
Introduction
Tobacco smoking is a major public health problem, associated with substantial preventable morbidity globally [1]. In particular, active smoking in adults accounts for a large proportion of age-related diseases, including various forms of cancer, respiratory and cardiovascular diseases [2]. Recent studies have demonstrated a role of DNA methylation, one of the main forms of epigenetic modification, in the pathways of smoking and smoking-induced diseases via regulating gene expression and genome stability [3]. An increasing number of smoking related CpG sites in various genes, such as AHRR, F2RL3 and GPR15, have been discovered by epigenome-wide association studies (EWASs) based on whole blood samples, and have been shown to be useful as quantitive biomarkers of current and past smoking exposure and predictors of smoking-associated health risks [4, 5]. Recently, Teschendorff et al. constructed a smoking index based on 1501 smoking-related loci and showed that smoking-related methylation indices could be useful risk indicators of smoking-induced health disorders [6].
Recent studies have also disclosed age-related alterations of DNA methylation [7], and an “epigenetic clock” for DNA methylation age based on known age-related biomarkers has been shown to predict an individual’s chronological age with high accuracy [8]. Horvath and Hannum et al. developed two broadly accepted measurements for determining DNA methylation age in multiple tissues and blood samples, respectively [9, 10]. The discrepancy between methylation age and chronological age (defined as age acceleration, denoted AA) was found to be heritable and has been suggested to be applied as an index of disproportionate aging. A positive AA indicates that an individual is ahead of his or her chronological age, and a negative one suggests an individual is biologically “younger” than reflected by the chronological age [7, 9]. Follow-up investigations linked AA to lifestyle factors, environmental hazards, as well as stressful life events, and further revealed that AA was a biologically meaningful biomarker associated with aging-related diseases [11-21].
Given the association of smoking with multiple age-related diseases [2], it would appear plausible that smoking may have an impact on AA. However, the few studies assessing this relationship have reported conflicting findings. Horvath et al. and Marioni et al. did not find significant associations of self-reported smoking with DNA methylation age determined in peripheral blood samples [11, 14], while Beach et al. recently reported such an association for the most robust smoking-related locus, cg05575921 (AHRR), as a biomarker of smoking exposure [15]. To further explore a possible role of smoking in AA, we conducted a comprehensive analysis of the associations of self-reported smoking, serum cotinine levels (an established biomarker of current smoking exposure) and smoking-associated methylation signatures with AA in a large population-based study.
Results
Participant characteristics
Characteristics of the study population in the discovery and validation panels were comparable with respect to chronological age, DNA methylation ages, smoking behaviors, as well as lifestyle factors, and are summarized in Table 1. Average age in the two subsets was about 62 years, and chronological ages were highly correlated with corresponding methylation ages (r ≥0.75, Figure S1). Hannum et al.’s methylation ages of both panels were higher than chronological ages and ages computed by Horvath’s approach. More than half of the participants in each subset were ever smokers (current /former smokers), and around 18% still smoked at the time of recruitment. In both subsets, the proportion of men was much higher in current smokers than that in never smokers: 60.8% vs. 29.4% in the discovery panel and 48.0% vs. 21.1% in the validation panel (data not included in the table). Average cumulative smoking exposure in current smokers was considerably higher than that of former smokers in both panels. Average cessation time for former smokers in the two subsets was also similar, approximately 17 years. Cotinine levels of current smokers (64.1 ng/ml) were much higher than levels of never (4.1 ng/ml) and former (7.3 ng/ml) smokers in the discovery panel.
