K, Walters RJ, Parry S, Jenkins MA, Win AK, Southey MC, Hopper JL, Giles GG, Williamson EJ, English DR and Buchanan DD. PIK3CA activating mutation in colorectal carcinoma: associations with molecular features and survival. PloS one. 2013; 8:e65479.
178. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B and Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304:554.
179. Day FL, Jorissen RN, Lipton L, Mouradov D, Sakthianandeswaren A, Christie M, Li S, Tsui C, Tie J, Desai J, Xu ZZ, Molloy P, Whitehall V, Leggett BA, Jones IT, McLaughlin S, et al. PIK3CA and PTEN gene and exon mutation-specific clinicopathologic and molecular associations in colorectal cancer. Clinical cancer research. 2013; 19:3285-3296.
180. Liao X, Morikawa T, Lochhead P, Imamura Y, Kuchiba A, Yamauchi M, Nosho K, Qian ZR, Nishihara R, Meyerhardt JA, Fuchs CS and Ogino S. Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clinical cancer research. 2012; 18:2257-2268.
181. Mao C, Yang ZY, Hu XF, Chen Q and Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Annals of oncology. 2012; 23:1518-1525.
182. Farina Sarasqueta A, Zeestraten EC, van Wezel T, van Lijnschoten G, van Eijk R, Dekker JW, Kuppen PJ, Goossens-Beumer IJ, Lemmens VE, van de Velde CJ, Rutten HJ, Morreau H and van den Brule AJ. PIK3CA kinase domain mutation identifies a subgroup of stage III colon cancer patients with poor prognosis. Cellular oncology. 2011; 34:523-531.
183. Cathomas G. PIK3CA in Colorectal Cancer. Frontiers in oncology. 2014; 4:35.
184. Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, Davidson B, Kerr DJ, Tomlinson IP and Midgley R. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. Journal of clinical oncology. 2013; 31:4297-4305.
185. Paleari L, Puntoni M, Clavarezza M, DeCensi M, Cuzick J and DeCensi A. PIK3CA Mutation, Aspirin Use after Diagnosis and Survival of Colorectal Cancer. A Systematic Review and Meta-analysis of Epidemiological Studies. Clinical oncology. 2016; 28:317-326.
186. Chan AT, Ogino S and Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. Jama. 2009; 302:649-658.
187. Li P, Wu H, Zhang H, Shi Y, Xu J, Ye Y, Xia D, Yang J, Cai J and Wu Y. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut. 2015; 64:1419-1425.
188. Schror K. Aspirin and platelets: the antiplatelet action of aspirin and its role in thrombosis treatment and prophylaxis. Seminars in thrombosis and hemostasis. 1997; 23:349-356.
189. Roth GJ and Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. The Journal of clinical investigation. 1975; 56:624-632.
190. Ogino S, Lochhead P, Giovannucci E, Meyerhardt JA, Fuchs CS and Chan AT. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: power and promise of molecular pathological epidemiology. Oncogene. 2014; 33:2949-2955.
191. Banu SK, Lee J, Speights VO, Jr., Starzinski-Powitz A and Arosh JA. Selective inhibition of prostaglandin E2 receptors EP2 and EP4 induces apoptosis of human endometriotic cells through suppression of ERK1/2, AKT, NFkappaB, and beta-catenin pathways and activation of intrinsic apoptotic mechanisms. Molecular endocrinology. 2009; 23:1291-1305.
192. Regan JW. EP2 and EP4 prostanoid receptor signaling. Life sciences. 2003; 74:143-153.
193. De Keijzer S, Meddens MB, Torensma R and Cambi A. The multiple faces of prostaglandin E2 G-protein coupled receptor signaling during the dendritic cell life cycle. International journal of molecular sciences. 2013; 14:6542-6555.
194. Chang J, Vacher J, Yao B, Fan X, Zhang B, Harris RC and Zhang MZ. Prostaglandin E receptor 4 (EP4) promotes colonic tumorigenesis. Oncotarget. 2015; 6:33500-33511. doi: 10.18632/oncotarget.5589.
195. Yano T, Zissel G, Muller-Qernheim J, Jae Shin S, Satoh H and Ichikawa T. Prostaglandin E2 reinforces the activation of Ras signal pathway in lung adenocarcinoma cells via EP3. FEBS letters. 2002; 518:154-158.
196. Castellone MD, Teramoto H, Williams BO, Druey KM and Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005; 310:1504-1510.
197. Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM and DuBois RN. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:1492-1497.
198. Wang D, Xia D and Dubois RN. The Crosstalk of PTGS2 and EGF Signaling Pathways in Colorectal Cancer. Cancers. 2011; 3:3894-3908.
199. Zhang Y and Daaka Y. PGE2 promotes angiogenesis through EP4 and PKA Cgamma pathway. Blood. 2011; 118:5355-5364.
200. Bergman M, Djaldetti M, Salman H and Bessler H. Inflammation and colorectal cancer: does aspirin affect the interaction between cancer and immune cells? Inflammation. 2011; 34:22-28.
