
INTRODUCTION
Kaposi sarcoma-associated herpesvirus (KSHV) is one of the most common etiologic agents for cancers arising in the setting of immune suppression, including Kaposi sarcoma (KS)—the most common HIV/AIDS-associated tumor worldwide and a leading cause of morbidity and mortality in this population [1]. Oral involvement occurs in a substantial proportion of patients with KS [2]. Published literatures suggest that KSHV dissemination within and from the oral cavity are critical factors for KSHV infection and oral KS progression in HIV-infected patients [3–7]. Person-to-person transmission of KSHV is thought to occur primarily through exchange of oropharyngeal secretions [3, 4], and epidemiologic data indicate that sexual practices involving contact with the oral cavity may promote KSHV transmission [5]. Furthermore, people have found that combination antiretroviral therapy (cART) cannot reduce KSHV replication within the oropharynx [3, 5] or KSHV transmission [7].
Oral KS lesions usually display higher KSHV viral loads and may portend more ominous prognoses relative to KS in other anatomic locations [8, 9]. We recently reported that KSHV successfully established latent infection in primary human gingival fibroblasts (HGF) or periodontal ligament fibroblasts (PDLF) in vitro, and virus de novo infection induced a tumor-associated fibroblast (TAF)-like phenotype within these cells [10]. Others also demonstrated that fibroblasts represented an imporant component within KS lesions and supported de novo KSHV infection [11, 12]. In addition, we recently reported that some pathogen-associated molecular patterns (PAMPs) molecules from periodontal pathogenic bacteria increased KSHV entry and subsequent viral latent gene expression within oral fibroblasts [13]. Despite this knowledge, the global altered gene expression profile in KSHV-infected oral fibroblasts has never been reported. KSHV needs to manipulate a number of host genes to facilitate the establishment of lifelong latent infection. In the current study, we used Illumina microarray to assess the altered gene profile in KSHV-infected PDLF and HGF relative to unifected mock cells. We found that the expression of various gene sets are significantly changed in virus-infected cells. In particular, KSHV de novo infection strongly up-regulates a series of interferon-induced gene in these oral cells, which are closely related to the maintenancy of virus latency.
RESULTS AND DISCUSSION
Microarray analysis of the global gene expression changes in KSHV-infected primary oral fibroblasts
We first used the HumanHT-12 v4 Expression BeadChip (Illumina), which contains more than 47,000 probes derived from the NCBI RefSeq Release 38 and other sources, to study global gene expression changes altered within KSHV-infected PDLF or HGF cells. We found that in PDLF cells, 134 genes were significantly up-regulated and 80 were down-regulated (≥ 2 fold and p < 0.05); in HGF cells, 166 genes were up-regulated and 268 down-regulated (Figure 1A). Intersection analysis indicated that 39 “common” genes were significantly up-regulated and 3 were down-regulated in both cell lines (listed in Table 1). We also performed enrichment analysis of these “common” genes in both cell lines by using the Pathway map, Gene Ontology (GO) Processes and Process Networks modules from Metacore Software (Thompson Reuters) [14]. Our analysis showed that most genes belong to several major cellular function categories, such as cellular response to type I interferon (IFN), inflammatory cytokine production, and other innate immune responses (Figure 2A–2B). The top 2 scored pathway maps (immune response_IFN α/β signaling pathway and immune response_Thymic stromal lymphopoietin [TSLP] signaling pathway) for these “common” genes are shown in Supplementary Figure S1. Interestingly, aberrant TSLP/TSLPR signaling has been associated with a variety of human diseases including asthma, atopic dermatitis, inflammatory bowel disease, eosinophilic esophagitis and acute lymphoblastic leukemia [15], but it has never been reported in KSHV infection and/or related malignancies.