Table 1: Study population characteristics in discovery and validation panels a

a: Mean values (SD) for continuous variables and n (%) for categorical variables;
b: Former smokers only, data missing for 9 and 3 participants, respectively, in discovery and validation panels; cessation time equals age at recruitment minus age at cessation;
c: Only measured in the discovery panel, not applicable (NA) in validation panel;
d: Data missing for 3 participants in discovery panel;
e: Data missing for 66 and 40 participants, respectively, in discovery and validation panels. Categories defined as follows: abstainer, low [women: 0 -<20 g/d, men: 0 -<40 g/d], intermediate [20 -<40 g/d and 40 -<60 g/d, respectively], high [<40 g/d and <60 g/d, respectively];
f: Categories defined as follows: inactive [ < 1h of physical activity/week], medium or high [≥2 h of vigorous and ≥ 2 h of light physical activity/week], low [other];
Associations between smoking indicators and age accelerations
In the analyses of associations of self-reported measures of smoking and serum cotinine levels with AA, two linear regression models were employed (details are presented in Methods), controlling for potential confounding factors. None of the self-reported smoking indicators (smoking status, cumulative exposure and smoking cessation time) or serum cotinine levels was significantly associated with AA in the discovery panel (Table 2, Figure S2). Furthermore, we selected a total of 150 loci related to active smoking, which were identified ≥2 times in previous smoking EWASs, as biomarkers of smoking exposure [4], excluding one locus (cg11314684) which was part of Horvath’s predictor of methylation age [9]. Associations between AA according to Horvath’s and Hannum et al.’s algorithms (dependent variable) and methylation levels of these candidates (independent variable) were assessed by two mixed linear regression models (Models 1, 2) with methylation assay batch as random effect and increasing adjustment for potential confounders (details are presented in Methods). However, even after fully controlling for confounding factors (Model 2), 103 and 94 of the 150 CpG candidates passed the threshold of FDR < 0.05 and thus demonstrated significant associations with AA according to the Horvath’s and Hannum et al.’s algorithms in the discovery phase, respectively (Figure S3). Subsequently, we selected 83 AA-related loci based on both algorithms and then verified them in the validation samples (Table S1, Figure S3, FDR < 0.05). 74 and 70 of these loci were confirmed as significantly related loci for AA derived according to Horvath’s and Hannum et al.’s algorithms by the fully-adjusted model, respectively. Eventually, a total of 66 smoking-related CpG sites that were statistically significant in both algorithms (Table S1, Figure S3, FDR < 0.05). We additionally conducted a sensitivity analysis in the validation panel adjusting for covariates of Model 2 plus the prevalence of cardiovascular diseases (yes/no), diabetes (yes/no) and cancer (yes/no). In this sensitivity analysis, associations remained statistically significant for all of the 66 loci with similar results (data not shown). The 66 CpG sites were eventually designated as the loci associated with DNA methylation aging in whole blood samples. Four hypermethylated smoking-related loci in smokers also showed positive correlations with AA (Table S1). Among the remaining negatively correlated CpG sites, 12 loci had Spearman’s coefficients less than or equal to -0.20 for both AA algorithms (Table 3). They are located at seven genes: 2q37.1 (n = 1), AHRR (n = 3), AVPR1B (n = 1), HUS1 (n = 1), KCNQ1 (n = 2), NCRNA00114 (n = 1), NFE2 (n = 1) and two unnamed genomic regions. Among these, methylation differentials in the locus cg07123182 (KCNQ1) were associated with the largest alterations in AA in regression analyses for both AA algorithms.
Table 2: Associations of self-reported smoking indicators and cotinine levels with age acceleration in the discovery panel
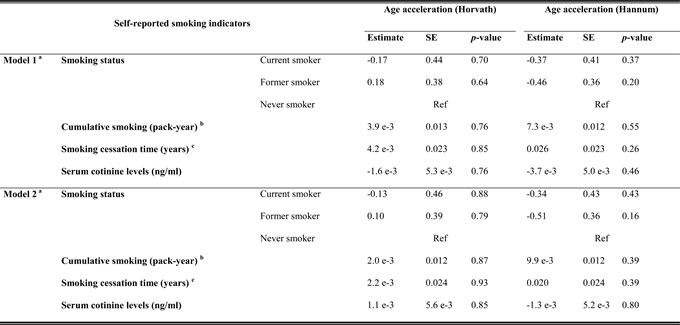
