
INTRODUCTION
Signal transducer and activator of transcription 3 (STAT3) is a critical signaling intermediate for many important cellular and biological processes in carcinogenesis, including inflammation, cellular transformation, proliferation, survival, invasion, angiogenesis, and metastasis [1–3]. Aberrant STAT3 signaling has been implicated in carcinogenesis: STAT3 is constitutively activated in cells transformed by diverse oncoproteins or tumor viruses, and elevated STAT3 signaling prevents apoptosis and confers survival advantages like chemo-resistance or radio-resistance [4]. STAT3 can be activated by non-receptor-associated kinases (eg, Src) or receptor-associated kinases (eg, JAK) via phosphorylation of tyrosine 705 (pYSTAT3). STAT3 tyrosine 705 phosphorylation can occur by stimulation of growth factor receptors with tyrosine kinase activity (eg, epidermal growth factor receptor [EGFR], platelet-derived growth factor receptor [PDGFR], hepatocyte growth factor receptor [HGFR or cMET], as well as cytokine receptors lacking intrinsic tyrosine kinase activity (eg, gp130 cytokine family) [5–9]. Activated STAT3 forms homo- or heterodimers with STAT1 through reciprocal Src Homology 2 (SH2)-pTyr interactions. The two known splice forms of STAT3 α/β differ in their transactivation domain and DNA binding affinity, which impacts also pYSTAT3 stability. Tyrosine phosphorylation of STAT3 results in translocation of STAT3 to the nucleus, where it regulates expression of target genes harboring STAT3 binding sites in their transcriptional regulatory region [10].
STAT3 can also be phosphorylated on a critical serine residue at position S727, used by the H-Ras oncoprotein and activated for example by the MEK-ERK pathway, essential for mitochondrial STAT3 traffic and function [11]. Mitochondrial serine-phosphorylated STAT3 is essential for RAS transformation via control of oxidative phosphorylation, which has an impact on overall reactive oxygen species (ROS) production and energy supply. ROS production is involved in cancer stem cell renewal, differentiation of epithelial cells, DNA double strand breaks and repair processes, lipid or protein oxidation, and inactivation of the catalytic center of tyrosine phosphatases prolonging the action of the JAK-STAT pathway [12, 13].
Moreover, STAT3 expression has been shown to correlate with PDGF-B expression, a well-described initiator of brain cancer. STAT3 activation alone was insufficient to induce brain cancer formation, but co-expression of STAT3 with PDGF-B in a transgenic mouse model resulted in efficient glioma multiforme formation [14]. PDGF promotes cell migration, proliferation and survival by binding to its cognate tyrosine kinase receptor PDGFR, which consists of α and β chains [15]. The homodimer PDGF-BB is the only PDGF that can bind both homo- and heterodimers of PDGFR with high affinity [16]. Also, Src kinase activation has been reported to contribute to PDGF-dependent cell-cycle progression, mitogenesis, and chemotaxis through its association with PDGFR-β in vitro [17, 18].
Our lab originally identified AF1q as an MLL fusion partner in acute myeloid leukemia patients with a t(1; 11)(q21; q23) translocation. We demonstrated that AF1q expression is associated with poor clinical outcomes in myeloid malignancies, and a number of studies have shown that AF1q plays a role in lung and breast cancer metastasis [19–23]. However, other reports indicated that AF1q could also influence pro-apoptotic effects mediated by BAD or fenretinide-induced ROS production [24, 25]. The consensus today is that AF1q plays an important role in malignancy of solid tumors, but the molecular mechanisms by which AF1q interacts with oncoproteins or influences tumor suppressor gene loss are incompletely understood. We previously demonstrated that AF1q physically interacts with the HMG box protein TCF7, a key factor in Wnt signaling, and AF1q enhances expression of Wnt target genes [26]. The AF1q-TCF7 interaction results in enhanced expression of CD44, a ubiquitous multi-structural and multi-functional cell surface glycoprotein involved in adhesion, migration, and homing of cells. We also demonstrated that elevated AF1q expression is significantly associated with breast cancer tumorigenesis and metastasis using patient-derived analysis and in vivo xenograft mouse models, combined with paired breast cancer cell line studies with enforced or suppressed AF1q.
In our earlier studies, we observed that STAT3 is activated when AF1q is expressed in breast cancer cells. We questioned whether the STAT3 pathway is influenced by AF1q expression, because in colorectal cancer progression it was convincingly shown that both STAT3 and Wnt signaling are needed for full malignancy and cancer progression [27]. To date, the mechanism of STAT3 activation by AF1q has not been studied. Here, we investigate how AF1q induces the activation of STAT3, and whether AF1q-induced STAT3 activation involves Srckinase and PDGFR signaling.
RESULTS
Enhancing AF1q expression activates STAT3 and promotes tumor growth and metastasis in immunodeficient Nod/Scid/Gamma chain null (NSG) mice
We recently demonstrated the contribution of AF1q to breast carcinoma formation in vivo using xenograft models of paired human breast cancer cell lines with enforced or suppressed AF1q expression injected into immunodeficient NSG mice [26]. We used the same system in this study to determine the mechanistic impact of AF1q expression on breast cancer progression. MDA-MB-231LN breast cancer cells stably expressing AF1q (MDA-MB-231LN/AF1q) or empty vector-transduced cells (MDA-MB-231LN/Ctrl) were injected into mammary fat pads of NSG mice. For the loss assay, MDA-MB-231LN cells stably expressing shRNA against AF1q (MDA-MB-231LN/shAF1q) and scramble-transduced cells (MDA-MB-231LN/shCtrl) were used. MDA-MB-231LN/AF1q-injected mice had significantly more aggressive tumor growth in mammary fat pads and metastasis compared with MDA-MB-231LN/Ctrl-injected mice, while MDA-MB-231LN/shAF1q-injected mice had attenuated tumor growth and metastasis compared with MDA-MB-231LN/shCtrl-injected mice (Figure 1A, 1B and Supplementary Figure S1), in line with our published results [26]. Previously we showed that AF1q plays a critical role in breast cancer initiation, progression, and metastasis through activation of the Wnt signaling pathway [26]. Entangled crosstalk between Wnt and other signaling pathways (i.e. Notch and STAT3) triggered other signaling pathways involved in cancer initiation, progression, and metastasis [28, 29]. Thus, alterations in Wnt signaling can lead to a variety of disorders, including cancer. Among signaling pathways which crosstalk with the Wnt signaling pathway, STAT3 activation has been particularly linked with breast cancer progression [30–34]. To evaluate STAT3 activation, tissues from our xenograft model were isolated, sectioned, and stained with anti-pYSTAT3 4 weeks post-injection. Lung metastasis nodules from MDA-MB-231LN/AF1q-injected mice had very strongly activated pYSTAT3, whereas normal lung and lung metastasis nodules from MDA-MB-231LN/shAF1q-injected mice did not (Figure 1C). Collectively, enforced AF1q expression promoted increased tumor growth and metastasis associated with elevated pYSTAT3 induction.