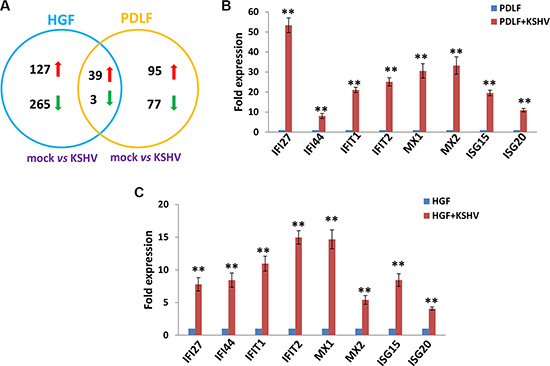
Figure 1: Intersection analysis and experimental validation of gene profile alterations in KSHV-infected primary oral fibroblast cells. (A) The HumanHT-12 v4 Expression BeadChip (Illumina) was used to detect alterations in gene profile in PDLF or HGF cells infected by KSHV (MOI~10, vs respective mock cells). Intersection analysis of significantly altered genes (up/down ≥ 2 fold and p < 0.05) was performed using the Illumina GenomeStudio Software. (B–C) The transcriptional levels of 8 selected ‘common’ candidate genes that were up-regulated in both sets of microarray data were validated by using qRT-PCR. Error bars represent the S.E.M. for 3 independent experiments. ** = p < 0.01 (vs PDLF or HGF).
Table 1: The “common” genes set altered within KSHV-infected HGF and PDLF cells (vs mock cells)
Gene Symbol |
Gene Description |
PDLF (folds) |
HGF (folds) |
|---|---|---|---|
IFI27 |
Interferon alpha-inducible protein 27, mitochondrial |
57.85 |
6.07 |
RSAD2 |
Radical S-adenosyl methionine domain-containing protein 2 |
43.99 |
4.48 |
MX1 |
Interferon-induced GTP-binding protein Mx1 |
36.48 |
15.76 |
MX2 |
Interferon-induced GTP-binding protein Mx2 |
36.48 |
3.26 |
IFIT2 |
Interferon-induced protein with tetratricopeptide repeats 2 |
28.42 |
12.6 |
ISG15 |
Ubiquitin-like protein ISG15 |
23.67 |
7.46 |
IFIT1 |
Interferon-induced protein with tetratricopeptide repeats 1 |
19.65 |
11.95 |
IFITM1 |
Interferon-induced transmembrane protein 1 |
16.44 |
3.16 |
HERC6 |
Probable E3 ubiquitin-protein ligase HERC6 |
14.97 |
4.47 |
IFIT3 |
Interferon-induced protein with tetratricopeptide repeats 3 |
14.77 |
3.84 |
ISG20 |
Interferon-stimulated gene 20 kDa protein |
12.03 |
3.38 |
IFI6 |
Interferon alpha-inducible protein 6 |
9.5 |
4.89 |
IFI44 |
Interferon-induced protein 44 |
9.13 |
9.24 |
SAMD9 |
Sterile alpha motif domain-containing protein 9 |
8.07 |
3.16 |
EPSTI1 |
Epithelial-stromal interaction protein 1 |
7.02 |
3.53 |
RARRES3 |
Retinoic acid receptor responder protein 3 |
5.71 |
2.19 |
IFI35 |
Interferon-induced 35 kDa protein |
5.64 |