a: Model 1: Adjusted for age (years) and sex; Model 2: Adjusted for age (years), sex, alcohol consumption (abstainer/ low/ intermediate/ high), body mass index (BMI, underweight or normal weight/ overweight/ obese), physical activity (inactive/ low/ medium or high), the prevalence of cardiovascular diseases (yes/no), diabetes (yes/no) and cancer (yes/no).;
b: A pack-year was defined as having smoked 20 cigarettes per day for 1 year, including current and former smokers from discovery panel;
c: Cessation time defined as age at the time of recruitment minus age at cessation, only including former smokers from discovery panel;
Table 3: Top 12 significantly age acceleration related CpG sites in validation panel a
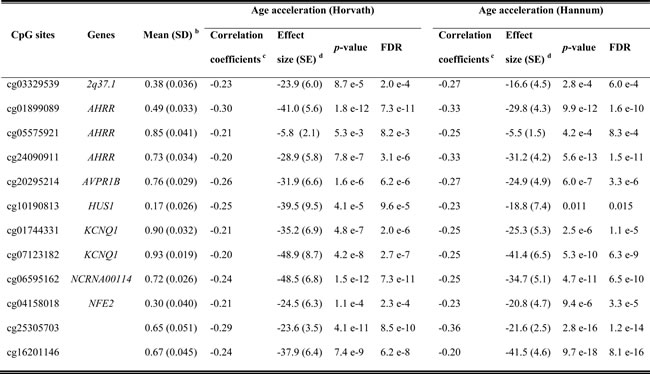
a: 12 loci with correlation coefficients ≤ -0.20;
b: Data of never smokers;
c: Spearman’s Rank-Order Correlation coefficients;
d: Adjusted for age (years), sex, random batch effects, leukocyte distribution (Houseman algorithm [41]), alcohol consumption (abstainer/ low/ intermediate/ high), body mass index (BMI, underweight or normal weight/ overweight/ obese) and physical activity (inactive/ low/ medium or high); The beta coefficients from regression models were reported as effect sizes;
Smoking index (SI) and cg05575921 (AHRR)
We constructed a SI based on the 66 selected smoking-related loci and compared this indicator to one of the most robust smoking-related biomarkers cg05575921 (AHRR), which is known to be hypomethylated under smoking exposure, and the SI estimated based on 1501 loci identified in the study by Teschendorff et al. (Teschendorff SI) [6]. First, as shown in Figure 1, both cg05575921 and SI based on 66 loci were strongly associated with smoking status: levels in current smokers were lower (for cg05575921)/ higher (for SI) than those in never smokers and levels of former smokers were in the intermediate position. Furthermore, the results of mixed linear regression models showed that both methylation markers were significantly associated with both AA algorithms (Table 4). However, the Teschendorff SI was associated with AA according to Horvath’s algorithm, but not with AA according to Hannum et al.’s algorithm (Table 4). Its correlations with both AA algorithms were much weaker than that of SI based on 66 loci (Table S2). In addition, the positive correlations of SI with the AA algorithms were stronger than the negative correlations between cg05575921 and the AA algorithms (Table S2). Another index based on 58 CpG sites without the eight AHRR loci further demonstrated similar correlations with AA and cg05575921 as SI (Table S2). The SI was also associated with the prevalence of cardiovascular diseases (p = 0.014, OR = 1.7 (95CI: 1.2 - 2.6, per unit of SI)), but not with the prevalence of diabetes (p = 0.19) or cancer (p = 0.39) in logistic regression models in the validation panel. Lastly, we explored the dose-response relationships of both smoking indicators with the AA algorithms. For both smoking indicators (Figures 2 and S4), monotonic associations with the AA algorithms were observed (monotonic decrease for cg05575921, monotonic increase for SI). An increase in the SI by one standard deviation was roughly associated an one-year increase in AA derived according to the Horvath’s algorithm, and with a 0.5 -1 year increase in AA derived according to the Hannum et al.’s algorithm.
Table 4: Associations of age accelerations with epigenetic smoking indicators
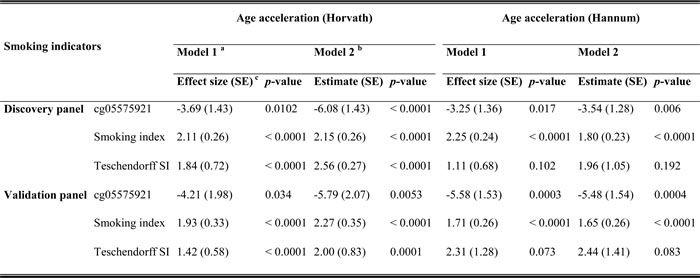
a: Adjusted for age (years), sex and random batch effects;
b: Adjusted for age (years), sex, random batch effects, leukocyte distribution (Houseman algorithm [41]), alcohol consumption (abstainer/ low/ intermediate/ high), body mass index (BMI, underweight or normal weight/ overweight/ obese) and physical activity (inactive/ low/ medium or high);
c: The beta coefficients from regression models were reported as effect sizes;
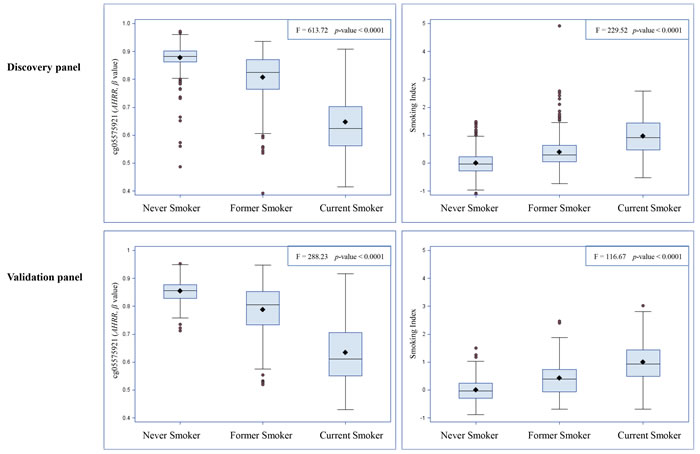
Figure 1: Distributions of cg05575921 and smoking index according to self-reported smoking status.
