
INTRODUCTION
The global burden of cancer continues to increase largely because of an aging population and an increasing adoption of cancer-causing life style behaviours [1]. Among the different kinds of cancer, gastric cancer (GC) is one of the most common malignancies, remaining a major public health issue as the fifth most common cancer and the second leading cause of cancer death worldwide [2, 3]. Gastric cancer is an aggressive disease, and its prognosis remains poor, generally owing to the absence of specific symptoms that renders early diagnosis of this disease difficult [4]. About half of GC patients have recurrent disease after curative surgery, but the outcome for these patients is still very poor, with an estimated 5-year survival rate for locoregional disease of 25% - 35% [5, 6]. Although various treatment modalities have been developed, many of them have failed to eliminate GC cells curatively [7]. The current standard of care for metastatic GC patients is palliative chemotherapy providing the best supportive care, aimed at extending survival of the patients from 3 months with no therapy to 7–15 months with modern combination chemotherapy, and a dismal 2% 5-year survival rate with chemotherapy [8]. Clinical trials evaluating novel targeted therapies identified HER-2 as a target for GC. A recent ‘Trastuzumab for Gastric Cancer’ (ToGA) trial successfully demonstrated trastuzumab (Trastuzumab®) a recombinant humanized monoclonal antibody that targets the extracellular domain IV of the HER2 protein, as the first biologic therapy to have activity in advanced GC. However, only 10–25% of GC samples showed HER-2 amplification and even in those cases the ToGA study showed an improvement of a significant, but poor median survival by 2.5 months for patients treated with classical chemotherapy plus trastuzumab [9]. Therefore, there is an urgent need to introduce new therapeutic agents for the clinical management of gastric cancer.
Epigenetic modifications play a key role in several types of cancer, including gastric cancer [10, 11], which ultimately contributes to the development and progression of this malignancy [12, 13]. The stomach has been described as one of the organs with increased frequency of hypermethylation of CpG islands, which has been associated with age and inflammatory processes [14], potentially related to the exposure of the tissue to exogenous agents. Targeting of epigenetic processes may therefore provide a new method in the treatment of gastric cancer.
Bromodomains (BRDs) are conserved chromatin binding domains found in many nuclear proteins. BRD containing proteins are specifically recruited to ε-N-acetylated lysine residues present in histones and other proteins. BRD containing proteins function as regulators of gene transcription and by modulating and modifying chromatin structure [15-18]. The dysfunction of BRD containing proteins has been linked to the development of various types of cancer [19]. In particular, proteins of the bromo and extra terminal (BET) family, namely BRD2, BRD3, BRD4 and BRDT, have been identified as regulators for the expression of key oncogenes and the first clinical trials using BET inhibitors have recently commenced [15, 18, 20-24]. However, also the first resistance mechanisms have been reported [25, 26].
Given the dire need for anticancer agents active against gastric cancer, the effect of an epigenetic inhibitor library on GC cells was assessed in both 2D and 3D in vitro models. BET family inhibitors (Figure 1A) were identified as the first potent epigenetic inhibitors of gastric cancer cells. Both the pan-BET inhibitor (+)-JQ1 as well as a newly developed isoxazole, PNZ5, showed potent inhibition of GC cells providing a starting point for future therapy (Figure 1B).
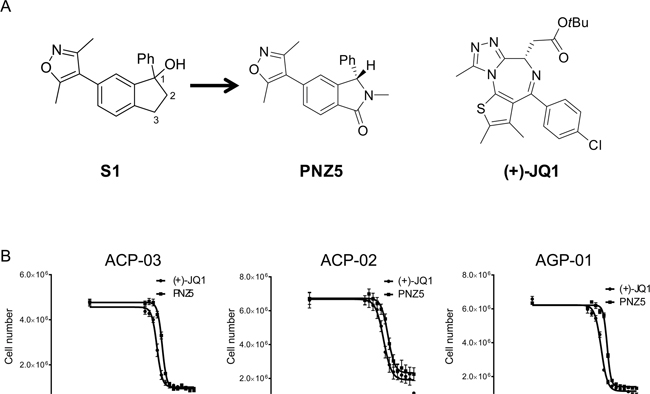
Figure 1: A. BET inhibitors used in the study (+)-JQ1 and (PNZ5) as well as lead compound S1. B. Growth inhibition curves of three gastric cancer cell lines derived from Brazilian patients after treatment with BET inhibitors (+)-JQ1 and PNZ5 for 72h.
RESULTS
Profiling of epigenetic probes in gastric cancer cell lines
Initial screening with a small library of 19 epigenetic probes was performed in three GC cell lines, but only 5 compounds (Bromosporine, UN1999, UNC0638, (+)-JQ1 and PNZ5) inhibited the growth of the cells at a concentration of 10 μM after 72h incubation (Supplementary Table 1). (+)-JQ1 and PNZ5 were the most active compounds, with strong antiproliferative activity and were therefore chosen for further investigation (Figure 1B).
PNZ5 is a potent pan-BET inhibitor
Isoxazoles have been explored previously as BET bromodomain inhibitors [27-29]. We have developed PNZ5, a new isoxazole-based inhibitor originating from the lead compound S1 (Figure 1). PNZ5 was developed as the result of a structure-based lead optimisation program. Dihydroindene S1 was identified as a ligand against BRD4(1) (pIC50 5.9) [30]. Investigation of the co-crystal structure of S1 with BRD4(1) [PDB ID 4GPJ] identified a number of positions for potential optimisation: addition of a carbonyl group at C-3 was envisaged to benefit from hydrogen-bonds (H-bonds) to a network of conserved water molecules; replacement of C-2 with a nitrogen would minimise interactions in the narrow ZA channel of BRD4(1) and N-substitution could provide further binding interactions with the protein; and alternative substitution of the hydroxyl at C-1 would provide a handle for further optimisation. Incorporation of a N-methyl lactam motif (S2) resulted in a signficant increase in the affinity, validating the new scaffold. Substitution at both C-2 and in place of the C-1 hydroxyl were found to be largely detrimental to affinity for BRD4(1), with proto-substituents in each position optimal for binding. The three best compounds, S3, S5 and S7, were destined to be separated into their component enantiomers for further evaluation (Supplementary Table 2). However, S7 was found to be unstable under the separation conditions and S5 was inseparable with the equipment available to us, but, pleasingly, S3 was readily resolved by preparative chiral HPLC. Biophysical assays on the two enantiomers revealed the S-enantiomer, PNZ5, as the biologically active species, which was then used for further biological interrogation of BRD4(1). The synthesis and analytical data are summarized in Scheme S1.