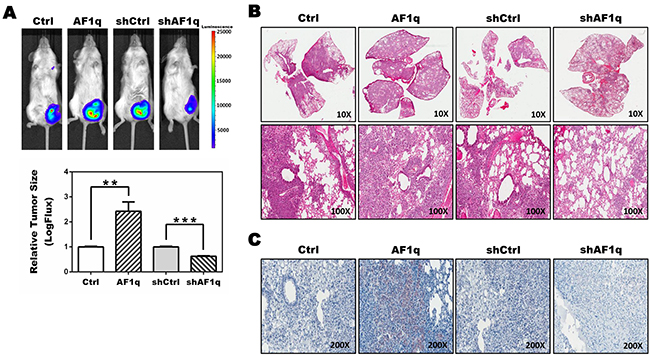
Figure 1: NSG xenograft mouse models demonstrate an association between AF1q expression and STAT3 activation. A. Representative images of NSG mice (n=5) 4 weeks after injection with MDA-MB-231LN/Ctrl, /AF1q, /shCtrl or /shAF1q cells in mammary fat pads (left panel). Quantification was carried out by measuring tumor size (right panel). Tumor size is shown as mean ± SD. P values were calculated using a student t test (*, P<0.05; **, P<0.01; ***, P<0.001). B. The lungs were harvested from NSG xenograft mouse models at 6 weeks after injection. MDA-MB-231LN/AF1q injected mice grew significantly larger metastasis tumor masses compared to /Ctrl mice. Suppressed the endogenous AF1q expression with shRNA in MDA-MB-231LN were significantly smaller tumors. C. Formalin-fixed sections of lung metastasis nodules from NSG xenograft mouse models were evaluated by immunohistochemistry for pYSTAT3 expression.
AF1q regulates PDGF-B expression through activation of its transcriptional pathway
To investigate how AF1q expression alters STAT3 activation, we examined the expression of cytokines, growth factors, and chemokines in MDA-MB-231LN/Ctrl, /AF1q, /shCtrl, and /shAF1q cells using a human cytokine array. We found that PDGF-B was differentially regulated in response to AF1q expression (Figure 2A). To confirm whether PDGF-B mRNA and protein expression were elevated, we used mRNA qPCR and ELISA, respectively, in each group of MDA-MB-231LN cells and cultured medium. We determined that the concentration of PDGF-B increased about three-fold when AF1q expression was enforced and decreased about two-fold when AF1q expression was suppressed (Figure 2B-2C). To investigate the mechanism underlying enhanced PDGF-B expression, we evaluated the activity of the PDGF-B promoter, determined as the ratio of the activity of firefly luciferase to the internal control Renilla luciferase. Enforced AF1q expression activated the PDGF-B promoter, while suppressed AF1q expression attenuated the activity of the PDGF-B promoter (Figure 2D).
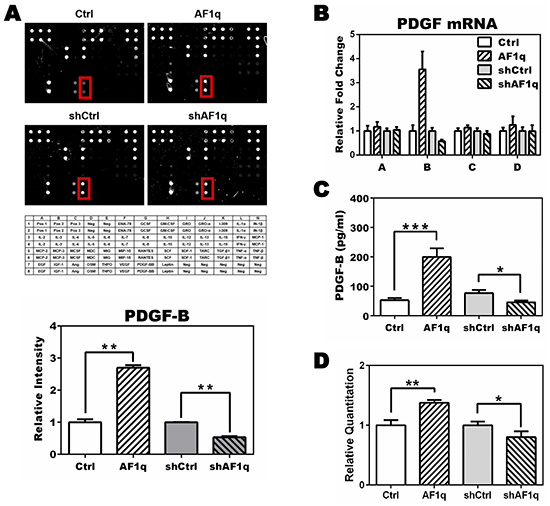
Figure 2: PDGF-B expression is altered by AF1q expression. A. The human cytokine array G3 (RayBiotech) was used to detect 42 human cytokines in lysate from MDA-MB-231LN/Ctrl, /AF1q, /shCtrl, and /shAF1q cells. PDGF-B expression, indicated by the red rectangle in each panel, is quantified in the graph below. Mean expression in /Ctrl cells and /shCtrl cells was normalized to 1. The intensity of /AF1q was compared with /Ctrl and the intensity of /shAF1q was compared with /shCtrl, because those paired groups used different backbone viral vectors, pLUTdNB and pTRIPZ, respectively. Bars indicate the SD. The table shows each target protein on the array slides. B. mRNA expression of the four members of the PDGF family (A, B, C, D) was determined by qPCR. Enforced AF1q increased the expression of PDGF-B mRNA (~3.5 fold) and suppressed AF1q expression using shRNA attenuated PDGF-B mRNA expression by 2 fold. Only PDGF-B was regulated by AF1q. C. The culture medium was harvested 24 h after doxycycline treatment to induce AF1q or shAF1q expression, and subjected to quantitative ELISA to assess PDGF-B protein concentration. D. Luciferase activity in MDA-MB-231LN cells transfected with reporter constructs of the proximal promoter of PDGF-B. Firefly luciferase was normalized to Renillar luciferase activity. Values are means ± SD of three independent experiments. P values were calculated using a student t test (*, P<0.05; **, P<0.01; ***, P<0.001).
AF1q-induced PDGF-B stimulates the migration and invasion of breast cancer cells
To investigate whether AF1q-induced PDGF-B altered the behavior of MDA-MB-231LN cells, MDA-MB-231LN cells were incubated with conditioned medium (CM) from MDA-MB-231LN/Ctrl, /AF1q, /shCtrl, and /shAF1q cells, and levels of cell migration and invasion were examined using wound healing, transwell migration, and matrigel invasion assays, respectively. As shown in Figure 3A-3B, CM from MDA-MB-231LN/AF1q cells stimulated motility, migration and invasion of MDA-MB-231LN cells compared with controls, whereas CM from MDA-MB-231LN/shAF1q cells decreased migration and invasion of MDA-MB-231LN cells.