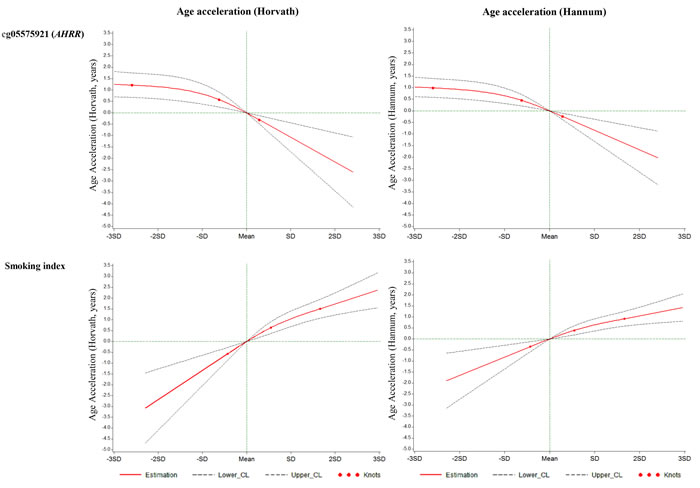
Figure 2: Graphs of the best-fitting models for the associations of cg05575921 and the smoking index with age accelerations in validation panel. Red lines: Estimation; Dashed lines: Confidence limits; Red dots: Knots (25th, 50th and 75th quartiles); Green lines: reference lines.
Discussion
To our knowledge, this is the first systematic investigation exploring the association between active smoking exposure and its biological correlates with DNA methylation age in whole blood samples, based on two independent subgroups of a population-based cohort of older adults from Germany. None of the self-reported smoking indicators, including smoking status, cumulative exposure and time since smoking cessation, or serum cotinine levels was significantly associated with AA. However, we found 66 previously confirmed smoking-related CpG sites to be also associated with AA. A smoking index (SI) based on these loci and methylation at a robust mono-biomarker of active smoking cg05575921 (AHRR) showed monotonic associations with AA. An association with Horvath’s algorithm of AA was also found for the Teschendorff SI.
Smoking has been considered as a critical factor in the risk of a number of age-related adverse health outcomes [2, 22, 23]. However, none of the genomic regions that become either hypermethylated or hypomethylated with aging has been identified in smoking EWASs [4, 7], even though the AA derived according to Hannum et al.’s algorithm is linked closely to one CpG site, cg05575921, which had been identified as an epigenetic indicator of smoking exposure in previous EWASs [15, 24, 25]. Our study confirmed this locus and additionally identified 65 loci that were associated with both smoking and AA as well. It appears plausible that these smoking-related loci might contribute to some of the aging-related health outcomes. In particular, eight out of the 66 loci were located at AHRR, a well-known tumor suppressor gene, which was suggested to be involved in or is involved in the metabolism of endogenous toxins from smoking [26]. We also identified another three smoking-related genomic regions with more than two AA-related sites that were associated with aging-related diseases: AVPR1B (Arginine Vasopressin Receptor 1B) contributes to overweight and might related with diabetes development [27], CNTNAP2 (Contactin Associated Protein-Like 2) is demonstrated to be associated with several mental diseases (e.g. autism, schizophrenia, epilepsy and depression) [28-30], and KCNQ1 (Voltage Gated KQT-Like Subfamily Q, Member 1) is another well-known gene for type 2 diabetes [31]. Additionally, the identified AA-related locus cg19713429 was located at CAPZB (Capping Protein Actin Filament Muscle Z-Line, Beta), which contains a locus cg13319175 that was used as an indicator in Horvath’s algorithm [9]. No associations were found with other well-established smoking-related loci, like cg03636183 (F2RL3) and cg19859270 (GPR15) [4]. The strongest association with AA, in particular a strong monotonic dose-response relationship based on restrict cubic spline regression, was found for a smoking index encompassing all 66 smoking-related CpG sites.
Although our findings of a lack of association between self-reported measures of smoking and AA, along with robust associations between smoking-related methylation markers and AA appear to be inconsistent and hard to reconcile at first sight, there are multiple mechanisms that might explain the observed patterns. First, it is well known that susceptibility of individuals to adverse health effects of environmental hazards strongly varies between individuals [32, 33]. For example, despite the fact that smoking strongly increases the risk of multiple age-related diseases, some proportion of smokers (especially light smokers) stays relatively healthy up to old age [34], and the health risks associated with smoking may depend on a number of factors such as genetic polymorphisms in detoxifying enzymes or co-prevalence of other risk factors [35, 36]. It appears well conceivable that both smoking-related methylation markers as well as methylation defined AA might to some extent reflect increased susceptibility to environmental hazards such as smoking. Along the same lines, the possibility has to be kept in mind that smoking-related methylation changes may not only reflect smoking exposure, but also that similar methylation changes might be induced by other environmental hazards, such as alcohol consumption, nutritional or lifestyle factors [37, 38], or by potentially interactive or addictive effects between those factors and smoking, which may likewise be associated with increased risk of age-related diseases and age acceleration [7, 23]. Finally, self-reported smoking exposure is known to be subject to inaccuracies, e.g. by recall bias or willful underreporting [39]. Smoking-related methylation markers may more accurately reflect true smoking exposure and thereby facilitate disclosure of smoking-related adverse health effects. While our results of strong associations between smoking-related methylation markers and AA are intriguing, further research is needed to unravel the underlying mechanisms, such as those discussed above.