Potency of PNZ5 for the first bromodomain of BRD4(1) was assessed by isothermal titration calorimetry (ITC) revealing a dissociation constant (KD) of 5.43 nM with a favourable enthalpic contribution of -15.57 kcal/mol (Figure 2A and Supplementary Table 3). The compound is highly selective for the BET family as assessed by a comprehensive thermal shift assay (Figure 2B and Supplementary Table 5). Since its discovery in our own labs, similar inhibitors have also been disclosed by researchers at Boehringer Ingelheim [reference: patent, WO 2014/154760 [31]]. The inhibitor contains two possible acetyl-lysine mimetic moieties. In order to obtain molecular insights on the interaction of the developed inhibitor, the crystal structure of the first bromodomain of BRD4 in complex with PNZ5 was solved at 1.85 Å (Figure 2C and Supplementary Table 4, 5FBX.pdb). The inhibitor, coordinating residues and water molecules were well defined by the electron density. The dimethylisoxazole moiety mimicked the acetyl lysine side chain and formed two canonical hydrogen-bond interactions with Asn140 and water mediated hydrogen-bond with Tyr 97 (Figure 2C). The common network of waters within the binding site was also conserved in the BRD4(1)-PNZ5 complex. The compound was further stabilized by lipophilic interactions with residues forming the binding site Leu92, Leu94, Ile146. The phenyl substituent was well accommodated next to the WPF shelf, as well as the N1-methyl substituent that formed hydrophobic interactions with Trp81. The carbonyl on the isoindolinone established a water-mediated hydrogen bond with Lys91. Superposition of the apo structure of BRD4(1) (2OSS.pdb) with the structure in complex with PNZ5 did not reveal any major rearrangements in the ZA-loop segment.
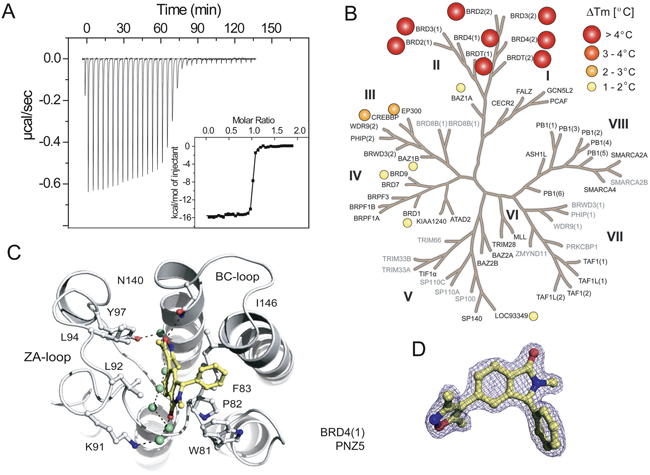
Figure 2: Characterization of PNZ5. A. Isothermal titration data of the interaction of PNZ5 with the first bromodomain of BRD4 (BRD4(1)). Shown are the raw binding heats for each injection as well as normalized binding enthalpies. B. Selectivity of PNZ5 calculated using thermal shift (ΔTm) assays. Screened targets (labelled in black) in the phylogenetic tree based on the BRD family structure based alignment [71]. C. Binding mode of PNZ5 bound to BRD4(1). Hydrogen bonds to the conserved asparagine (N140) and tyrosine (Y97) are shown as dotted green lines. D. Electron density map (2FoFc) contoured at 2σ around the inhibitor.
(+)-JQ1 and PNZ5 alter intracellular motility of BRD4
Cellular activity of PNZ5 was demonstrated using a full length BRD4 fluorescence recovery after photobleaching (FRAP) assay [32]. The data showed that at a concentration of ≤0.5 μM, PNZ5 effectively reduced the fluorescent half-time recovery of BRD4 fused to GFP to levels comparable to both the BET inhibitor (+)-JQ1 and the double mutant N140F/N433F, which is unable to bind to chromatin (Figure 3). The effect on fluorescence recovery was stronger than seen for the single domain mutant N140F of the first bromodomain or N433F of the second bromodomain, respectively. These findings suggested that PNZ5 effectively displaced both bromodomains of ectopically expressed BRD4 from chromatin.
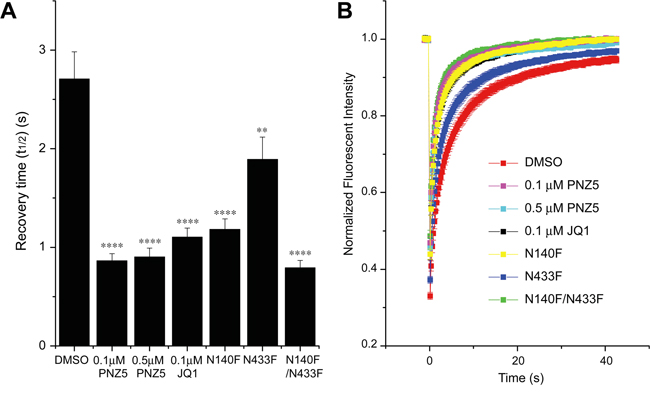
Figure 3: Effect of (+)-JQ1 and PNZ5 on intracellular motility of BRD4. A. Fluorescence recovery half-times for full-length BRD4. PNZ5 significantly decreased half recovery times of U2OS cells transfected with wild-type full-length GFP-BRD4, as did (+)-JQ1. Half recovery times were accelerated to the level of the double bromodomain mutant N140F/N1433F and faster than the single mutant. B. Time dependence of the fluorescence recovery of the bleached area. At least 20 nuclei were bleached for each experiment and the mean recovery time as well as the standard error of mean (SEM) are shown in A (****p<0.0001; ** p<0.002).
Diverse gastric cancer cell lines differ in BRD4 and c-MYC expression levels
We aimed to explore if sensitivity to BET inhibitors was a general feature of gastric cancer and assessed the effect of BET inhibition on GC lines of Asian and Brazilian origins. The 2 Asian cell lines are derived from metastatic sites. The Brazilian cell lines represent different tumors: ACP-02, is a diffuse type GC; ACP-03 an intestinal type and AGP-01 represents a malignant ascites. Interestingly, BET inhibitors did not have an effect on proliferation of the 2 cell lines originating from Asian patient cohorts (Table 1). In order to assess if different expression levels of BRD4 were present in these different cell lines qPCR experiments in the GC cell lines AGP-01, ACP-02, ACP-03 originating from patients in Brazil, the Asian GC lines SNU-16 and KATO III, and HEK 293T cells were performed. No large differences were observed between the cell lines, but the ACP-02 cell line was shown to have the highest amount of BRD4, whereas KATO III had the lowest amount of BRD4 (Figure 4A), thus expression of the target did not predict inhibitor sensitivity of the studied cell lines. Efficacy of BET inhibitors in cell proliferation has been linked to the transcriptional downregulation of c-MYC in several different cancer models [33-36]. In order to understand if modulation of c-MYC plays also a role in gastric cancer, c-MYC expression studies were performed on cells treated with (+)-JQ1 and PNZ5. The inhibitors (+)-JQ1 and PNZ5 did indeed reduce c-MYC expression in most of the cell lines, although the effect was not very pronounced in SNU-16, which is a cell line with known c-MYC amplification [37] (Figure 4B). Interestingly, there was also no downregulation of c-MYC mRNA levels in ACP-02, despite having high BRD4 levels and responding to BET inhibitor treatment. Treatment with the 2 BET inhibitors did not have an effect on c-MYC protein level (data not shown). Furthermore, there is no correlation between c-MYC expression levels and BRD4 levels. However, to our surprise we saw some differential response of c-Myc mRNA levels upon treatment with PNZ5 as compared to JQ1 although they bind to BRD4 with similar affinity (Supplementary Table 3).