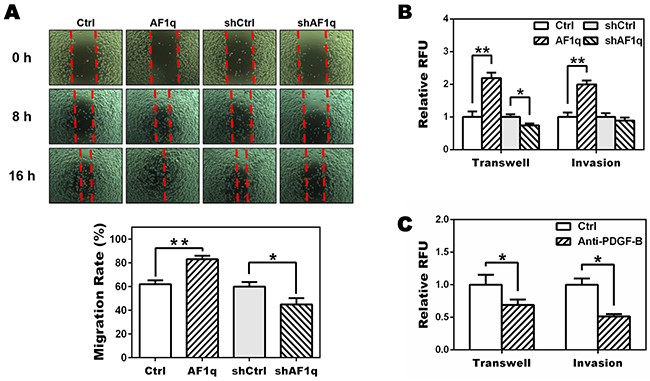
Figure 3: Targeting PDGF-B inhibits migration of MDA-MB-231LN cells. A. A wound healing assay was performed to determine the effect of AF1q-induced PDGF-B on motility of MDA-MB-231LN cells. Cell monolayers were wounded with a micropipette tip, washed twice with culture medium and cultured with conditioned medium (CM) from MDA-MB-231LN/Ctrl, /AF1q, /shCtrl, and /shAF1q cells, respectively, and then photographed at 0, 8 and 16 h. Quantification was carried out by measuring distance between the borders (bottom panel). B. The CM from MDA-MB-231LN/AF1q cells promoted transwell migration and matrigel invasion compared with CM from their corresponding controls (Ctrl), whereas CM from MDA-MB-231LN/shAF1q cells decreased migration and invasion of MDA-MB-231LN cells. C. MDA-MB-231LN cells were incubated with CM from MDA-MB-231LN/AF1q cells in the presence of a neutralizing PDGF-B antibody (5 μg/ml) or isotype antibody (5 μg/ml). Cell migration and invasion were attenuated by the PDGF-B antibody. P values were calculated using a student t test (*, P<0.05; **, P<0.01; ***, P<0.001).
To verify these findings, we used an antibody to neutralize AF1q-induced PDGF-B in CM from MDA-MB-231LN/AF1q cells and performed the transwell migration and matrigel invasion assay. As shown in Figure 3C, when AF1q-induced PDGF-B was neutralized, migration and invasion of MDA-MB-231LN cells was reduced. Taken together, these results show that AF1q expression in MDA-MB-231LN cells has paracrine activity through PDGF-B to promote motility, migration and invasion of MDA-MB-231LN cells.
AF1q activates STAT3 through PDGF-B/PDGFR tyrosine kinase signaling
To investigate further the mechanism underlying AF1q-induced PDGF-B, we evaluated the PDGF/PDGFR signaling pathway in MDA-MB-231LN cells using Western blot. Up-regulating PDGF-B through an AF1q-dependent transcriptional mechanism induced tyrosine phosphorylation of PDGFR in MDA-MB-231LN cells. Conversely, suppressing AF1q expression by shAF1q effectively down-regulated activation of the PDGF/PDGFR signaling pathway. These results indicate that PDGF-B/PDGFR signaling was up-regulated by AF1q (Figure 4A). To investigate the mechanism responsible for promoting migration and invasion, we examined whether signaling downstream from PDGF-B/PDFGR was altered in two carcinoma cell lines, MDA-MB-231LN breast cancer cells and SW620 colon cancer cells, transformed to enforce or suppress AF1q. It has been shown that constitutive STAT3 activity in both breast and colon cancer contributes to enhanced cell proliferation and metastasis [35, 36]. As shown in Figure 4B, AF1q-induced PDGF-B/PDGFR signaling effectively induced Src kinase activation, as measured with Y416 phospho-specific Src antibody Western. Then, we evaluated STAT3 expression and activation status. Interestingly, pYSTAT3 was strongly induced and activated in cells with enforced AF1q expression. Because MDA-MB-231LN breast cancer cell lines are known to possess constitutively activated Src kinases, we examined STAT3 phosphorylation levels in MCF10a normal human breast epithelial cells with enforced or suppressed AF1q expression. As shown in Figure 4C, pYSTAT3 and pSrc were effectively increased in MCF10a cells with enforced AF1q expression, but attenuated when AF1q expression was suppressed.
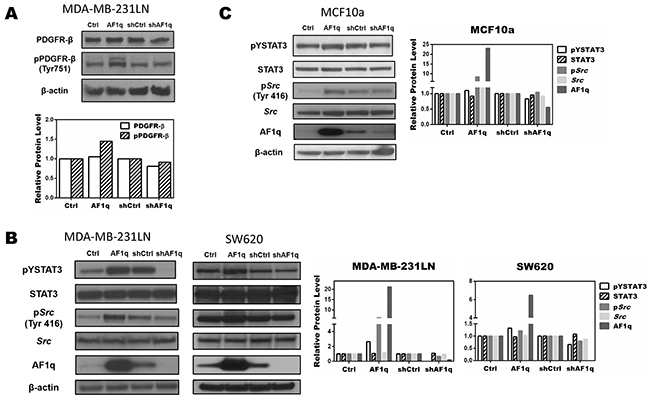
Figure 4: AF1q controls STAT3 activation by modulating Src phosphorylation through the PDGF-B/PDGFR signaling cascade. A. AF1q induced PDGFR-β phosphorylation in MDA-MB-231LN cells, as determined by Western blot. B. Constitutive expression and phosphorylation of Src and STAT3 protein in MDA-MB-231LN and SW620 cells transformed to enforce or suppress AF1q. C. Constitutive phosphorylation and expression levels of Src and STAT3 protein in normal breast epithelial MCF10a cells after transformation to enforce or suppress AF1q.Band density was determined by ImageJ analysis software.
STAT3 activation by AF1q requires the phosphorylation of Src kinase
Src has been shown to directly increase pYSTAT3 levels [37, 38]. To evaluate whether Src was responsible for AF1q-induced STAT3 activation, we treated MDA-MB-231LN/AF1q and SW620/AF1q cells with 0.1 μM of the selective Src inhibitor PP1 for 24 h [39]. The phosphorylation of Src decreased significantly, and STAT3 phosphorylation also decreased (Figure 5A). Conversely, suppressing AF1q expression by shAF1q effectively down-regulated activation of the PDGF/PDGFR signaling pathway. These results indicate that AF1q-induced PDGF-B/PDGFR signaling triggered the activation of STAT3 by Src kinase.
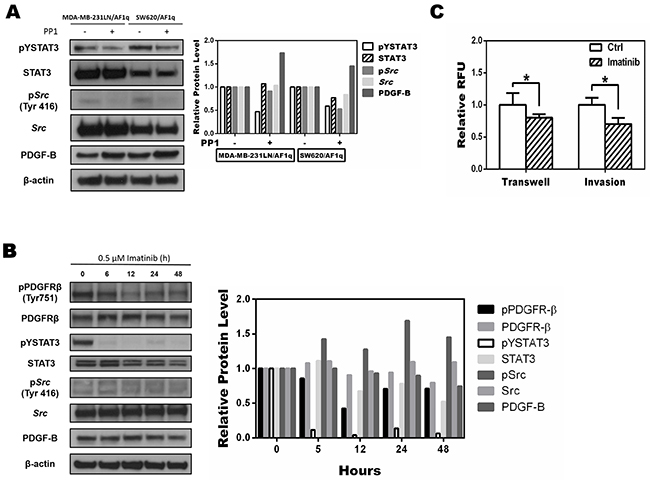
Figure 5: Targeting the phosphorylation of Src inhibits AF1q-induced STAT3 activation A. Src phosphorylation of MDA-MB-231LN/AF1q and SW620/AF1q cells was inhibited by PP1, a selective Src inhibitor, and pYSTAT3 expression also decreased. B. MDA-MB-231LN/AF1q cells were treated with imatinib (0.5 μM) and expression of PDGFR-β, STAT3, Src, and PDGF-B were assessed by Western blot 0, 6, 12, 24, and 48 h later. Band density was determined by ImageJ analysis software. C. Quantification of transwell migration and invasion of MDA-MB-231LN/AF1q after imatinib treatment for 24h. P values were calculated using a student t test (*, P<0.05; **, P<0.01; ***, P<0.001).