2.35 |
XAF1 |
XIAP-associated factor 1 |
5.32 |
2.22 |
DDX58 |
Probable ATP-dependent RNA helicase DDX58 |
4.85 |
2.19 |
SAMD9L |
Sterile alpha motif domain-containing protein 9-like |
4.7 |
3.08 |
STAT1 |
Signal transducer and activator of transcription 1-alpha/beta |
4.6 |
2.29 |
PARP12 |
Poly [ADP-ribose] polymerase 12 |
4.38 |
2.06 |
DDX60 |
Probable ATP-dependent RNA helicase DDX60 |
4.04 |
2.25 |
MYPN |
Myopalladin |
3.26 |
2.3 |
IL12A |
Interleukin-12 subunit alpha |
3.03 |
5.81 |
PSG7 |
Putative pregnancy-specific beta-1-glycoprotein 7 |
3.02 |
4.46 |
COL4A1 |
Collagen alpha-1(IV) chain |
2.72 |
2.56 |
PSG1 |
Pregnancy-specific beta-1-glycoprotein 1 |
2.67 |
4.46 |
PSG2 |
Pregnancy-specific beta-1-glycoprotein 2 |
2.67 |
6.05 |
ANO3 |
Anoctamin-3 |
2.54 |
6.2 |
IL7R |
Interleukin-7 receptor subunit alpha |
2.48 |
6.46 |
NR2C1 |
Nuclear receptor subfamily 2 group C member 1 |
2.39 |
6.34 |
PSG4 |
Pregnancy-specific beta-1-glycoprotein 4 |
2.39 |
6.34 |
GBP2 |
Interferon-induced guanylate-binding protein 2 |
2.38 |
2.44 |
KRTAP1-1 |
Keratin-associated protein 1-1 |
2.24 |
2.48 |
VEGFC |
Vascular endothelial growth factor C |
2.23 |
3.04 |
MT1M |
Metallothionein-1M |
2.21 |
2.05 |
KRT34 |
Keratin, type I cuticular Ha4 |
2.1 |
9.24 |
PSME2 |
Proteasome activator complex subunit 2 |
2.02 |
2.19 |
RCAN2 |
Calcipressin-2 |
0.47 |
0.41 |
CEMIP |
Cell migration-inducing and hyaluronan-binding protein |
0.4 |
0.07 |
ATP8B4 |
Probable phospholipid-transporting ATPase IM |
0.38 |
0.25 |
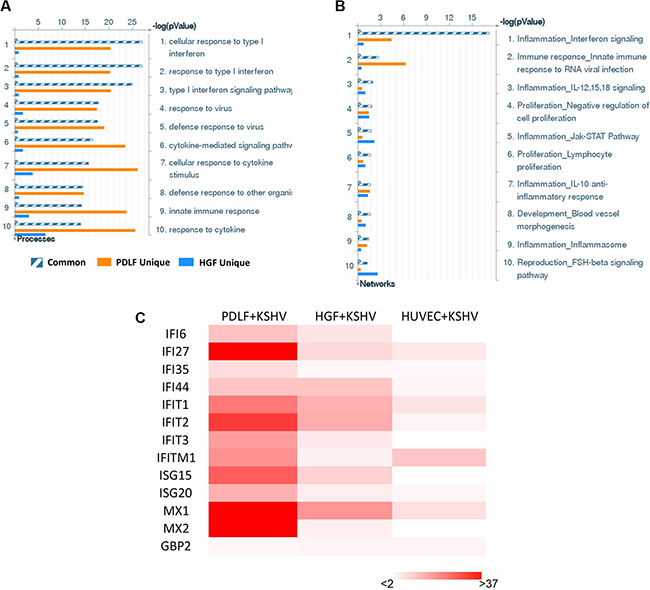
Figure 2: The enrichment analysis of gene profile alterations in KSHV-infected primary oral fibroblast cells. (A–B) The enrichment analysis of gene profile significantly altered (up/down ≥ 2 fold and p < 0.05) in KSHV-infected PDLF or HGF cells (vs mock cells) was performed using the Metacore Software (Thompson Reuters) Modules: Gene Ontology Processes (A) and Process Networks (B). (C) Heat map of interferon-induced genes signature altered in KSHV-infected PDLF, HGF and HUVEC cells (vs respective mock cells) was made by using Microsoft Excel 2010.