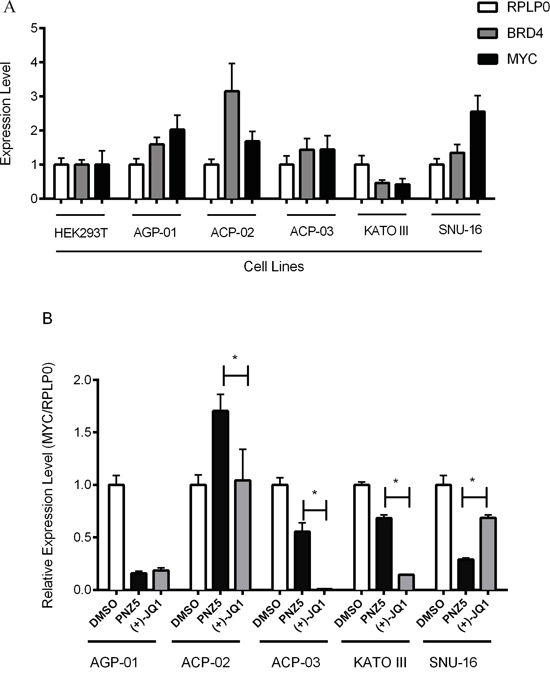
Figure 4: Expression profile of BRD4 and c-MYC mRNA levels by qRT–PCR in a panel of gastric cancer cell lines. A. Expression of BRD4 was evaluated in a panel of human gastric cancer cell lines and compared with HEK293T cells. B. Expression of c-MYC was evaluated in all gastric cell lines after 6 h treatment with (+)-JQ1 and PNZ5 (500 nM). The mRNA level for BRD4 and c-MYC was normalized to RPLP0 mRNA level in each cell line. Average and standard deviation were calculated from two independent experiments in triplicate.
Table 1: In vitro cytotoxic activity of (+)-JQ-1 and PNZ5
IC50 (μM) |
||
|---|---|---|
(+)JQ-1 |
PNZ5 |
|
AGP-01 |
||
24h |
0.60 |
0.95 |
48h |
0.21 |
0.47 |
72h |
0.12 |
0.34 |
ACP-02 |
||
24h |
0.58 |
1.33 |
48h |
0.56 |
0.85 |
72h |
0.29 |
0.61 |
ACP-03 |
||
24h |
0.15 |
0.47 |
48h |
0.12 |
0.29 |
72h |
0.09 |
0.22 |
Kato III |
||
24h |
>5 |
>5 |
48h |
>5 |
>5 |
72h |
>5 |
>5 |
SNU-16 |
||
24h |
>5 |
>5 |
48h |
>5 |
>5 |
72h |
0.66 |
1.34 |
Five cell lines were exposed to (+)-JQ1 and DDX (0.01 - 50 μM). IC50 values (μM) and confidence interval of 95% obtained by the Resazurin assay in gastric cancer cell lines after 24, 48 and 72 hours of exposure. Experiments were performed in triplicates and means from three independent experiments are reported.
BET inhibitors display cytotoxicity and induce apoptosis in gastric cancer cell lines
PNZ5 and (+)-JQ1 are both potent and selective BET inhibitors, but belong to different chemical classes. We therefore performed a more detailed analysis of the antiproliferative effect of these 2 compounds in a high-content image screen (Howarth et al. unpublished data). (+)-JQ1 was the most potent compound in all cell lines (Table 1). Although at early time points there seemed to be small differences in the sensitivity of the cells treated with (+)-JQ1 or PNZ5, after 72 h there was no longer a statistical difference between the inhibitors. Based on our results, there was a tendency of the effect of PNZ5 to be time-dependent in all tested cell lines as the IC50 decreased over time, whereas (+)-JQ1 activity appeared to be time- and cell type-dependent (Table 1). The most sensitive cell line for both compounds was ACP-03, a primary intestinal-type adenocarcinoma and the most resistant was ACP-02, a diffuse-type adenocarcinoma cell line (Table 1). The compounds were then tested against 2 GC cell lines of Asian origin, which proved more resistant to BET inhibition. Growth of SNU-16 cells was only inhibited after 72h of treatment, with an IC50 of 0.66 and 1.34 μM for (+)-JQ1 and PNZ5 respectively, whereas proliferation of KATO III was unaffected by BET inhibition (Table 1).
In order to determine if the two compounds lead to the same cell death pattern, we performed a triple staining protocol using a high content imaging system (Figure 5). Cells treated with (+)-JQ1 and PNZ5 induced apoptosis in the three cell lines; AGP-01, ACP-02 and ACP-03 (Figure 5). After 72h, (+)-JQ1 (400 nM) induced apoptosis and necrosis in 62% and 31% of AGP-01cells, whereas PNZ5 (400 nM) induced 19% and 44% respectively. As for ACP-02, (+)-JQ1 induced 19% and 74% and PNZ5 induced 4% and 45% of apoptosis and necrosis, respectively. In ACP-03, (+)-JQ1 induced 56% and 42% and PNZ5 induced 24% and 35% of apoptosis and necrosis, respectively. There was no significant difference between the two compounds in inducing either apoptosis or necrosis apart from induction of necrosis in the ACP-02 cell line, consistent with their identical mode of action. As expected, there was a significant reduction in persistent cells in all cell lines when compared to the DMSO control.