Imatinib, a FDA-approved tyrosine kinase inhibitor specific for oncogenic fusions or mutations of PDGFRs, c-KIT, and ABL tyrosine kinase oncogenes, has been an effective or even curative treatment for certain leukemias [40]. We applied imatinib (0.5 μM) to MDA-MB-231LN/AF1q cells and evaluated the activity status of key proteins associated with PDGF/PDGFR signaling using Western blot. Phosphorylation of pYPDGFR-β, pYSTAT3, and pYSrc in MDA-MB-231LN/AF1q cells was inhibited by imatinib, but PDGF-B expression was not changed (Figure 5B). Notably, imatinib inhibited the migration and invasion of MDA-MB-231LN/AF1q cells (Figure 5C). Our results indicate that imatinib exerts a significant inhibitory effect on breast cancer cell invasion and migration through inhibition of PDGFR and STAT3 phosphorylation, although the expression of PDGF-B was unchanged. This result is in line with data we observed with the anti-PDGF-B inhibitory antibody (Figure 3C). Collectively, our data suggest that the AF1q-induced PDGF-B/PDGFR signaling cascade is directly associated with altered cell invasion and migration.
AF1q-induced STAT3 activation via Src-PDGFR tyrosine kinase signaling increases binding activity
To verify whether an increase in AF1q-induced STAT3 phosphorylation was accompanied by an increase in STAT3 binding activity, we performed electrophoretic mobility shift assays (EMSA) using a hSIE oligonucleotide probe. The oligonucleotide Alexa 488-labeled probe was incubated with nuclear protein from MDA-MB-231LN cells. The nuclear protein formed a protein-DNA complex with the hSIE oligonucleotide, but no protein-DNA complex formed with the competitor hSIE oligonucleotide or when nuclear protein extract was omitted. In addition, band intensity reflects the level of DNA binding activity in response to STAT3 phosphorylation. As shown in Figure 6A, we found that enforced AF1q expression in MDA-MB-231LN cells enhanced the binding affinity of STAT3 for the hSIE oligonucleotide probe. Conversely, the binding affinity of STAT3 was decreased when AF1q expression was suppressed in MDA-MB-231LN cells. Moreover, a specific C-terminal STAT3 antibody caused gel retardation of the hSIE-containing complexes in a supershift assay, but a STAT1 antibody did not cause severe gel retardation (Figure 6B). Taken together, the PDGF-B/PDGFR signaling cascade was activated upon enforced AF1q expression and this caused an increase in STAT3 DNA binding activity through Src kinase action in MDA-MB-231LN cells.
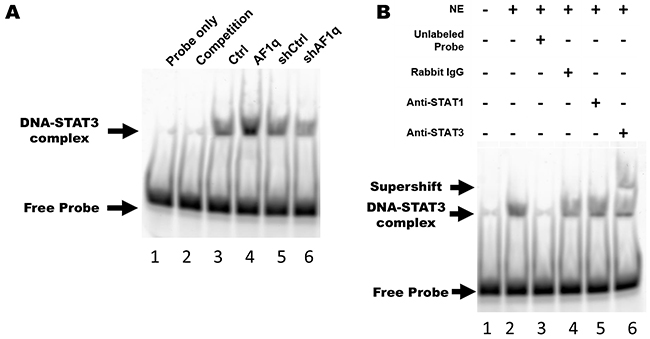
Figure 6: AF1q-induced PDGF-B enhances STAT3 binding activity through the PDGFR signaling cascade. A. Nuclear protein was isolated from MDA-MB-231LN/Ctrl, /AF1q, /shCtrl, and /shAF1q cells, and EMSA was performed using Alexa 488-labeled STAT3 hSIE probes. Lane 1, free labeled probes without nuclear extract (negative control); lane 2, labeled probes and 10 fold molar excess unlabeled probes (competitor) with nuclear extract of MDA-MB-231/Ctrl; lane 3-6, labeled probe with nuclear extract from each cell type. The upper arrow points to the position of the STAT3-hSIE probe complex. The intensity of this band is proportional to the amount of STAT3 translocated to the nucleus and bound to DNA. B. Supershift assay using MDA-MB-231LN/AF1q nuclear extracts revealed that STAT3 bound to probe, not STAT1. Lane 1, free labeled probes without nuclear extract (NE); lane 2, labeled probes with NE; lane 3, labeled probes and 10 fold molar excess unlabeled probes (competitor) with nuclear extract; lane 4, labeled probes and isotype rabbit IgG antibody with nuclear extract; lane 5, labeled probes and anti-STAT1 antibody with nuclear extract; lane 6, labeled probes and anti-STAT3 antibody with nuclear extract.
DISCUSSION
STAT3 is constitutively activated in many human cancers with poor prognosis or chemo- or radio-resistance, which have been molecularly linked through cancer genome landscape studies to increased tyrosine kinase signaling in particular [41–43]. Aberrant STAT3 signaling is an important process in malignant progression. Hence, greater understanding of the regulatory mechanism of STAT3 is important to improve treatment. We found that AF1q activates STAT3 in human breast cancer cells by activating Src kinase through activation of the PDGF-B/PDGFR cascade. The transcriptionally amplified PDGF-B regulated by AF1q accelerated activation of the PDGFR-Src kinase cascade, driving persistent STAT3 activation (pYSTAT3) in breast cancer cells (Figure 7). A number of reports have suggested that phosphorylation of serine 727 (pSSTAT3) promotes breast cancer growth by localizing to mitochondria [44]. We tested whether pSSTAT3 is also activated by AF1q using western blot. We found that pSSTAT3 was not changed by expression of AF1q (Supplementary Figure S2).