Major strengths of the present study include the relatively large sample size with detailed information on a broad range of covariates in a large population-based cohort and the comprehensive validation in an independent group, as well as the estimation of DNA methylation ages by two widely accepted methods. There are also several limitations that have to be considered in the interpretation of our study. Associations of smoking with DNA methylation in whole blood might be influenced by smoking-induced shifts in leukocyte distribution [40]. In order to remove potential confounding by this factor, our analyses adjusted for leukocyte distribution by the Houseman algorithm [41]. Stressful life events, another potential determinant of epigenetic aging [17, 18], could not be controlled for as information on this potential confounder was not collected in our study. In addition, our study was undertaken in an almost exclusively Caucasian population and results may not be generalized to other populations. For instance, different smoking associated CpG sites have been identified in Asian and African populations [42-44]. Hence, additional studies in other ethnic groups are required to get a more comprehensive picture of the potential role of smoking and smoking-related DNA methylation in age acceleration. Finally, due to the lack of potential genetic predictors of SI or mQTLs for smoking-related loci, we were not able to disentangle causal pathways via Mendelian Randomisation-type approaches which should be followed in further research [45, 46].
Along with the modernization of human society, expanding environmental hazards, beyond conventional factors like smoking and alcohol consumption, i.e. emerging factors like novel chemicals, biohazards and diseases, may be accelerating our biological aging in silence [7]. As the reliability of self-reported or externally measured exposure to such hazards remains limited, measurement of biologically relevant internal doses in epigenetic assays might be a promising approach for establishing related health hazards [47], and monitoring DNA methylation age may provide a window to target early interventions in high-risk individuals. Beyond advancing the understanding of AA and its association with active smoking, our study highlights the potential of surrogate epigenetic indicators, such as the smoking index and DNA methylation age, to quantify biologically relevant exposures and health outcomes. Further research should explore whether and to what extent such epigenetic signatures can be of value in clinical practice to enhance risk stratification and evaluation of preventive and therapeutic interventions.
materials and Methods
Study population
Study subjects were selected from the ESTHER study, an ongoing statewide population-based cohort study conducted in Saarland, a state located in southwest Germany. Details of the study design have been reported previously [48]. Briefly, 9949 older adults (aged 50-75 years) were enrolled by their general practitioners during a routine health check-up between July 2000 and December 2002, and followed up thereafter. Two independent subgroups were selected as discovery panel and validation panel, respectively, for epigenetic analyses. The discovery panel included 1000 participants recruited consecutively at the start of ESTHER study between July and October 2000. The validation panel included 548 participants randomly selected from participants recruited between October 2000 and March 2001. The study was approved by the ethics committees of the University of Heidelberg and the state medical board of Saarland, Germany. Written informed consent was obtained from all participants.
Data collection
Information on socio-demographic characteristics, lifestyle factors and health status at baseline was obtained by standardized self-administered questionnaires. Participants were asked about past and present cigarette, cigar and pipe smoking behaviors and were then categorized into current, former and never smokers. Detailed information on smoking history was also obtained from questionnaires, including age at initiation and smoking intensities at various ages, as well as age of quitting smoking for former smokers. 22 and 17 participants were excluded from the discovery and the validation panel, respectively, due to missing information on smoking status. Additional information on body mass index (BMI) was extracted from a standardized form filled by the general practitioners during the health check-ups. Blood samples were taken during the health check-up and stored at -80°C until further processing. DNA from whole blood samples was extracted using a salting out procedure [49].
Laboratory data
DNA methylation profiles were assessed by the Illumina Infinium Human Methylation 450 Beadchip array (Illumina, San Diego, CA, USA). As previously described [50], samples were analyzed following the manufacturer’s instruction at the Genomics and Proteomics Core Facility of the German Cancer Research Center, Heidelberg, Germany. Illumina’sGenomeStudio® (version 2011.1; Illumina.Inc.) was employed to extract DNA methylation signals from the scanned arrays (Module version 1.9.0; Illumina.Inc.). The methylation status of a specific CpG site was quantified as a β value ranging from 0 (no methylation) to 1 (full methylation). According to the manufacturer’s protocol, no background correction was done and data were normalized to internal controls provided by the manufacturer. All controls were checked for inconsistencies in each measured plate. Signals of probes with a detection p-value > 0.05 were excluded from analysis. We used the Illumina normalization and preprocessing method implemented in Illumina’s Genomestudio (“Illumina normalization”). In addition, as previously described [51], we measured the cotinine levels in serum samples of the discovery panel, using the customized version of an enzyme-linked immunosorbent assay (Inspec II-Cotinine-EIA; Mahsan Diagnostika).