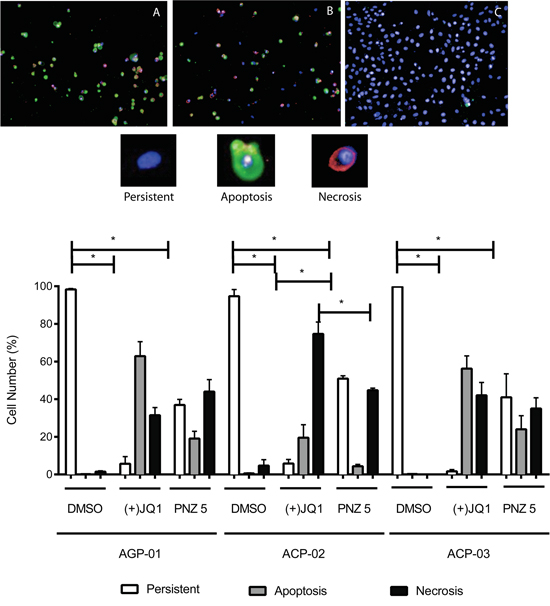
Figure 5: High content images of in vitro cytotoxic activity of (+)-JQ1 A. and PNZ5 B. on cell death pattern in gastric cancer cell lines compared to DMSO control C. (upper panel). Apoptosis increased after 72 hours exposure (lower panel). Apoptotic cells were defined as Annexin V positive with- or without Yo-Pro 3 uptake; Necrotic cells were defined as Yo-Pro 3 positive; and Persistent cells were defined as Annexin V and Yo-Pro 3 negative. Hoechst was used to identify the nuclei. Results are shown as mean +/- SEM from triplicates of two independent experiments.*p<0.01
BET inhibitors display cytotoxicity in 3D multicellular spheroids
In addition to 2D cell monolayers, we also validated the cytotoxic effect of PNZ5 and (+)-JQ1 on a well-accepted three-dimensional (3D) in vitro model of cancer multicellular spheroids formed from AGP-01 cells. With the increase of drug dosage from 0.01μM to 1μM, the cells in the spheroids showed higher cell death, indicating an effective drug penetration and cytotoxicity in the 3D culture, which mimic the in vivo tumor tissue (Figure 6A and 6C).
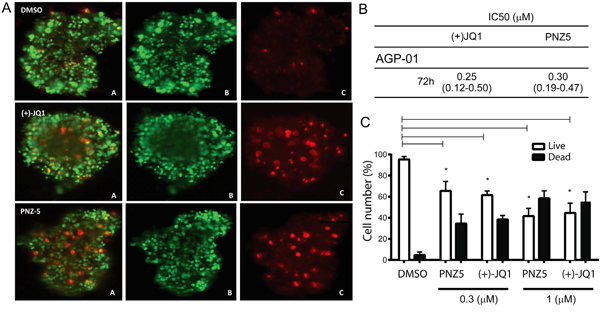
Figure 6: In vitro cytotoxic activity of (+)JQ-1 and PNZ5 on AGP-01 spheroids. A. Live/Dead cell staining for AGP-01 spheroids treated with (+)JQ-1 (300nM) and PNZ-5 (300 nM). A. Merge; B. Calcein; C. EthD-1; B. IC50 values (μM) and confidence interval of 95% obtained by the Resazurin assay in AGP-01 spheroids 72 h of exposure; C. Live/Dead cell quantification. Experiments were performed in triplicates and means from three independent experiments are reported. *p<0.01
The IC50 of PNZ5 and (+)-JQ1 in multicellular spheroids formed by AGP-01 cells were estimated to be 0.3 μM and 0.2 μM, respectively (Figure 6B) after 72 h of treatment. The IC50 values obtained from 3D multicellular spheroids were higher than the values from 2D monolayers of cell culture, however they were not statistically different, which implied that the two compounds are efficient in both 2D and 3D cell culture models.
DISCUSSION
Herein, we describe inhibition of BET family members as a new possible strategy for the treatment/management of gastric cancer. Despite an overall decrease in incidence over the past decades, gastric cancer is still associated with poor survival. This is mainly due to the delayed signs and symptoms, such that at the time of diagnosis, 35% of GC patients have signs of metastases, 31% with peritoneal disease, 14% with liver metastases, and 16% with lung metastases [8]. Treatment of GC remains a major clinical challenge, especially for patients with metastasis. There is an urgent need for adjuvant and neoadjuvant trials evaluating novel targeted therapies in gastric cancer treatment.
In this study, BET inhibitors of two different scaffolds showed strong antiproliferative effects on three GC cell lines. PNZ5 is a newly identified BET isoxazole-based inhibitor with high selectivity and potency similar to the well-established (+)-JQ1, which is one of a range of diazepines that have been identified as potent BET ligands. A second class of key BET inhibitors are focused around a 3,5-dimethylisoxazole moiety, which was identified as an alternative acetyl-lysine mimic to replicate the same hydrogen-bond to a conserved arginine residue as does the native ligand. The most notable example to date is I-BET151, a highly potent and specific BET inhibitor, which has the advantage of improved pharmacokinetic properties compared to the triazolobenzodiazepine scaffolds [38-40]. PNZ5 is another example of a potent and selective isoxazole-based BET inhibitor with improved ligand efficacy. Interestingly, we observed some differences in the c-MYC expression levels when cells were treated with JQ1 or PNZ5. This effect was not expected and reasons for these variances are currently not well understood, but there might be differences in affinities for the different members of the BET family between JQ1 and PNZ5, which may mediate differential activation of antiproliferative pathways.
BET proteins are key mediators in the process of transcription of many genes involved in proliferation and anti-apoptotic and cell cycle progression [41]. BET specific inhibitors have shown therapeutic potential in many different tumor models through multiple mechanisms, including down-regulation of c-MYC expression [42, 15, 22, 36, 38, 43, 44]. c-MYC is a transcriptional factor involved in cell cycle regulation and has been described as a key element of gastric carcinogenesis [45-47]. The cells used in this study have been described previously to present chromosome 8 trisomy - the chromosome where the MYC oncogene is located [48]. Furthermore, frequency of MYC gain in tumors of patients with advanced GC seems to be higher in the Brazilian population [48-53] than in Asia, where Deng et al. [54] demonstrated the existence of five distinct gastric cancer patient subgroups, defined by the signature genomic alterations (FGFR2 [9% of tumours], KRAS [9%], EGFR [8%], ERBB2 [7%] and MET [4%]); however MYC was not one of them. The data obtained here is in line with these findings as both tested BET inhibitors only had little if any effect in the Asian cell lines tested, but showed potent antiproliferative effects in the cells of Brazilian origin, pointing to a potential genetic and demographic factor of GC.
Clonal high amplification of c-MYC is less frequent in diffuse-type gastric adenocarcinoma (ACP-02) than in ACP-03 and AGP-01 cells, originated from intestinal-type gastric adenocarcinoma [49, 50, 55]. In our study, we show that ACP-03 and AGP-01 were more sensitive to PNZ5 and (+)-JQ1 when compared to the ACP-02 cell line. Also, it seems that (+)-JQ1 and PNZ5 induce more apoptosis in ACP-03 and AGP-01 cell lines as compared to ACP-02 cells, where necrosis is the predominant modus of cell death.