IFN-induced genes are highly up-regulated in KSHV-infected primary oral fibroblasts
Among these “common” genes, we noticed that a series of IFN-induced genes were strongly up-regulated in KSHV-infected primary oral fibroblasts (Table 1). We next selected 8 IFN-induced genes from Table 1 for validation of their transcriptional changes by using qRT-PCR analysis. Our results indicated that all of these genes (IFI27, IFI44, IFIT1, IFIT2, MX1, MX2, ISG15 and ISG20) were significantly up-regulated in a manner comparable to those found in the microarray data (Figure 1B–1C), demonstrating the credibility of our microarray analysis. Interestingly, when compared to the microarray data in KSHV-infected primary endothelial cells (HUVEC) we recently published [16], we found that the up-regulation of IFN-induced genes were much stronger in KSHV-infected PDLF/HGF than those in KSHV-infected HUVEC cells (Figure 2C). Production of IFN, in particular type I IFN is one of the most important host anti-viral immune responses, which can induce an anti-viral transcriptional program, producing proteins that cooperate to inhibit the spread of infection. Therefore, our data indicate that oral fibroblasts may represent an important cellular resource for type I IFN production during KSHV infection stimulus in the microenvironment of oral cavity. However, KSHV has successfully established the escape mechanisms from host immune responses, including the type I IFN response. For example, KSHV encodes 4 viral homologs of cellular interferon regulatory factors (named as vIRF1, −2, −3, and −4) with pleiotropic functions such as evasion of cell death, increased proliferation and evasion of immune responses [17]. For example, previous data have demonstrated that the expression of vIRF1 and vIRF2 can inhibit increases in IFN-β mediated by Toll-like receptor 3 (TLR3) [18]. In addition to their immunoregulatory effects, KSHV-encoded vIRFs were also shown to modulate cell growth by targeting the function of the tumor suppressor p53 and enhancing the activity of the c-Myc proto-oncogene [19]. While the KSHV-encoded vIRFs share an ability to block IFN or p53 signaling, each vIRF demonstrates a unique ability to block specific cellular functions [17].
IFN-induced genes, ISG15 and ISG20, are required for maintenance of KSHV latency in oral fibroblasts
Like other herpesviruses, KSHV can establish a lifelong infection in the host, and in more than 90% of infected host cells, the virus exists in a latency stage. Here we found that at least 2 IFN-induced genes, ISG15 and ISG20, are required for maintenance of KSHV latency in oral fibroblasts. Our data indicated that directly targeting ISG15 or ISG20 by RNAi significantly caused viral lytic gene (e.g. Rta, vGpcr and K8.1) transcripts from latently-infected PDLF with qRT-PCR analysis (Figure 3A and Supplementary Figure S2). We also confirmed the strong up-regulation of lytic K8.1 protein expression in either ISG15 or ISG20 “knock-down” KSHV-infected PDLF by using immunoblots (Figure 3B). Next, we isolated the KSHV virions from conditioned medium of ISG15 or ISG20 “knock-down” or control cells, then infected fresh PDLF cells. We found that silencing of either ISG15 or ISG20 greatly increased the virion release (there were more Lana transcripts in these infected groups compared to controls) (Figure 3C). Interestingly, one very recent study also reported that silencing of ISG15 in KSHV latently infected iSLK.219 cells resulted in a higher level of virus reactivation and an increase in infectious virus production [20]. They also found that KSHV-encoded vIRF1 protein can inhibit IFN activation in response to viral infection, through interaction with HERC5, an ISG15 E3 ligase, to alter ISG15 modification of cellular proteins [20]. Interestingly, vIRF1 itself was also a target of ISG15 conjugation. KSHV-infected cells exhibited increased ISG15 conjugation upon reactivation from latency in coordination with increased IFN [20].
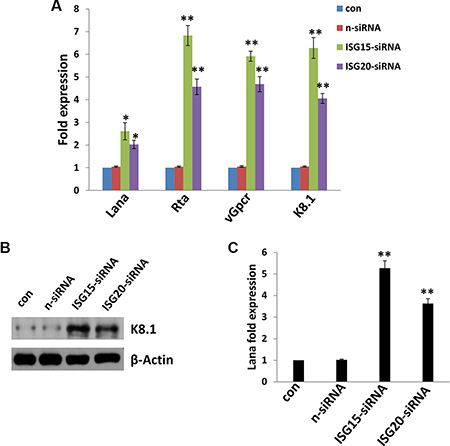
Figure 3: Targeting ISG15 and/or ISG20 induces KSHV lytic reactivation from infected primary oral fibroblast cells. (A–B) PDLF were first incubated with purified KSHV (MOI~10) for 2 h, then after 24 h p.i. transfected with either control non-target (n-siRNA), ISG15-siRNA or ISG20-siRNA for additional 48 h. Viral representative latent (Lana) and lytic gene (Rta, vGpcr, K8.1) transcripts were quantified using qRT-PCR. Protein expression was measured by immunoblots. (C) Released virions was isolated and purified from supernatant from groups in (A), then used to infect fresh PDLF cells. After 24 h p.i., Lana transcripts were quantified using qRT-PCR. Error bars represent the S.E.M for three independent experiments. * = p < 0.05, ** = p < 0.01 (vs n-siRNA group).