DNA methylation age
DNA methylation age of each participant was calculated by two algorithms proposed by Horvath [9] and Hannum et al. [10]. Horvath’s algorithm, which was derived from a range of tissues and cell types, uses 353 probes targeted in the Illumina 27k and 450k methylation arrays. Methylation ages of study participants according to Horvath’s algorithm were estimated by online calculator (http:// labs.genetics.ucla.edu /horvath/dnamage/), where background-corrected beta values were pre-processed using the calculator’s internal normalization method [9]. Hannum’s algorithm is based on 71 methylation probes from the Illumina 450k methylation array which were derived as the best age predictors with data generated from whole blood DNA [10]. Hannum’s methylation age was determined as the sum of the methylation beta values multiplied by the reported effect sizes of the predictors. Age accelerations (AAs) were determined as discrepancies between methylation and chronological age in the form of residuals, which have a mean of 0 and thus represent positive and negative deviations from chronological age in years. The residuals were calculated by a linear regression procedure in which methylation age was the outcome and chronological age was the independent variable.
Statistical analyses
Study populations in the discovery and validation panels were described with respect to major
socio-demographic characteristics, DNA methylation age, lifestyle factors, smoking behavior and serum cotinine levels.
Initially, we investigated the associations of self-reported smoking indicators (smoking status [current/ former/ never smoker], cumulative smoking exposure [pack-years, in current and former smokers] and smoking cessation time [years, in former smokers only], independent variables) and cotinine levels (ng/ml, independent variable) with AA (dependent variable) derived according to both algorithms (Horvath & Hannum et al.) in the discovery panel. Two linear regression models were employed, controlling for potential confounding factors. Model 1 was adjusted for age (years) and sex, and Model 2 was additionally adjusted for alcohol consumption (abstainer, low [women: 0 - < 20 g/d, men: 0 - < 40 g/d], intermediate [20 - < 40 g/d and 40 - < 60 g/d, respectively], high [≥40 g/d and ≥60 g/d, respectively]), body mass index (BMI, kg/m2, underweight or normal weight [ < 25], overweight [25 - < 30], obese [≥30]), physical activity (inactive [ < 1h of physical activity/week], medium or high [≥2 h of vigorous and ≥2 h of light physical activity/week], low [other]), the prevalence of cardiovascular diseases (yes/no), diabetes (yes/no) and cancer (yes/no). Indicators with a p-value < 0.05 were considered as AA-associated factors.
Furthermore, we selected a total of 150 loci related to active smoking, which were identified ≥2 times in previous smoking EWASs, as biomarkers of smoking exposure [4], excluding one locus (cg11314684) which was part of Horvath’s predictor of methylation age [9]. Associations of their methylation levels (independent variables) with AA (dependent variable) were analyzed by two mixed linear regression models with methylation assay batch as random effect, controlling for potential confounding factors in both panels. Model 1 was adjusted for age (years) and sex. Model 2 was additionally adjusted for the leukocyte distribution estimated by the Houseman algorithm [41], alcohol consumption, body mass index and physical activity. After correction for multiple testing by the false discovery rate (FDR, Benjamini-Hochberg method [52]), CpG sites with corrected p-values < 0.05 were selected from the discovery panel and then replicated in the validation panel. Loci with FDR < 0.05 in the validation panel were eventually considered as AA-associated loci. We additionally conducted a sensitivity analysis in the validation panel adjusting for covariates of Model 2 plus the prevalence of cardiovascular diseases (yes/no), diabetes (yes/no) and cancer (yes/no) to confirm the identified AA-associated loci.
Finally, we used the identified AA associated loci to construct a smoking index (SI) according to Teschendorff et al.’s algorithm [6], to measure the deviation of DNA methylation in a given sample from a normal reference, with the mean taken over the identified loci. In more detail, we computed the mean β value (μc) and standard deviation (σc) across the never smokers of the given dataset, and then defined the SI as
![]()
where Wc is +1(-1) if the smoking-associated CpG, c, is hypermethylated (hypomethylated) in smokers and where βc is the β value of this CpG in samples s [6]. We calculated the SI for each participant in both panels based on the validated AA associated loci, and then compared it with the single epigenetic smoking indicator cg05575921 (AHRR) used in the study by Beach et al., [15] and the SI estimated based on 1501 loci identified in the study by Teschendorff et al. (Teschendorff SI) [6]. Mutual correlations of these indicators and AA were assessed by Spearman’s correlation coefficients, and the associations of the smoking indicators with AA were assessed by mixed linear regression (Models 1 and 2). The associations of SI with the prevalence of aging-related diseases (Yes/No), including cardiovascular diseases, diabetes and cancer, were analyzed by logistic regression with adjustment for potential covariates in the validation panel. Additionally, we employed restricted cubic spline functions using the SAS macro from Desquilbet et al. to evaluate the dose-response relationships of both indicators with AAs [53], controlling for age (years), sex, the leukocyte distribution estimated by Houseman’s algorithm, alcohol consumption, body mass index and physical activity (categorical variables were transformed into dummy variables). The 25th, 50th and 75th percentiles of the SI were chosen as the knots. Data cleaning and all aforementioned analyses were performed by SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).