Furthermore, our study shows that (+)-JQ1 and PNZ5 exposure reduces the expression of c-MYC in AGP-01 and ACP-03 cells, but not in ACP-02 cells, reinforcing the presence of different pathways involved in intestinal-type and diffuse-type gastric carcinogenesis [56]. Interestingly, although c-MYC expression is downregulated in the Asian derived-cell line KATO III and to a lesser extent in SNU-16, a cell line with high c-MYC amplification, there was a complete lack of antiproliferative activity of (+)-JQ1 and PNZ5, indicating that the antiproliferative effect is independent of c-MYC levels, a finding that has also been reported for leukemia cell lines [57]. The observed differences in antiproliferative response to BET inhibition may result from different genetic and/or epigenetic make up of these cell lines from different ethnic origin. The observations in the Brazilian cell lines corroborate previous work by Wyce et al. (2013) who demonstrated that prostate cancer cells with high c-MYC protein expression are more sensitive to I-BET762 compared to cell lines with low c-MYC protein expression [58]. The positive effect of BET inhibitors might be due to c-MYC transcriptional suppression since AGP-01 and ACP-03 cell lines present high amplification of MYC as shown previously [59].
Furthermore, Fowler et al. [60] demonstrate that cells with BRD4 amplification are more sensitive to (+)-JQ1. However, our own data indicate that ACP-02, which has the highest BRD4 and c-MYC levels are less sensitive to BET inhibition (Figure 4) and there seems to be little correlation between BRD4 levels and MYC regulation and antiproliferative effect in the Asian gastric cancer cell lines. MYC independent mechanisms of tumor cell killing by BET inhibitors have also been reported in other tumors like osteosarcoma cells [61]. Taken together, we have identified BET inhibition as an alternative avenue for the treatment/management of gastric cancer in a selective group of patients and/or histological type of gastric cancer. The mechanisms leading to the sensitivity of gastric cell lines to BET inhibition and suitable biomarkes for patient stratification remain however to be established.
MATERIALS AND METHODS
Synthesis of PNZ5
See supporting information (Scheme 1)
Protein expression and purification
BRD4 and other bromodomains were cloned, expressed and purified as described previously [21].
Thermal shift assays
The recombinant bromodomains at 2 μM were mixed with 10 μM inhibitors. The assays and data evaluation for melting temperatures were performed using a Real-Time PCR Mx3005p machine (Stratagene) and the protocols previously described [62]. Experiments profiling the final compound (PNZ5) were performed in quadruplicates.
Isothermal titration calorimetry
Calorimetric experiments were performed on a VP-ITC micro-calorimeter (MicroCal™, LLC Northampton, MA). Protein solution was buffer exchanged by dialysis into buffer 20 mM Hepes pH 7.5, 150 mM NaCl, and 0.5 mM TCEP. All measurements were carried out at 293.15 K while stirring at 286 rpm. The micro syringe was loaded with a protein solution of 250 μM, the compound solution was prepared at 20 μM and 2mL for the cell. All injections were performed using an initial injection of 2 μl followed by 34 injections of 8 μl with a duration of 16 sec per injection and a spacing of 240 sec between injection. The data were analysed with the MicroCal ORIGIN software package employing a single binding site model. The first data point was excluded from the analysis. Thermodynamic parameters were calculated (ΔG = ΔH - TΔS = -RTlnKB where ΔG, ΔH and ΔS are the changes in free energy, enthalpy and entropy of binding, respectively).
Crystallization
BRD4 construct (Uniprot identifier as BRD4_HUMAN O60884-1 fragment 44-168) was used for crystallographic studies in complex with the inhibitor. Aliquots of the purified protein were set up for crystallization using a mosquito® crystallization robot (TTP Labtech). Coarse screens were typically setup onto Greiner 3-well plates using three different drop ratios of precipitant to protein per condition (200+100 nL, 150+150 nL and 100+200 nL). All crystallizations were carried out using the sitting drop vapour diffusion method at 277.15 K. Small rod co-crystals of BRD4(1) in complex with the inhibitor (4 mM final concentration) were obtained by mixing 150 nL of the protein (12.5 mg/ml) and 150 nL crystallization buffer (20% PEG3350, 0.2 M zinc acetate, 0.1 M imidazole pH 7.8). A complete dataset was collected for BRD4(1) co-crystals at Diamond Light Source (beamline I04) and processed to 1.85 Å.
Data collection and structure solution
Complex crystal was cryo-protected using the well solution supplemented with additional 20% ethylene glycol and was flash frozen in liquid nitrogen. Data was collected at Diamond Light Source beamline I04 at a wavelength of 0.9795 Å. Indexing and integration was carried out using XDS [63] and scaling was performed with AIMLESS [64]. Initial phases were calculated by molecular replacement with PHASER [65] using the apo template structure 2OSS.pdb. Unique and initial solutions were improved in a total of 50 cycles of automated protein chain tracing starting from existing model and computed using ARP/wARP [66]. Further manual building with COOT [67] and refinement against maximum likelihood target using REFMAC5 [68]. Thermal motions were analysed using TLSMD and hydrogen atoms were included in late refinement cycles. GRADE [69] was used to generate compound coordinates and cif files. All model validations were carried out using MolProbity [70]. Data collection and refinement statistics are compiled in Supplementary Table 4 The models and structure factors have been deposited with PDB accession codes: 5FBX.pdb
Ethics statement
All samples were derived with written informed consent and approval from the University Hospital (Belém, Pará, Brazil) ethical review boards (protocol number: 142004).
Gastric cancer cells
Our group established and characterized cytogenetically three new GC cell lines obtained from primary gastric adenocarcinoma (ACP02, diffuse type and ACP03, intestinal type) and peritoneal carcinomatosis (AGP01, ascitic fluid) from a patient with primary tumor on stomach (Intestinal-type adenocarcinoma), each of which exhibited a composite karyotype with several clonal chromosome alterations similar to primary tumor from the stomach as described previously [48]. All cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) medium supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine at 37°C with 5% CO2. Asian ethnicity cell lines, SNU-16 cell line (CRL-5974™), derived from a metastatic ascites of the stomach and Kato III (HTB-103™), derived from a metastatic site from a stomach primary tumor, were obtain from ATCC collection and cultured according to ATCC protocols.
Chemicals
A library of Epigenetic compounds (Supplementary Table 1) was dissolved in DMSO to a concentration of 50 mM and stored at –20°C as a master stock solution. DMEM, FBS, GLUTAMAX, Annexin V, Yo-Pro 3 and Hoechst 33342 stain were purchased from Life Technologies, UK. Resazurin as sodium salt was purchased from Sigma, UK.