201. Wang K and Karin M. Tumor-Elicited Inflammation and Colorectal Cancer. Advances in cancer research. 2015; 128:173-196.
202. Vogel LK, Saebo M, Hoyer H, Kopp TI, Vogel U, Godiksen S, Frenzel FB, Hamfjord J, Bowitz-Lothe IM, Johnson E, Kure EH and Andersen V. Intestinal PTGS2 mRNA levels, PTGS2 gene polymorphisms, and colorectal carcinogenesis. PloS one. 2014; 9:e105254.
203. Guillem-Llobat P, Dovizio M, Bruno A, Ricciotti E, Cufino V, Sacco A, Grande R, Alberti S, Arena V, Cirillo M, Patrono C, FitzGerald GA, Steinhilber D, Sgambato A and Patrignani P. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget. 2016; doi: 10.18632/oncotarget.8655.
204. Nieswandt B, Hafner M, Echtenacher B and Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer research. 1999; 59:1295-1300.
205. Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Jirouskova M and Degen JL. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005; 105:178-185.
206. Gay LJ and Felding-Habermann B. Contribution of platelets to tumour metastasis. Nature reviews Cancer. 2011; 11:123-134.
207. Degen JL and Palumbo JS. Hemostatic factors, innate immunity and malignancy. Thrombosis research. 2012; 129 Suppl 1:S1-5.
208. Labelle M and Hynes RO. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer discovery. 2012; 2:1091-1099.
209. Liao D, Zhong L, Duan T, Zhang RH, Wang X, Wang G, Hu K, Lv X and Kang T. Aspirin Suppresses the Growth and Metastasis of Osteosarcoma through the NF-kappaB Pathway. Clinical cancer research. 2015; 21:5349-5359.
210. Lu L, Sun HC, Zhang W, Chai ZT, Zhu XD, Kong LQ, Wang WQ, Zhang KZ, Zhang YY, Zhang QB, Ao JY, Li JQ, Wang L, Wu WZ and Tang ZY. Aspirin minimized the pro-metastasis effect of sorafenib and improved survival by up-regulating HTATIP2 in hepatocellular carcinoma. PloS one. 2013; 8:e65023.
211. Reddy BS, Wang CX, Kong AN, Khor TO, Zheng X, Steele VE, Kopelovich L and Rao CV. Prevention of azoxymethane-induced colon cancer by combination of low doses of atorvastatin, aspirin, and celecoxib in F 344 rats. Cancer research. 2006; 66:4542-4546.
212. Miliani de Marval PL and Zhang Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget. 2011; 2:234-238. doi: 10.18632/oncotarget.228.
213. Pei D, Zhang Y and Zheng J. Regulation of TP53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012; 3:228-235. doi: 10.18632/oncotarget.443.
214. Li XL, Zhou J, Chen ZR and Chng WJ. TP53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World journal of gastroenterology. 2015; 21:84-93.
215. Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA and Kudryavtseva AV. Targeting VDAC-bound hexokinase II: a promising approach for concomitant anti-cancer therapy. Expert opinion on therapeutic targets. 2013; 17:1221-1233.
216. Shoshan-Barmatz V and Mizrachi D. VDAC1: from structure to cancer therapy. Frontiers in oncology. 2012; 2:164.
217. Ryan KM, Phillips AC and Vousden KH. Regulation and function of the TP53 tumor suppressor protein. Current opinion in cell biology. 2001; 13:332-337.
218. Wang X, Simpson ER and Brown KA. TP53: Protection against Tumor Growth beyond Effects on Cell Cycle and Apoptosis. Cancer research. 2015; 75:5001-5007.
219. Ladelfa MF, Toledo MF, Laiseca JE and Monte M. Interaction of TP53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxidants & redox signaling. 2011; 15:1749-1761.
220. Liu J, Zhang C, Hu W and Feng Z. Tumor suppressor TP53 and its mutants in cancer metabolism. Cancer letters. 2015; 356:197-203.
221. Yeung SJ, Pan J and Lee MH. Roles of TP53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cellular and molecular life sciences : CMLS. 2008; 65:3981-3999.
222. Nakamura Y. Isolation of TP53-target genes and their functional analysis. Cancer science. 2004; 95:7-11.
223. Slebos RJ, Lee MH, Plunkett BS, Kessis TD, Williams BO, Jacks T, Hedrick L, Kastan MB and Cho KR. TP53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:5320-5324.
224. Krasnov GS, Dmitriev AA, Snezhkina AV and Kudryavtseva AV. Deregulation of glycolysis in cancer: glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target. Expert opinion on therapeutic targets. 2013; 17:681-693.
225. Mathupala SP, Heese C and Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor TP53. The Journal of biological chemistry. 1997; 272:22776-22780.
226. Blandino G, Levine AJ and Oren M. Mutant TP53 gain of function: differential effects of different TP53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999; 18:477-485.