Acknowledgments
The work of Xu Gao is supported by the grant from the China Scholarship Council (CSC). We thank Mr. Jonathan Heiss of DKFZ for providing the estimation of leukocyte distribution.
Conflicts of Interest
The authors declare that they have no competing interests.
Grant Support
The ESTHER study was supported in part by the Baden-Württemberg state Ministry of Science, Research and Arts (Stuttgart, Germany) and from the German Federal Ministry of Education and Research (Berlin, Germany).
References
1. Mathers CD and Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS medicine. 2006; 3:e442.
2. Lee KW and Pausova Z. Cigarette smoking and DNA methylation. Frontiers in genetics. 2013; 4:132.
3. Bakulski KM and Fallin MD. Epigenetic epidemiology: promises for public health research. Environmental and molecular mutagenesis. 2014; 55:171-183.
4. Gao X, Jia M, Zhang Y, Breitling LP and Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clinical epigenetics. 2015; 7:113.
5. Zhang Y, Schöttker B, Florath I, Stock C, Butterbach K, Holleczek B, Mons U and Brenner H. Smoking-Associated DNA Methylation Biomarkers and Their Predictive Value for All-Cause and Cardiovascular Mortality. Environmental health perspectives. 2016; 124:67-74.
6. Teschendorff AE, Yang Z, Wong A, Pipinikas CP, Jiao Y, Jones A, Anjum S, Hardy R, Salvesen HB, Thirlwell C, Janes SM, Kuh D and Widschwendter M. Correlation of Smoking-Associated DNA Methylation Changes in Buccal Cells With DNA Methylation Changes in Epithelial Cancer. JAMA Oncol. 2015; 1:476-485.
7. Jones MJ, Goodman SJ and Kobor MS. DNA methylation and healthy human aging. Aging cell. 2015; 14:924-932.
8. Mitteldorf JJ. How does the body know how old it is? Introducing the epigenetic clock hypothesis. Biochemistry Biokhimiia. 2013; 78:1048-1053.
9. Horvath S. DNA methylation age of human tissues and cell types. Genome biology. 2013; 14:R115.
10. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell. 2013; 49:359-367.
11. Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome biology. 2015; 16:25.
12. Levine ME, Lu AT, Bennett DA and Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). 2015; 7:1198-1211. doi: 10.18632/aging.100864.
13. Marioni RE, Shah S, McRae AF, Ritchie SJ, Muniz-Terrera G, Harris SE, Gibson J, Redmond P, Cox SR, Pattie A, Corley J, Taylor A, Murphy L, et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. International journal of epidemiology. 2015; 44:1388-1396.
14. Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, et al. Obesity accelerates epigenetic aging of human liver. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:15538-15543.
15. Beach SR, Dogan MV, Lei MK, Cutrona CE, Gerrard M, Gibbons FX, Simons RL, Brody GH and Philibert RA. Methylomic Aging as a Window onto the Influence of Lifestyle: Tobacco and Alcohol Use Alter the Rate of Biological Aging. J Am Geriatr Soc. 2015; 63:2519-2525.
16. Breitling LP, Saum KU, Perna L, Schottker B, Holleczek B and Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clinical epigenetics. 2016; 8:21.
17. Boks MP, van Mierlo HC, Rutten BP, Radstake TR, De Witte L, Geuze E, Horvath S, Schalkwyk LC, Vinkers CH, Broen JC and Vermetten E. Longitudinal changes of telomere length and epigenetic age related to traumatic stress and post-traumatic stress disorder. Psychoneuroendocrinology. 2015; 51:506-512.
18. Zannas AS, Arloth J, Carrillo-Roa T, Iurato S, Roh S, Ressler KJ, Nemeroff CB, Smith AK, Bradley B, Heim C, Menke A, Lange JF, Bruckl T, et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome biology. 2015; 16:266.
19. Wolf EJ, Logue MW, Hayes JP, Sadeh N, Schichman SA, Stone A, Salat DH, Milberg W, McGlinchey R and Miller MW. Accelerated DNA methylation age: Associations with PTSD and neural integrity. Psychoneuroendocrinology. 2016; 63:155-162.
20. Brody GH, Yu T, Chen E, Beach SR and Miller GE. Family-centered prevention ameliorates the longitudinal association between risky family processes and epigenetic aging. J Child Psychol Psychiatry. 2016; 57:566-574.
21. Horvath S and Ritz BR. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY). 2015; 7:1130-1142. doi: 10.18632/aging.100859.
22. Breitling LP, Salzmann K, Rothenbacher D, Burwinkel B and Brenner H. Smoking, F2RL3 methylation, and prognosis in stable coronary heart disease. European heart journal. 2012; 33:2841-2848.
23. Benayoun BA, Pollina EA and Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nature reviews Molecular cell biology. 2015; 16:593-610.
24. Philibert RA, Beach SR, Lei MK and Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clinical epigenetics. 2013; 5:19.
25. Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, Weidinger S, Lattka E, Adamski J, Peters A, Strauch K, Waldenberger M and Illig T. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PloS one. 2013; 8:e63812.
26. Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH and Philibert RA. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2012; 159:141-151.
27. Enhorning S, Sjogren M, Hedblad B, Nilsson PM, Struck J and Melander O. Genetic vasopressin 1b receptor variance in overweight and diabetes mellitus. European journal of endocrinology. 2016; 174:69-75.
28. Alarcón M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL and Ledbetter DH. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. The American Journal of Human Genetics. 2008; 82:150-159.
29. Ji W, Li T, Pan Y, Tao H, Ju K, Wen Z, Fu Y, An Z, Zhao Q and Wang T. CNTNAP2 is significantly associated with schizophrenia and major depression in the Han Chinese population. Psychiatry research. 2013; 207:225-228.
30. Friedman J, Vrijenhoek T, Markx S, Janssen I, Van Der Vliet W, Faas B, Knoers N, Cahn W, Kahn R and Edelmann L. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Molecular psychiatry. 2008; 13:261-266.
31. Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A and Sato Y. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature genetics. 2008; 40:1092-1097.
32. Philibert RA, Beach SR and Brody GH. The DNA methylation signature of smoking: an archetype for the identification of biomarkers for behavioral illness. Nebraska Symposium on Motivation. 2014; 61:109-127.
33. Sun YV. The Influences of Genetic and Environmental Factors on Methylome-wide Association Studies for Human Diseases. Current genetic medicine reports. 2014; 2:261-270.
34. Strandberg AY, Strandberg TE, Pitkala K, Salomaa VV, Tilvis RS and Miettinen TA. The effect of smoking in midlife on health-related quality of life in old age: a 26-year prospective study. Archives of internal medicine. 2008; 168:1968-1974.
35. Autrup H. Genetic polymorphisms in human xenobiotica metabolizing enzymes as susceptibility factors in toxic response. Mutat Res-Gen Tox En. 2000; 464:65-76.
36. Kelada SN, Eaton DL, Wang SS, Rothman NR and Khoury MJ. The role of genetic polymorphisms in environmental health. Environmental health perspectives. 2003; 111:1055-1064.
37. Andersen AM, Dogan MV, Beach SR and Philibert RA. Current and Future Prospects for Epigenetic Biomarkers of Substance Use Disorders. Genes (Basel). 2015; 6:991-1022.
38. Ladd-Acosta C and Fallin MD. The role of epigenetics in genetic and environmental epidemiology. Epigenomics. 2016; 8:271-283.
39. Connor Gorber S, Schofield-Hurwitz S, Hardt J, Levasseur G and Tremblay M. The accuracy of self-reported smoking: a systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine & tobacco research. 2009; 11:12-24.
40. Schwartz J and Weiss ST. Cigarette smoking and peripheral blood leukocyte differentials. Annals of epidemiology. 1994; 4:236-242.
41. Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK and Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC bioinformatics. 2012; 13:86.
42. Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q, Sun H, Zhang X, He M, Guo H, Chen W, Yuan J, Zhang B, et al. Genome-Wide Analysis of DNA Methylation and Cigarette Smoking in Chinese. Environmental health perspectives. 2016.
43. Zaghlool SB, Al-Shafai M, Al Muftah WA, Kumar P, Falchi M and Suhre K. Association of DNA methylation with age, gender, and smoking in an Arab population. Clinical epigenetics. 2015; 7:6.
44. Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR and Philibert RA. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC genomics. 2014; 15:151.
45. Didelez V and Sheehan N. Mendelian randomization as an instrumental variable approach to causal inference. Statistical methods in medical research. 2007; 16:309-330.
46. Relton CL and Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. International journal of epidemiology. 2012; 41:161-176.
47. Jung M and Pfeifer GP. Aging and DNA methylation. BMC biology. 2015; 13:7.
48. Schöttker B, Haug U, Schomburg L, Kohrle J, Perna L, Muller H, Holleczek B and Brenner H. Strong associations of 25-hydroxyvitamin D concentrations with all-cause, cardiovascular, cancer, and respiratory disease mortality in a large cohort study. The American journal of clinical nutrition. 2013; 97:782-793.
49. Miller SA, Dykes DD and Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic acids research. 1988; 16:1215.
50. Florath I, Butterbach K, Heiss J, Bewerunge-Hudler M, Zhang Y, Schöttker B and Brenner H. Type 2 diabetes and leucocyte DNA methylation: an epigenome-wide association study in over 1,500 older adults. Diabetologia. 2015; 59:130-138.
51. Zhang Y, Florath I, Saum KU and Brenner H. Self-reported smoking, serum cotinine, and blood DNA methylation. Environmental research. 2016; 146:395-403.
52. Benjamini Y and Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B Stat Methodol. 1995; 57:289-300.
53. Desquilbet L and Mariotti F. Dose-response analyses using restricted cubic spline functions in public health research. Statistics in medicine. 2010; 29:1037-1057.