KSHV microRNAs are involved in viral lytic reactivation caused by silencing of ISG15 or ISG20
KSHV-microRNAs (mostly miR-K12-1, 3, 4, 5, 7, 9 and 11) have been shown to positively or negatively regulate viral latency in a variety of infected host cells, through either directly targeting the viral lytic reactivation activator, Rta [21, 22], or through indirect mechanisms including targeting host factors such as IκBα, nuclear factor I/B (NFIB), Rbl2, BCLAF1 and IKKε [23–27]. By using qRT-PCR screening analysis, we found that silencing of ISG15 prominently reduced the transcripts of miR-K12-1 and miR-K12-11, while silencing of ISG20 caused a significant reduction of miR-K12-1 and miR-K12-3 in PDLF cells (Figure 4A–4B). To further confirm the role of specific viral microRNA in ISG15- or ISG20-mediated virus latency, we used individual recombinant construct encoding miR-K12-1 as described previously [28] to restore its expressional level. We found that this overexpression of miR-K12-1 significantly repressed KSHV lytic gene expression for infected PDLF cells during “knock-down” ISG15 or ISG20 with RNAi (Figure 4C). Published data have shown that miR-K12-1 can targets IκBα, an inhibitor of NF-κB complexes, thereby promoting NF-κB-dependent viral latency and cell survival [23]. Our recent data also demonstrate that the NF-κB pathway is important to KSHV-positive lymphoma cell survival and viral latency [29]. Therefore, ongoing work will try to understand the involvement of NF-κB pathway in either ISG15- or ISG20-mediated virus latency for oral cells.
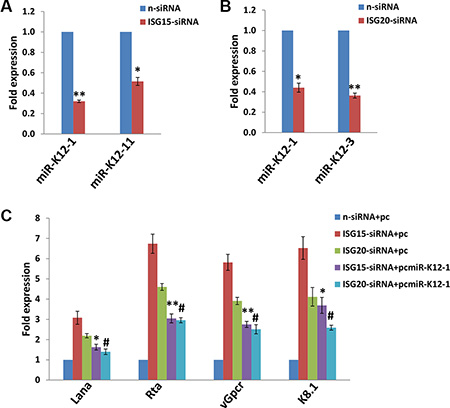
Figure 4: Targeting ISG15 and/or ISG20 induces KSHV lytic gene expression through suppression of KSHV microRNAs. (A–B) PDLF were first incubated with purified KSHV (MOI~10) for 2 h, then after 24 h p.i. transfected with either control non-target (n-siRNA), ISG15-siRNA or ISG20-siRNA for additional 48 h. KSHV microRNA transcripts were quantified using qRT-PCR as described in Methods. (C) PDLF were incubated with purified KSHV for 2 h, then transfected with control vector (pc), or vectors encoding miR-K12-1 (pcmiR-K12-1) for additional 24 h. Thereafter, cells were transfected with either control non-target (n-siRNA), ISG15-siRNA or ISG20-siRNA for additional 48 h. Viral representative latent (Lana) and lytic gene (Rta, vGpcr, K8.1) transcripts were quantified using qRT-PCR. Error bars represent the S.E.M for three independent experiments. */# = p < 0.05, ** = p < 0.01 (vs ISG15-siRNA+pc or ISG20-siRNA+pc groups, respectively).
In summary, we provide for the first time global gene expression profile alterations in KSHV-infected oral fibroblasts by microarray analysis. Among the altered candidates, many interferon-induced genes are strongly up-regulated in these oral cells, while some of them such as ISG15 and ISG20 are required for the maintenance of virus latency. Our data indicate that oral fibroblasts may represent one of the important host cellular defense components for anti-viral infection, as well as acting as a reservoir for herpesvirus lifelong infection in the oral cavity.