227. Li R, Sutphin PD, Schwartz D, Matas D, Almog N, Wolkowicz R, Goldfinger N, Pei H, Prokocimer M and Rotter V. Mutant TP53 protein expression interferes with TP53-independent apoptotic pathways. Oncogene. 1998; 16:3269-3277.
228. Almazov VP, Kochetkov DV and Chumakov PM. Use of TP53 for therapy of human cancer. Mol Biol+. 2007; 41:863-877.
229. Godai TI, Suda T, Sugano N, Tsuchida K, Shiozawa M, Sekiguchi H, Sekiyama A, Yoshihara M, Matsukuma S, Sakuma Y, Tsuchiya E, Kameda Y, Akaike M and Miyagi Y. Identification of colorectal cancer patients with tumors carrying the TP53 mutation on the codon 72 proline allele that benefited most from 5-fluorouracil (5-FU) based postoperative chemotherapy. BMC cancer. 2009; 9:420.
230. Offer SM, Wegner NJ, Fossum C, Wang K and Diasio RB. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer research. 2013; 73:1958-1968.
231. Li Q, Liu Y, Zhang HM, Huang YP, Wang TY, Li DS and Sun HZ. Influence of DPYD Genetic Polymorphisms on 5-Fluorouracil Toxicities in Patients with Colorectal Cancer: A Meta-Analysis. Gastroenterology research and practice. 2014; 2014:827989.
232. Yen JL and McLeod HL. Should DPD analysis be required prior to prescribing fluoropyrimidines? European journal of cancer. 2007; 43:1011-1016.
233. Caudle KE, Thorn CF, Klein TE, Swen JJ, McLeod HL, Diasio RB and Schwab M. Clinical Pharmacogenetics Implementation Consortium guidelines for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing. Clinical pharmacology and therapeutics. 2013; 94:640-645.
234. Rosmarin D, Palles C, Pagnamenta A, Kaur K, Pita G, Martin M, Domingo E, Jones A, Howarth K, Freeman-Mills L, Johnstone E, Wang H, Love S, Scudder C, Julier P, Fernandez-Rozadilla C, et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut. 2015; 64:111-120.
235. Falvella FS, Caporale M, Cheli S, Martinetti A, Berenato R, Maggi C, Niger M, Ricchini F, Bossi I, Di Bartolomeo M, Sottotetti E, Bernardi FF, de Braud F, Clementi E and Pietrantonio F. Undetected toxicity risk in pharmacogenetic testing for dihydropyrimidine dehydrogenase. International journal of molecular sciences. 2015; 16:8884-8895.
236. Koopman M, Venderbosch S, van Tinteren H, Ligtenberg MJ, Nagtegaal I, Van Krieken JH and Punt CJ. Predictive and prognostic markers for the outcome of chemotherapy in advanced colorectal cancer, a retrospective analysis of the phase III randomised CAIRO study. European journal of cancer. 2009; 45:1999-2006.
237. Sasaki S, Watanabe T and Nakayama H. Analysis of the mRNA expression of chemotherapy-related genes in colorectal carcinoma using the danenberg tumor profile method. Journal of oncology. 2013; 2013:386906.
238. Farrugia DC, Ford HE, Cunningham D, Danenberg KD, Danenberg PV, Brabender J, McVicar AD, Aherne GW, Hardcastle A, McCarthy K and Jackman AL. Thymidylate synthase expression in advanced colorectal cancer predicts for response to raltitrexed. Clinical cancer research. 2003; 9:792-801.
239. Vallbohmer D, Yang DY, Kuramochi H, Shimizu D, Danenberg KD, Lindebjerg J, Nielsen JN, Jakobsen A and Danenberg PV. DPD is a molecular determinant of capecitabine efficacy in colorectal cancer. International journal of oncology. 2007; 31:413-418.
240. Cheng L, Li M, Hu J, Ren W, Xie L, Sun ZP, Liu BR, Xu GX, Dong XL and Qian XP. UGT1A1*6 polymorphisms are correlated with irinotecan-induced toxicity: a system review and meta-analysis in Asians. Cancer chemotherapy and pharmacology. 2014; 73:551-560.
241. Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL and Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. The pharmacogenomics journal. 2002; 2:43-47.
242. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007; 50:113-130.
243. Fearon ER and Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759-767.
244. Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S and Vogelstein B. TP53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer research. 1990; 50:7717-7722.
245. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr. and Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546-1558.
246. Tomasetti C, Marchionni L, Nowak MA, Parmigiani G and Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America. 2015; 112:118-123.
247. Sveen A, Agesen TH, Nesbakken A, Rognum TO, Lothe RA and Skotheim RI. Transcriptome instability in colorectal cancer identified by exon microarray analyses: Associations with splicing factor expression levels and patient survival. Genome medicine. 2011; 3:32.
248. Sveen A, Johannessen B, Teixeira MR, Lothe RA and Skotheim RI. Transcriptome instability as a molecular pan-cancer characteristic of carcinomas. BMC genomics. 2014; 15:672.