Cytotoxicity against cancer cell lines
Initially, the epigenetic library compounds were tested in three cancer cell lines, at a single concentration of 10 μM for 72 h. (+)-JQ1 and PNZ5 (0.01-50 μM) were tested for cytotoxic activity against a panel of six gastric cancer cell lines: ACP-02 (Diffuse-type adenocarcinoma), ACP-03 (Intestinal-type adenocarcinoma) and AGP-01 (Malignant ascites) from Brazil and Kato III (Gastric metastasis), SNU- 16 (Gastric Ascites) from Asia. Each compound was dissolved in DMSO and diluted with media to obtain a maximum concentration of 50 μM. Cells (300 cells/well) were plated in 384-well flat-bottomed plates and cultured for 24h. Cells were exposed to a serial dilution of the compounds in DMEM with 1% FBS, for an additional 72 h. After the incubation time, the plates were centrifuged (1500rpm/3min), the supernatant removed and resazurin (10ng/ml) was added for 2h at 37°C before measurement of fluorescence at Ex 579nm/Em 584nm on an Envision microplate spectrophotometer (PerkinElmer). The concentration of compounds resulting in 50% growth inhibition (IC50) was calculated for each cell line in GraphPad Prism 5.0.
Spheroids formation and drug treatment
GC cells AGP-01 in exponential growth phase were chosen for the spheroid formation. Briefly, 100 μL cell suspensions with the seeding density 1×103 cells/μL were added to each well of a 96-well plate with an ultra-low cell adherence coating surface (Corning-Costar, UK), and were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM, Lonza, UK) supplemented with 10% (v/v) foetal bovine serum (FBS, Life Technologies, US) and 100 U/mL penicillin - 100 μg/mL streptomycin (PAA, US), in a humidified incubator at 37 ºC and in an atmosphere of 5% CO2 in air. The spheroids were imaged and monitored using an In Cell Analyser (GE, UK). Spheroids with stable structures and diameters had formed after three days. On the day commencing the treatment, to obtain the final concentration, drug was diluted into 100 μL solution at twice the desired dosage with DMEM without FBS before adding to the 100 μL media of the well, and incubated with the spheroids under standard culture condition for 72 h.
Cell live/dead imaging and image process of spheroids
A viability/cytotoxicity kit (Invitrogen, UK) was used for imaging the live and dead cells. The red fluorescent ethidium homodimer-1 (EthD-1) only permeated through the cell membrane of the dead cells and stained their nuclei, while living cells allowed the penetration of non-fluorescent acetomethoxy derivative of calcein (calcein AM) and degraded it into the green fluorescent calcein. The spheroid in each well of the 96-well plates was visualised and processed using a high-throughput imaging system, the In Cell Analyser (GE, UK). To process the 3D images, In Cell Investigator software (GE, UK) was used. Briefly, a background threshold was set for all the fluorescent images. The segmentation was selected using an iterative process of erosions and dilations. This allowed us to calculate a cell death percentage for the whole spheroid, indicated by the segmented red fluorescent areas.
Hoechst 33342/Yo-Pro 3/annexin triple staining and live cell death pattern analysis
Cells exposed to (+)-JQ1 and PNZ5 (10μM) for 72 h were stained with Hoechst 33342 (1μM), Yo-Pro 3 (1μM) and Annexin V (0.3 μL per well) for 1 h. Cellular fluorescence was measured using the Operetta®, a high content imaging system (PerkinElmer, USA) using the following setup parameters: Brightfield transmitted light at 50% for 15 ms; Hoechst 33342 was excited by 35 ms exposure; Ex 360-400 nm/Em 410-480 nm, Yo-Pro 3 by 35 ms exposure; Ex 560-580 nm/Em 650-760 nm and Annexin V (Alexa 488) by 45 ms exposure; Ex 460-490 nm/Em 500-550 nm. All the generated data were analyzed by Harmony software and three categories were distinguished: persistent cells, apoptosis and necrosis - calculated as percentage of each class for every concentration used.
BRD4 and c-MYC expression level
c-MYC expression was performed on AGP-01, ACP-02, ACP-03, SNU-16 and Kato III cell lines treated with (+)JQ1 and PNZ5 (500 nM) for 6h and collected for RNA extractions. For BRD4 expression levels, cells were grown to 70% confluence and collected for RNA extractions. Total RNA was prepared by using a Qiagen RNEasy kit according to the manufacturer’s instructions. cDNA were prepared by using The High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s manual. For cell line samples, an appropriate dilution of cDNA and gene-specific primers were combined with FAST SYBR® Green MasterMix (Applied Bioscience) and amplified in a Light Cycler® 480 II (Roche). All qPCR reactions were performed in triplicates. Ct (threshold cycle number) and expression values with standard deviations were calculated. RPLP0 (human ribosomal protein, large, P0) was used as a housekeeping gene. Primer sequences for real-time PCRs were as follows: c-MYC forward, 5’-GCTGCTTAGACGCTGGATTT-3’; c-MYC reverse, 5’-TAACGTTGAGGGGCATCG-3’; BRD4 forward, 5'-AGGCAAAAGGAAGAGGA-3'; BRD4 reverse, 5'-CGATGCTTGAGTTGTGTT-3'; RPLP0 forward, 5'-AGCCCAGAACACTGGTC-3'; RPLP0 reverse, 5'-ACTCAGGATTTCAATGGT-3'. BRD4, c-MYC and RPLP0 primers were used at 0.25 μM final and exhibited PCR efficiencies of over 90%. Real-time amplification was performed with initial denaturation at 95°C for 30 sec, followed by 40 cycles of two-step amplification (95°C for 3 s, 60°C for 30 s).
Fluorescence recovery after photobleaching (FRAP) assay
FRAP studies were performed using U2OS cells (purchased from ATCC) expressing a full-length BRD4 protein with an N-terminal eGFP as previously described [32]. In brief, six hours post-transfection the medium was replaced and 24 hours later imaging took place. The inhibitors were incubated with the cells for one hour before imaging. Half times of fluorescence recovery (t1/2) were calculated from individual recovery curves of twenty cells per group. Results were analysed according to their means and standard errors in GraphPad Prism 5.0. Data obtained from different experiments are presented as mean ± SEM from at least three independent experiments in triplicate and evaluated by analysis of variance (ANOVA) followed by Tukey test using a significance level of 5%.
ACKNOWLEDGMENTS
The authors would like to express thanks to CAPES and CNPq (Brazil) for the fellowship in the Program Science Without Borders (211488/2013-5). PGKC would like to thank the Woolf Fisher Trust for his fellowship. We would like to thank Dr. Jens Peter von Kries at the FMP, Berlin for advice and helpful discussion.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
The authors are supported by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z].