MATERIALS AND METHODS
Cell culture, reagents and infection protocol
Body cavity-based lymphoma cells (BCBL-1, KSHV+/EBV−) were maintained in RPMI 1640 medium (Gibco) with supplements as described previously [29]. Primary human gingival fibroblasts (HGF) and periodontal ligament fibroblasts (PDLF) were purchased from ScienCell. These cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Mediatech) supplemented with 10% FBS, 10 mM HEPES (pH 7.5), 100 U/mL of penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B. All cells were incubated at 37°C in 5% CO2. All experiments were carried out using cells harvested at low (< 20) passages. To obtain KSHV for infection experiments, BCBL-1 cells were incubated with 0.6 mM valproic acid for 6 days, and purified virus was concentrated from culture supernatants and infectious titers were determined as described previously [28].
Microarray
Total RNA was isolated using Qiagen RNeasy kit (Qiagen), and 500 ng of total RNA was used to synthesize dscDNA. Biotin-labeled RNA was generated using the TargetAmp-Nano Labeling Kit for Illumina Expression BeadChip (Epicentre), according to the manufacturers’ instructions, and hybridized to the HumanHT-12 v4 Expression BeadChip (Illumina), which contains more than 47,000 probes derived from the NCBI RefSeq Release 38 and other sources, at 58°C for 16 h. The chip was washed, stained with streptavadin-Cy3, and scanned with the Illumina BeadStation 500 and BeadScan. Using the Illumina’s GenomeStudio software, we normalized the signals using the “cubic spline algorithm” that assumes that the distribution of transcript abundance is similar in all samples, according to the method proposed by Workman et al. [30]. The background signal was removed using the “detection p-value algorithm” to remove targets with signal intensities equal or lower than that of irrelevant probes (with no known targets in the human genome but thermodynamically similar to the relevant probes). Common and unique sets of genes and enrichment analysis were performed using the MetaCore Software (Thompson Reuters) as previously reported [14]. The microarray original data have been submitted to Gene Expression Omnibus (GEO) database (Accession number: GSE79548).
RNA interference and plasmid transfection
ISG15 and ISG20 ON-TARGET plus SMART pool siRNA, or negative control siRNA (n-siRNA) (Dharmacon), were delivered using the DharmaFECT transfection reagent according to the manufacturer’s instructions. For plasmid transfection, PDLF were transfected in 12-well plates with miR-K12-1 recombinant construct or control vector as previously described [28] by using Lipofectamine 3000 (Invitrogen) for 48 h. Transfection efficiency was normalized through co-transfection of a lacZ reporter construct and determination of β-galactosidase activity using a commercial β-galactosidase enzyme assay system according to the manufacturer’s instructions (Promega).
Immunoblotting
Total cell lysates (20 μg) were resolved by 10% SDS–PAGE, transferred to nitrocellulose membranes, and immunoblotted with antibodies for K8.1 (ABI) and β-Actin (Sigma) for loading controls. Immunoreactive bands were identified using an enhanced chemiluminescence reaction (Perkin-Elmer), visualized by autoradiography and quantitated using Image-J software.
qRT-PCR
Total RNA was isolated using the RNeasy Mini kit (QIAGEN), and cDNA was synthesized from equivalent total RNA using a SuperScript III First-Strand Synthesis SuperMix Kit (Invitrogen) according to the manufacturer’s instructions. Primers used for amplification of target genes are displayed in Supplementary Table S1. Amplification was carried out using an iCycler IQ Real-Time PCR Detection System, and cycle threshold (Ct) values were tabulated in duplicate for each gene of interest in each experiment. “No template” (water) controls were used to ensure minimal background contamination. Using mean Ct values tabulated for each gene, and paired Ct values for β-actin as a loading control, fold changes for experimental groups relative to assigned controls were calculated using automated iQ5 2.0 software (Bio-rad). For amplification of viral miRNAs, cDNA was synthesized using the Taqman miRNA RT kit (Applied Biosystems), and qPCR was performed using the Taqman MicroRNA Assays kit (Applied Biosystems) and a 7500 Real Time PCR System. Fold changes for microRNA were calculated using paired Ct values for RNU6B as recommended by the manufacturer (Applied Biosystems).