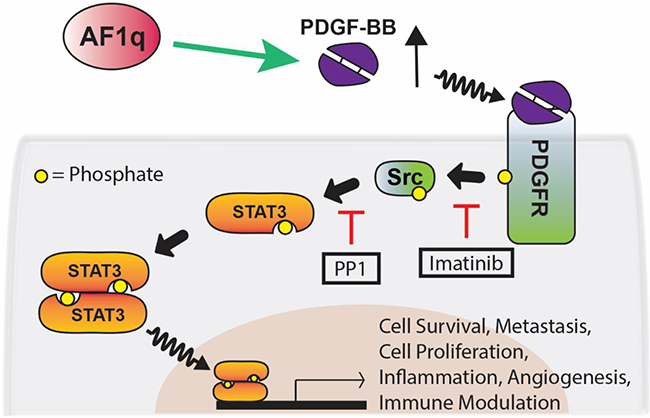
Figure 7: Proposed network for AF1q-induced PDGF-B/PFGFR/Src/STAT3 signaling. AF1q enhances the expression of PDGF-B, which forms homodimers (PDGF-BB) that bind to the protein tyrosine kinase receptor PDGFR. Binding of PDGF-BB to PDGFR results in tyrosine phosphorylation of PDGFR. Src is recruited to phosphorated PDGFR and activated by phosphorylation of tyrosine 416. Activation of STAT3 occurs via phosphorylation of tyrosine 705 through the Src tyrosine kinase. Using selective inhibitors of tyrosine kinases (PP1 and imatinib), we showed that PDGFR and Src cooperate to mediate STAT3 activation in breast cancer cell lines. This signaling pathway affects multiple cellular processes including cell survival, metastasis, cell proliferation, inflammation, angiogenesis, and immune modulation. Green arrows and red lines indicate activation and suppression, respectively.
We observed that MDA-MB-231LN/AF1q breast cancer cells show about 2.5-fold more aggressive tumor growth (Figure 1A) and approximately 4 times the number of lung metastatic colonies (data not shown) than control cells in NSG mice. Also, we observed that pYSTAT3 levels were significantly elevated in lung metastatic colonies (Figure 1B). We also provided mechanistic insight into the effects of AF1q-induced STAT3 activation at the cellular level. To explain these observations, we investigated kinase signaling upstream of the STAT3 signaling pathway and proved that AF1q activated the 1,095 bp promoter region of PDGF-B, a growth factor ligand that binds to PDGFR to enhance autoregulation and stimulation of PDGFR signaling. In the PDGF family, only PDGF-B was associated with AF1q. To identify the mechanism of enhanced PDGF-B expression induced by AF1q, we performed aPDGF-B promoter assay using the luciferase reporter system. It is clear that AF1q boosts PDGF-B expression through activation of the PDGF-B promoter region. We next examined whether PDGF-B regulates cell migration. PDGF and PDGFR may participate in cellular migration and PDGF-B is the strongest inducer in that process [45–47]. As expected, PDGF-B regulated cell migration and we confirmed this finding by an inhibition assay. Our studies showed that imatinib inhibited migration of MDA-MB-231LN cells (Figure 5C). One interpretation of these results is that disruption of PDGFR-β/PDGF-B signaling may be important to block activation of STAT3 signaling, due to the effects of imatinib on inhibition of PDGFR phosphorylation in breast cancer (Figure 5B) [48].
We also investigated whether PDGF-B promoted phosphorylation of PDGFR-α and PDGFR-β by Western blot. However, we detected a PDGF-B-dependent increase in the phosphorylation of PDGFR-β only. Repeated attempts failed to detect significant phosphorylation of PDGFR-α, which could have been due to problems with the antibodies we used or to exceptionally low PDGFR- α levels.
Because activation of STAT3 occurs in cancers with activated Src kinase, we examined regulation of Src kinase in MDA-MB-231 cells overexpressing or suppressing AF1q. We observed a cooperative activation of Src kinase with AF1q expression in MDA-MB-231LN cells, which correlates with cooperative oncogenic signaling by kinases in breast cancer. Furthermore, we observed that the activation of STAT3 was inhibited by PP1, a selective Src kinase inhibitor, in MDA-MB-231LN cells overexpressing AF1q (Figure 5A). These results demonstrate a fundamental mechanistic regulation of STAT3 by AF1q. Using EMSA analysis, we also determined that constitutive activation of STAT3 DNA-binding activity was enhanced by AF1q expression (Figure 6).
We earlier showed that the coactivator peptide AF1q activates Wnt signaling via direct interaction with TCF7, forming a transcriptional complex in conjunction with LEF1 and β-catenin [26]. Other groups have shown that the gp130-Jak-STAT3 signaling pathway promotes intestinal tumor growth and regeneration [27], as does Wnt signaling. Interestingly, the tumor growth and regeneration mediated by the Wnt signaling pathway can be prevented by inhibition of gp130-Jak-STAT3 signaling [27]. However, it was not clear if these two pathways share any common components, or how they interact. Here, we showed that inhibition of AF1q-induced PDGF-B limits the migration response of breast carcinoma cells. Thus, AF1q is a more general cofactor for both Wnt and STAT signaling, with the latter activated through the Src-PDGFR kinase cascade. In both cases, binding of AF1q to transcription factors results in higher transcriptional rates of genes required for tumorigenesis or metastasis. Thus, AF1q is an acidic cofactor peptide that boosts the transactivation potential of proto-oncogene transcription factors associated with invasive properties of cancer cells.
We earlier demonstrated that AF1q-positive cancer cells have significantly greater prevalence at metastatic sites than in primary breast tumors [26]. Together with our current results, these observations suggest that AF1q-positive breast cancer cells might contain persistently high levels of pYSTAT3 due to high levels of PDGF-B, which translates into a migration advantage. To investigate that possibility, we plan to evaluate AF1q, pYSTAT3, and PDGF-B expression in paired human samples of primary breast tumors and matched metastatic cancers.
In summary, our results suggest that inhibition of AF1q-induced PDGF-B might hold therapeutic promise for metastatic breast cancer. Our studies here have addressed for the first time the impact of AF1q on PDGF-B-driven STAT3 signaling.
MATERIALS AND METHODS
Cell culture
MCF10a and SW620 cells were purchased from the American Type Culture Collection (ATCC). MDA-MB-231-luc-D3H2LN (MDA-MB-231LN) cells were purchased from Caliper Life Science. MCF10a cells were maintained in DMEM/F-12 (1:1) medium (Invitrogen) supplemented with 5% horse serum (Gibco), 0.5 μg/ml hydrocortisone, 10 μg/ml insulin, 20 ng/ml epidermal growth factor, and 0.1 μg/ml cholera endotoxin (Sigma-Aldrich). MDA-MB-231LN and SW620 cells were cultured in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Gibco). They were cultured at 37°C in a 5% CO2 humidified atmosphere.