REFERENCES
1. Romani M, Pistillo MP, Banelli B. Environmental Epigenetics: Crossroad between Public Health, Lifestyle, and Cancer Prevention. BioMed research international. 2015; 2015:587983.
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011; 61:69-90.
3. Globocan. (2012). Section of Cancer Surveillance.
4. Catalano V, Labianca R, Beretta GD, Gatta G, de Braud F, Van Cutsem E. Gastric cancer. Critical reviews in oncology/hematology. 2005; 54:209-241.
5. Smalley SR, Benedetti JK, Haller DG, Hundahl SA, Estes NC, Ajani JA, Gunderson LL, Goldman B, Martenson JA, Jessup JM, Stemmermann GN, Blanke CD, Macdonald JS. Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012; 30:2327-2333.
6. van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, Cuesta MA, Blaisse RJ, Busch OR, ten Kate FJ, Creemers GJ, Punt CJ, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. The New England journal of medicine. 2012; 366:2074-2084.
7. Gallo A, Cha C. Updates on esophageal and gastric cancers. World journal of gastroenterology : WJG. 2006; 12:3237-3242.
8. Gadde R, Tamariz L, Hanna M, Avisar E, Livingstone A, Franceschi D, Yakoub D. Metastatic gastric cancer (MGC) patients: Can we improve survival by metastasectomy? A systematic review and meta-analysis. Journal of surgical oncology. 2015; 112:38-45.
9. Gunturu KS, Woo Y, Beaubier N, Remotti HE, Saif MW. Gastric cancer and trastuzumab: first biologic therapy in gastric cancer. Therapeutic advances in medical oncology. 2013; 5:143-151.
10. Panani AD. Cytogenetic and molecular aspects of gastric cancer: clinical implications. Cancer letters. 2008; 266:99-115.
11. Kang C, Song JJ, Lee J, Kim MY. Epigenetics: an emerging player in gastric cancer. World journal of gastroenterology : WJG. 2014; 20:6433-6447.
12. Gigek CO, Chen ES, Calcagno DQ, Wisnieski F, Burbano RR, Smith MA. Epigenetic mechanisms in gastric cancer. Epigenomics. 2012; 4:279-294.
13. Calcagno DQ, Gigek CO, Chen ES, Burbano RR, Smith Mde A. DNA and histone methylation in gastric carcinogenesis. World journal of gastroenterology : WJG. 2013; 19:1182-1192.
14. Selaru FM, David S, Meltzer SJ, Hamilton JP. Epigenetic events in gastrointestinal cancer. The American journal of gastroenterology. 2009; 104:1910-1912.
15. Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, Natsume H, Yao H, Ozato K. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. The Journal of biological chemistry. 2008; 283:9040-9048.
16. Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Current opinion in drug discovery & development. 2009; 12:659-665.
17. Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert reviews in molecular medicine. 2011; 13:e29.
18. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews Drug discovery. 2014; 13:337-356.
19. Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Molecular cell. 2014; 54:728-736.
20. Zhao Y, Yang CY, Wang S. The making of I-BET762, a BET bromodomain inhibitor now in clinical development. Journal of medicinal chemistry. 2013; 56:7498-7500.
21. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067-1073.
22. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011; 478:524-528.
23. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, Nekritz EA, Zeid R, Gustafson WC, Greninger P, Garnett MJ, McDermott U, Benes CH, Kung AL, Weiss WA, Bradner JE, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer discovery. 2013; 3:308-323.
24. Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, Gonzalez-Gomez P, Morante M, Jubierre L, Zhang W, Darvishian F, Ohlmeyer M, Osman I, Zhou MM, Hernando E. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer research. 2013; 73:6264-6276.
25. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Peluffo G, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016; 529:413-417.
26. Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee SC, Carpenter C, Gregory R, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015; 525:538-542.
27. Hewings DS, Wang M, Philpott M, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Vuppusetty C, Marsden B, Knapp S, Conway SJ, Heightman TD. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. Journal of medicinal chemistry. 2011; 54:6761-6770.
28. Mirguet O, Gosmini R, Toum J, Clement CA, Barnathan M, Brusq JM, Mordaunt JE, Grimes RM, Crowe M, Pineau O, Ajakane M, Daugan A, Jeffrey P, Cutler L, Haynes AC, Smithers NN, et al. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. Journal of medicinal chemistry. 2013; 56:7501-7515.
29. Muller S, Knapp S. Discovery of BET bromodomain inhibitors and their role in target validation. Med Chem Comm. 2014; 5:288-296.
30. Hay D, Fedorov O, Filippakopoulos P, Martin S, Philpott M, Hewings DS, Uttarkar S, Heightman TD, Conway SJ, Knapp S, Brennan P. The design and synthesis of 5- and 6-isoxazolylbenzimidazoles as selective inhibitors of the BET bromodomains. Med Chem Comm. 2013; 4:140-144.
31. Gianni D, MANTOULIDIS A, SMETHURST C. (2014). INDOLINONE ANALOGUES AS BRD4 INHIBITORS.
32. Philpott M, Rogers CM, Yapp C, Wells C, Lambert JP, Strain-Damerell C, Burgess-Brown NA, Gingras AC, Knapp S, Muller S. Assessing cellular efficacy of bromodomain inhibitors using fluorescence recovery after photobleaching. Epigenetics & chromatin. 2014; 7:14.
33. Li GQ, Guo WZ, Zhang Y, Seng JJ, Zhang HP, Ma XX, Zhang G, Li J, Yan B, Tang HW, Li SS, Wang LD, Zhang SJ. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget. 2015. doi: 10.18632/oncotarget.6275.
34. Shao Q, Kannan A, Lin Z, Stack BC, Jr., Suen JY, Gao L. BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo. Cancer research. 2014; 74:7090-7102.
35. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, Bolin S, Schumacher SE, Zeid R, Masoud S, Yu F, Vue N, Gibson WJ, Paolella BR, Mitra SS, Cheshier SH, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014; 20:912-925.
36. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011; 146:904-917.
37. Park IC, Park MJ, Lee SH, Choe TB, Jang JJ, Hong SI. Increased susceptibility of the c-Myc overexpressing cell line, SNU-16, to TNF-alpha. Cancer letters. 1998; 125:17-23.
38. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011; 478:529-533.
39. Hewings DS, Fedorov O, Filippakopoulos P, Martin S, Picaud S, Tumber A, Wells C, Olcina MM, Freeman K, Gill A, Ritchie AJ, Sheppard DW, Russell AJ, Hammond EM, Knapp S, Brennan PE, et al. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. Journal of medicinal chemistry. 2013; 56:3217-3227.