Viral production and infection
Replication of incomplete lentiviral particles was performed by co-transfecting three plasmids into HEK293T cells (ATCC) with the lentiviral constructs pVSV-G and psPAX2 (Addgene) using Lipofectamine 2000 (Invitrogen). Lentivirus in the supernatant was harvested 48, 72, and 96 h after transfection and concentrated by 10% PEG-8000 (Sigma-Aldrich). Cells were transduced with concentrated lentivirus using 8 μg/ml Polybrene (Santa Cruz). Cells which underwent lentiviral transduction were selected using 1 μg/ml puromycin (Thermo Fisher) for 1 week as previously described [49]. After antibiotic selection, cells were cultured in complete medium including 1 μg/ml doxycycline (Thermo Fisher) for AF1q or shRNA induction. The control lentiviral vector for pLUTdNB-AF1q, designated Ctrl, was pLUTdNB empty vector, while pTRIPZ-Scramble, designated shCtrl, stably expressed scramble (GE Dharmacon). In this study, all cells were divided into four groups, designated MDA-MB-231LN/Ctrl, /AF1q, /shCtrl or /shAF1q.
Measurement of cytokine, chemokine, and growth factor production
Expression of cytokines, chemokines, and growth factors by MDA-MB-231LN cells was evaluated using the human cytokine array G3 (RayBiotech). 100 μl of cell culture medium was incubated with the array antibody slide, following the manufacturer’s instructions. Signals were detected using Streptavidin and scanned to measure spot intensity. We compared the spot intensity of two pairs of cells: (1) MDA-MB-231LN/Ctrl and /AF1q and (2) /shCtrl and /shAF1q. The relative amount of each cytokine or growth factor is presented as fold change of spot intensity.
Western blots
Growing medium of the cells was exchanged with 0.5% FBS Opti-MEM (Invitrogen) including doxycycline (1 μg/ml) 2 h prior to lysis. To inhibit Src kinase activity, cells were treated with 0.1 μM PP1 (Sigma-Aldrich) for 24 h prior to lysis. Cells were lysed directly in M-PER lysis buffer with the Halt protease inhibitor cocktail and the Halt phosphatase inhibitor cocktail (Pierce) according to the manufacturer’s instructions. Cell lysates were separated by electrophoresis on a 4–12% SDS-PAGE gradient gel and blotted on to PVDF membranes. Anti-PDGFR-β, pPDGFR-β, Src, STAT3, pYSTAT3 and β-actin monoclonal antibodies were purchased from Cell Signaling Technology and anti-AF1q, pSrc (pTyr426) and PDGF-B monoclonal antibodies were purchased from Abcam. After incubation with the appropriate antibody, an enhanced chemiluminescence system (Denville) was used for developing blots. Western blot band quantifications were performed with ImageJ analysis software (NIH).
RNA extraction and qPCR analysis
Total RNA extracted with the mirVana miRNA isolation kit (Invitrogen) was reverse transcribed using high-capacity cDNA reverse transcription kits (Applied Biosystems) according to the manufacturer’s instructions. PCR amplification of cDNA encoding PDGF-B was carried out on an ABI 7500 System using TaqMan universal PCR master mix (Applied Biosystems). The following primers were used to amplify PDGF-BB: sense: 5′- TCTCTGCTGCTACCTGCGT-3′, antisense: 5′- CAAAGGAGCGGATCGAGTGG-3′ and HPRT1 (internal control); sense: 5′- CAGAGGG CTACAATGTGATGGC-3′, antisense: 5′- GCTGAGGA TTTGGAAAGGGTG-3′. All samples were run in triplicate and Ct values of internal control and PDGF-B were determined. The relative expression for the target gene was given by 2−ΔΔCt.
PDGF-B ELISA
MDA-MB-231LN cells were plated into 6-well plates at a density of 5×105 cells per well in 0.5% FBS Opti-MEM for 24 h. Supernatants were harvested and PDGF-B levels were measured using a PDGF-B ELISA kit (R&D Systems) according to the manufacturer’s instructions.
PDGF-B promoter assay
The PDGF-B promoter fragment (-1,220/-25 bp from the transcriptional start site; Chr22:39619685-39636914) was PCR-amplified from the genomic DNA of the MDA-MB-231LN cell line using these primers: sense: 5′- CTCGAGAATCCATGGCACAGACCAGC – 3′, antisense: 5′ – AAGTTTTTGGCATCGTGCGTGACAAT – 3′. The resulting PCR product (-1,220/-25) was digested with XhoI and HindIII and ligated into pGL4 luciferase reporter vectors (Promega), resulting in the PDGF-B luciferase promoter construct. Dual-luciferase reporter assays were performed with 1×104 cells plated in each well of a 96-well plate. The next day, the reporter (100 ng) and Renilla plasmid (10 ng, Promega) were co-transfected into cells with lipofectamine 2000 (Invitrogen). After 48 h, cells were washed with phosphate-buffered saline (PBS) and assayed with the Dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Luciferase activity was determined using the Synergy H1 multi-mode reader (Biotek). Quantitation of the luminescent signal from the reporter plasmid was normalized by quantitation of the luminescent signal from Renilla.
Collection of conditioned medium
When cells reached 70-80% confluency, the medium was changed to 0.5% FBS Opti-MEM, treated with doxycycline (1 μg/ml) for 24 h, and then the culture medium was collected. The collected supernatant was centrifuged for 5 min at 20,000 g at 4°C and used in other experiments as conditioned medium (CM).
Wound healing and invasion assay
MDA-MB-231LN cells from each group were seeded in 6-well plates at a density of 1×106 cells per well. A 1-mm-wide linear scratch (the wound zone) was established across the center of each well with a micropipette tip and washed three times with medium. We used TScratch software (http://www.cse-lab.ethz.ch/index.php?&option=com_content&view=article&id=363) to evaluate migration rate. We photographed the well under the microscope at the desired time point, and analyzed the open image area using TScratch. The migration rate was calculated as the open image area at 0 h divided by the open image area at 16 h.
For the invasion assay, we used the Fluoroblock HTS insert system (BD Biosciences). 100 μl of matrigel (200 μg/ml) was overlaid into inserts and incubated for 2 h at 37°C. The lower side chambers were filled with 750 μL of culture medium. The cells (2.5×104 cells per insert) were seeded in the upper side chambers with the appropriate CM. For neutralization, anti-PDGF-BB (5 μg/ml, Cell Signaling) or isotype IgG (5 μg/ml, Sigma-Aldrich) was used. For inhibition of PDGFR, MDA-MB-231LN/AF1q cells were treated with imatinib (0.5 μM). Cells were allowed to migrate 24 h and non-migrated cells were removed with cotton swabs. Migrated cells were fixed with ice-cold methanol and stained with calcein AM (4 μg/ml, Invitrogen) for 1 h. Fluorescence intensity was measured with the Synergy H1 multi-mode reader. Experiments were repeated three times independently.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared using the NE-PER™ Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific) according to the manufacturer’s instructions. The reaction mixtures (10 μl) contained 5 μg of nuclear extract, 1x binding buffer, 50 ng of poly (dI-dC) and 20 ng of Alexa 488-labeled STAT3 hSIE binding region probe (5′–CATTTCCCGTAAATC-3′, IDT), incubated for 30 min at room temperature. Reaction mixtures containing no nuclear extract were also incubated with labeled probes as negative control samples. Each sample was loaded onto a 6% polyacrylamide gel in 0.5x Tris/Borate/EDTA (TBE) buffer. For the antibody supershift analysis, 1 μg of anti-STAT3 antibody (Cell Signaling) was added to nuclear extracts of MDA-MB-231LN/AF1q cells prior to incubation with the Alexa-488 labeled probe for 30 min. In the competition analysis, 100-fold molar excess of the unlabeled double-strand oligonucleotide was added to the reaction mixture prior to the addition of labeled probe. The reaction mixtures were resolved by electrophoresis using 6% DNA retardation gel (Invitrogen). Samples were electrophoresed until the dye had reached 1 inch from the bottom of the gel and then scanned with the PharosFX Plus (Bio-Rad).
Tumor growth assay
We performed xenograft transplants to measure tumorigenicity and metastasis in vivo, according to the protocol approved by the University of Louisville Institutional Animal Care and Use Committee. The tumor growth assay was performed as previously described [26]. Briefly, MDA-MB-231LN cells labeled with firefly luciferase were diluted with PBS to a concentration of 1×107 cells/ml and 100 μl of cell suspension (1×106 cells) were injected into abdominal mammary fat pads of 6-week old NSG female mice (5 mice/group). For imaging, mice were injected intraperitoneally with a Luciferin solution (150 mg/kg; 15 mg/mL in PBS). The Luciferin solution was allowed to distribute for about 5 minutes, and then an image was taken (1-5 min). Mice were imaged weekly for 4 weeks using the IVIS imaging system. Mice were euthanized and necropsied 4 weeks post-cell injection. Mice were fed 200 mg/kg of doxycycline daily to induce AF1q or shAF1q expression.
Immunohistochemistry
Tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin, and then 4-μm sections were prepared. Sections were de-waxed and steam pre-treated in Tris/EDTA buffer (DAKO). Endogenous peroxidase activity was quenched by incubation in 3% hydrogen peroxide in PBS. For blocking steps, Avidin (Sigma-Aldrich), Biotin (Sigma-Aldrich) in PBS, and a Super Block (ID Labs Biotechnology) were used. Rabbit monoclonal pYSTAT3 antibody in a 1:200 dilution was incubated at 4°C overnight. IHC detection was performed with the IDetect Super Stain System HRP (ID Labs Biotechnology). Specific signals were amplified using 3-amino-9-ethylcarbazole (ID Labs Biotechnology) under visual control, followed by counterstaining with hematoxylin.
ACKNOWLEDGMENTS
We thank Helen Kim for her editorial assistance and Dr. Donghoon Chung for experimental design and technical advice. This work was supported by the use of the facilities of the James Graham Brown Cancer Center, University of Louisville, Louisville, Kentucky.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was partly supported by the NIH R01DK059380 (KB). RM was supported by grants SFB-F2807 and SFB-F4707 from the Austrian Science Fund (FWF). JJ was supported by grants from the University of Louisville School of Medicine and the Elsa U. Pardee Foundation.
REFERENCES
1. Yu H, Pardoll D and Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nature reviews Cancer. 2009; 9:798-809.
2. Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R and Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene. 2004; 23:3550-3560.
3. Hirano T, Ishihara K and Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000; 19:2548-2556.
4. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS and Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999; 10:105-115.
5. Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY and Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer cell. 2005; 7:575-589.
6. Yu YC, Yang PM, Chuah QY, Huang YH, Peng CW, Lee YJ and Chiu SJ. Radiation-induced senescence in securin-deficient cancer cells promotes cell invasion involving the IL-6/STAT3 and PDGF-BB/PDGFR pathways. Scientific reports. 2013; 3:1675.
7. Liu PS, Jong TH, Maa MC and Leu TH. The interplay between Eps8 and IRSp53 contributes to Src-mediated transformation. Oncogene. 2010; 29:3977-3989.
8. Schreiner SJ, Schiavone AP and Smithgall TE. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. The Journal of biological chemistry. 2002; 277:45680-45687.
9. Levy DE and Darnell JE, Jr. Stats: transcriptional control and biological impact. Nature reviews Molecular cell biology. 2002; 3:651-662.
10. Zhang X, Yue P, Fletcher S, Zhao W, Gunning PT and Turkson J. A novel small-molecule disrupts Stat3 SH2 domain-phosphotyrosine interactions and Stat3-dependent tumor processes. Biochemical pharmacology. 2010; 79:1398-1409.
11. Gough DJ, Koetz L and Levy DE. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PloS one. 2013; 8:e83395.
12. Zhou L and Too H-P. Mitochondrial Localized STAT3 Is Involved in NGF Induced Neurite Outgrowth. PloS one. 2011; 6:e21680.
13. Boengler K, Ungefug E, Heusch G and Schulz R. The STAT3 inhibitor stattic impairs cardiomyocyte mitochondrial function through increased reactive oxygen species formation. Current pharmaceutical design. 2013; 19:6890-6895.
14. Doucette TA, Kong L-Y, Yang Y, Ferguson SD, Yang J, Wei J, Qiao W, Fuller GN, Bhat KP, Aldape K, Priebe W, Bögler O, Heimberger AB and Rao G. Signal transducer and activator of transcription 3 promotes angiogenesis and drives malignant progression in glioma. Neuro-Oncology. 2012; 14:1136-1145.
15. Ostman A and Heldin CH. Involvement of platelet-derived growth factor in disease: development of specific antagonists. Advances in cancer research. 2001; 80:1-38.
16. Cao Y, Cao R and Hedlund EM. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. Journal of molecular medicine (Berlin, Germany). 2008; 86:785-789.
17. DeMali KA, Godwin SL, Soltoff SP and Kazlauskas A. Multiple roles for Src in a PDGF-stimulated cell. Exp Cell Res. 1999; 253:271-279.
18. Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA and Jove R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A. 2001; 98:7319-7324.
19. Yang S, Dong Q, Yao M, Shi M, Ye J, Zhao L, Su J, Gu W, Xie W, Wang K, Du Y, Li Y and Huang Y. Establishment of an experimental human lung adenocarcinoma cell line SPC-A-1BM with high bone metastases potency by (99m)Tc-MDP bone scintigraphy. Nuclear medicine and biology. 2009; 36:313-321.
20. Li DQ, Hou YF, Wu J, Chen Y, Lu JS, Di GH, Ou ZL, Shen ZZ, Ding J and Shao ZM. Gene expression profile analysis of an isogenic tumour metastasis model reveals a functional role for oncogene AF1Q in breast cancer metastasis. European journal of cancer (Oxford, England : 1990). 2006; 42:3274-3286.
21. Chang XZ, Li DQ, Hou YF, Wu J, Lu JS, Di GH, Jin W, Ou ZL, Shen ZZ and Shao ZM. Identification of the functional role of AF1Q in the progression of breast cancer. Breast cancer research and treatment. 2008; 111:65-78.