40. Bamborough P, Diallo H, Goodacre JD, Gordon L, Lewis A, Seal JT, Wilson DM, Woodrow MD, Chung CW. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. Journal of medicinal chemistry. 2012; 55:587-596.
41. Bres V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Current opinion in cell biology. 2008; 20:334-340.
42. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:8758-8763.
43. Dey A, Nishiyama A, Karpova T, McNally J, Ozato K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Molecular biology of the cell. 2009; 20:4899-4909.
44. Wyce A, Ganji G, Smitheman KN, Chung CW, Korenchuk S, Bai Y, Barbash O, Le B, Craggs PD, McCabe MT, Kennedy-Wilson KM, Sanchez LV, Gosmini RL, Parr N, McHugh CF, Dhanak D, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PloS one. 2013; 8:e72967.
45. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Seminars in cancer biology. 2006; 16:253-264.
46. Calcagno DQ, Leal MF, Assumpcao PP, Smith MA, Burbano RR. MYC and gastric adenocarcinoma carcinogenesis. World journal of gastroenterology : WJG. 2008; 14:5962-5968.
47. Calcagno DQ, Freitas VM, Leal MF, de Souza CR, Demachki S, Montenegro R, Assumpcao PP, Khayat AS, Smith Mde A, dos Santos AK, Burbano RR. MYC, FBXW7 and TP53 copy number variation and expression in gastric cancer. BMC gastroenterology. 2013; 13:141.
48. Leal MF, Martins do Nascimento JL, da Silva CE, Vita Lamarao MF, Calcagno DQ, Khayat AS, Assumpcao PP, Cabral IR, de Arruda Cardoso Smith M, Burbano RR. Establishment and conventional cytogenetic characterization of three gastric cancer cell lines. Cancer genetics and cytogenetics. 2009; 195:85-91.
49. Calcagno DQ, Leal MF, Taken SS, Assumpcao PP, Demachki S, Smith Mde A, Burbano RR. Aneuploidy of chromosome 8 and C-MYC amplification in individuals from northern Brazil with gastric adenocarcinoma. Anticancer research. 2005; 25:4069-4074.
50. Calcagno DQ, Leal MF, Seabra AD, Khayat AS, Chen ES, Demachki S, Assumpcao PP, Faria MH, Rabenhorst SH, Ferreira MV, de Arruda Cardoso Smith M, Burbano RR. Interrelationship between chromosome 8 aneuploidy, C-MYC amplification and increased expression in individuals from northern Brazil with gastric adenocarcinoma. World journal of gastroenterology : WJG. 2006; 12:6207-6211.
51. Assumpcao PP, Ishak G, Chen ES, Takeno SS, Leal MF, Guimaraes AC, Calcagno DQ, Khayat AS, Demachki S, Smith Mde A, Burbano RR. Numerical aberrations of chromosome 8 detected by conventional cytogenetics and fluorescence in situ hybridization in individuals from northern Brazil with gastric adenocarcinoma. Cancer genetics and cytogenetics. 2006; 169:45-49.
52. Burbano RR, Assumpcao PP, Leal MF, Calcagno DQ, Guimaraes AC, Khayat AS, Takeno SS, Chen ES, De Arruda Cardoso Smith M. C-MYC locus amplification as metastasis predictor in intestinal-type gastric adenocarcinomas: CGH study in Brazil. Anticancer research. 2006; 26:2909-2914.
53. Costa Guimaraes A, Goncalves Quintana L, Ferreira Leal M, Satomi Takeno S, Pimentel Assumpcao P, Moura Lima E, Salim Khayat A, Suchi Chen E, de Arruda Cardoso Smith M, Rodriguez Burbano R. Aneuploidy of chromosome 8 detected by fluorescence in situ hybridisation in ACP01 cell line gastric adenocarcinoma. Clinical and experimental medicine. 2006; 6:129-133.
54. Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, Zhang S, Lee M, Wu J, Lim KH, Lei Z, Goh G, Lim QY, Tan AL, Sin Poh DY, Riahi S, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012; 61:673-684.
55. Calcagno DQ, Guimaraes AC, Leal MF, Seabra AD, Khayat AS, Pontes TB, Assumpcao PP, De Arruda Cardoso Smith M, Burbano RR. MYC insertions in diffuse-type gastric adenocarcinoma. Anticancer research. 2009; 29:2479-2483.
56. de Souza CR, Leal MF, Calcagno DQ, Costa Sozinho EK, Borges Bdo N, Montenegro RC, Dos Santos AK, Dos Santos SE, Ribeiro HF, Assumpcao PP, de Arruda Cardoso Smith M, Burbano RR. MYC deregulation in gastric cancer and its clinicopathological implications. PloS one. 2013; 8:e64420.
57. Picaud S, Da Costa D, Thanasopoulou A, Filippakopoulos P, Fish PV, Philpott M, Fedorov O, Brennan P, Bunnage ME, Owen DR. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer research. 2013; 73:3336-3346.
58. Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, McHugh CF, Vessella R, Creasy CL, Tummino PJ, Barbash O. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013; 4:2419-2429. doi: 10.18632/oncotarget.1572.
59. Leal MF, Calcagno DQ, Borges da Costa Jde F, Silva TC, Khayat AS, Chen ES, Assumpcao PP, de Arruda Cardoso Smith M, Burbano RR. MYC, TP53, and chromosome 17 copy-number alterations in multiple gastric cancer cell lines and in their parental primary tumors. Journal of biomedicine & biotechnology. 2011; 2011:631268.
60. Fowler T, Ghatak P, Price DH, Conaway R, Conaway J, Chiang CM, Bradner JE, Shilatifard A, Roy AL. Regulation of MYC expression and differential JQ1 sensitivity in cancer cells. PloS one. 2014; 9:e87003.
61. Baker EK, Taylor S, Gupte A, Sharp PP, Walia M, Walsh NC, Zannettino AC, Chalk AM, Burns CJ, Walkley CR. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Scientific reports. 2015; 5:10120.
62. Fedorov O, Niesen FH, Knapp S. Kinase inhibitor selectivity profiling using differential scanning fluorimetry. Methods in molecular biology. 2012; 795:109-118.
63. Kabsch W. Xds. Acta crystallographica Section D, Biological crystallography. 2010; 66:125-132.
64. Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta crystallographica Section D, Biological crystallography. 2011; 67:282-292.
65. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of applied crystallography. 2007; 40:658-674.
66. Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature protocols. 2008; 3:1171-1179.
67. Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004; 60:2126-2132.
68. Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography. 1997; 53:240-255.
69. Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, Bricogne G. (2011). Grade v1.102. In: Cambridge UK, Global Phasing Ltd., ed.
70. Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological crystallography. 2010; 66:12-21.
71. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214-231.