22. Tse W, Zhu W, Chen HS and Cohen A. A novel gene, AF1q, fused to MLL in t(1;11) (q21;q23), is specifically expressed in leukemic and immature hematopoietic cells. Blood. 1995; 85:650-656.
23. Strunk CJ, Platzbecker U, Thiede C, Schaich M, Illmer T, Kang Z, Leahy P, Li C, Xie X, Laughlin MJ, Lazarus HM, Gerson SL, Bunting KD, Ehninger G and Tse W. Elevated AF1q expression is a poor prognostic marker for adult acute myeloid leukemia patients with normal cytogenetics. American journal of hematology. 2009; 84:308-309.
24. Co NN, Tsang WP, Wong TW, Cheung HH, Tsang TY, Kong SK and Kwok TT. Oncogene AF1q enhances doxorubicin-induced apoptosis through BAD-mediated mitochondrial apoptotic pathway. Molecular cancer therapeutics. 2008; 7:3160-3168.
25. Tiberio P, Cavadini E, Callari M, Daidone MG and Appierto V. AF1q: a novel mediator of basal and 4-HPR-induced apoptosis in ovarian cancer cells. PloS one. 2012; 7:e39968.
26. Park J, Schlederer M, Schreiber M, Ice R, Merkel O, Bilban M, Hofbauer S, Kim S, Addison J, Zou J, Ji C, Bunting ST, Wang Z, et al. AF1q is a novel TCF7 co-factor which activates CD44 and promotes breast cancer metastasis. Oncotarget. 2015. doi: 10.18632/oncotarget.4136.
27. Phesse TJ, Buchert M, Stuart E, Flanagan DJ, Faux M, Afshar-Sterle S, Walker F, Zhang HH, Nowell CJ, Jorissen R, Tan CW, Hirokawa Y, Eissmann MF, et al. Partial inhibition of gp130-Jak-Stat3 signaling prevents Wnt-beta-catenin-mediated intestinal tumor growth and regeneration. Science signaling. 2014; 7:ra92.
28. Morris S-AL and Huang S. Crosstalk of the Wnt/β-catenin pathway with other pathways in cancer cells. Genes & Diseases. 2016; 3:41-47.
29. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX and Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature reviews Clinical oncology. 2015; 12:445-464.
30. Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, Bornmann W and Bromberg JF. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast cancer research : BCR. 2007; 9:R32.
31. Bowman T, Garcia R, Turkson J and Jove R. STATs in oncogenesis. Oncogene. 2000; 19:2474-2488.
32. Buettner R, Mora LB and Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002; 8:945-954.
33. Guo S, Liu M and Gonzalez-Perez RR. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochimica et biophysica acta. 2011; 1815:197-213.
34. Chung SS, Aroh C and Vadgama JV. Constitutive Activation of STAT3 Signaling Regulates hTERT and Promotes Stem Cell-Like Traits in Human Breast Cancer Cells. PloS one. 2014; 8:e83971.
35. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, Baus D, Kaufmann R, Huberb LA, Zatloukal K, Beug H, Öhlschläger P, Schütz A, Halbhuber K-J and Friedrich K. Persistent STAT3 Activation in Colon Cancer Is Associated with Enhanced Cell Proliferation and Tumor Growth. Neoplasia (New York, NY). 2005; 7:545-555.
36. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, Tan BK, Sethi G and Bishayee A. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochimica et biophysica acta. 2014; 1845:136-154.
37. Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J and Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science (New York, NY). 1995; 269:81-83.
38. Cao X, Tay A, Guy GR and Tan YH. Activation and association of Stat3 with Src in v-Src-transformed cell lines. Molecular and cellular biology. 1996; 16:1595-1603.
39. Chatain N, Ziegler P, Fahrenkamp D, Jost E, Moriggl R, Schmitz-Van de Leur H and Muller-Newen G. Src family kinases mediate cytoplasmic retention of activated STAT5 in BCR-ABL-positive cells. Oncogene. 2013; 32:3587-3597.
40. Malavaki CJ, Roussidis AE, Gialeli C, Kletsas D, Tsegenidis T, Theocharis AD, Tzanakakis GN and Karamanos NK. Imatinib as a key inhibitor of the platelet-derived growth factor receptor mediated expression of cell surface heparan sulfate proteoglycans and functional properties of breast cancer cells. The FEBS journal. 2013; 280:2477-2489.
41. Yu H and Jove R. The STATs of cancer--new molecular targets come of age. Nature reviews Cancer. 2004; 4:97-105.
42. Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, Fujiwara T, Tsuta K, Nokihara H, Tamura T, Asamura H, Kawaishi M, Kuwano K and Ochiya T. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Molecular therapy : the journal of the American Society of Gene Therapy. 2015.
43. Zhang C, Yang X, Zhang Q, Guo Q, He J, Qin Q, Zhu H, Liu J, Zhan L, Lu J, Liu Z, Xu L, Ma J, Dai S, Cheng H and Sun X. STAT3 inhibitor NSC74859 radiosensitizes esophageal cancer via the downregulation of HIF-1alpha. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014; 35:9793-9799.
44. Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, Derecka M, Chen Q, Hu Y, Sisler J, Hamed H, Lesnefsky EJ, Valerie K, Dent P and Larner AC. Mitochondrial Localized Stat3 Promotes Breast Cancer Growth via Phosphorylation of Serine 727. The Journal of biological chemistry. 2013; 288:31280-31288.
45. Yokote K, Mori S, Siegbahn A, Ronnstrand L, Wernstedt C, Heldin CH and Claesson-Welsh L. Structural determinants in the platelet-derived growth factor alpha-receptor implicated in modulation of chemotaxis. The Journal of biological chemistry. 1996; 271:5101-5111.
46. Ustach CV, Taube ME, Hurst NJ, Jr., Bhagat S, Bonfil RD, Cher ML, Schuger L and Kim HR. A potential oncogenic activity of platelet-derived growth factor d in prostate cancer progression. Cancer research. 2004; 64:1722-1729.
47. Li X, Tjwa M, Moons L, Fons P, Noel A, Ny A, Zhou JM, Lennartsson J, Li H, Luttun A, Ponten A, Devy L, Bouche A, et al. Revascularization of ischemic tissues by PDGF-CC via effects on endothelial cells and their progenitors. J Clin Invest. 2005; 115:118-127.
48. McGary EC, Onn A, Mills L, Heimberger A, Eton O, Thomas GW, Shtivelband M and Bar-Eli M. Imatinib Mesylate Inhibits Platelet-Derived Growth Factor Receptor Phosphorylation of Melanoma Cells But Does Not Affect Tumorigenicity In Vivo. J Investig Dermatol. 2004; 122:400-405.
49. Park JO, Park SH and Hong ST. A new method for transduction of mesenchymal stem cells using mechanical agitation. Molecules and cells. 2009; 28:515-520.