Statistical analysis
Significance for differences between experimental and control groups was determined using the two-tailed Student’s t-test (Microsoft Excel 2010), and p values < 0.05 or <0.01 were considered significant or highly significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by grants from a DOD Career Development Award (CA140437), the SOM Research Enhancement Funding (2015–2016) to Z.Q., the National Institute of General Medical Sciences (NIGMS P20GM103501 subproject 2, P30GM114732, U54GM104940-01), the National Institute on Minority Health and Health Disparities (NIMHD P20MD004817, U54MD008176-01) and the Louisiana Cancer Research Consortium to J.Z., a Louisiana Clinical and Translational Science Center Pilot grant (U54GM104940 from NIH) to Z.L., R01AI101046 and R01AI106676 from NIH to E.K.F., as well as awards from the National Natural Science Foundation of China (81272191, 81472547 to Z.Q. and 81400164 to L.D.). J.Q. was supported by funding from Shanghai Science and Technology committee (14411971400) and Pudong Science and Technology committee, Shanghai (PK2013-17). Funding sources had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICTS OF INTEREST
All the authors declare no conflicts of interest.
REFERENCES
1. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994; 266:1865–1869.
2. Feller L, Lemmer J, Wood NH, Jadwat Y, Raubenheimer EJ. HIV-associated oral Kaposi sarcoma and HHV-8: a review. J Int Acad Periodontol. 2007; 9:129–136.
3. Pauk J, Huang ML, Brodie SJ, Wald A, Koelle DM, Schacker T, Celum C, Selke S, Corey L. Mucosal shedding of human herpesvirus 8 in men. N Engl J Med. 2000; 343:1369–1377.
4. Casper C, Redman M, Huang ML, Pauk J, Lampinen TM, Hawes SE, Critchlow CW, Morrow RA, Corey L, Kiviat N, Wald A. HIV infection and human herpesvirus-8 oral shedding among men who have sex with men. J Acquir Immune Defic Syndr. 2004; 35:233–238.
5. Dukers NH, Renwick N, Prins M, Geskus RB, Schulz TF, Weverling GJ, Coutinho RA, Goudsmit J. Risk factors for human herpesvirus 8 seropositivity and seroconversion in a cohort of homosexual men. Am J Epidemiol. 2000; 151:213–224.
6. Miller CS, Berger JR, Mootoor Y, Avdiushko SA, Zhu H, Kryscio RJ. High prevalence of multiple human herpesviruses in saliva from human immunodeficiency virus-infected persons in the era of highly active antiretroviral therapy. J Clin Microbiol. 2006; 44:2409–2415.
7. Osmond DH, Buchbinder S, Cheng A, Graves A, Vittinghoff E, Cossen CK, Forghani B, Martin JN. Prevalence of Kaposi sarcoma-associated herpesvirus infection in homosexual men at beginning of and during the HIV epidemic. JAMA. 2002; 287:221–225.
8. Rohrmus B, Thoma-Greber EM, Bogner JR, Rocken M. Outlook in oral and cutaneous Kaposi’s sarcoma. Lancet. 2000; 356:2160.
9. Gorsky M, Epstein JB. A case series of acquired immunodeficiency syndrome patients with initial neoplastic diagnoses of intraoral Kaposi’s sarcoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000; 90:612–617.
10. Dai L, Qin Z, Defee M, Toole BP, Kirkwood KL, Parsons C. Kaposi sarcoma-associated herpesvirus (KSHV) induces a functional tumor-associated phenotype for oral fibroblasts. Cancer Lett. 2012; 318:214–220.
11. Akula SM, Naranatt PP, Walia NS, Wang FZ, Fegley B, Chandran B. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J Virol. 2003; 77:7978–7990.
12. Offermann MK. Kaposi’s sarcoma and HHV-8. Trends Microbiol. 1996; 4:419.
13. Dai L, DeFee MR, Cao Y, Wen J, Wen X, Noverr MC, Qin Z. Lipoteichoic acid (LTA) and lipopolysaccharides (LPS) from periodontal pathogenic bacteria facilitate oncogenic herpesvirus infection within primary oral cells. PLoS One. 2014; 9:e101326.
14. Kim SH, Sierra RA, McGee DJ, Zabaleta J. Transcriptional profiling of gastric epithelial cells infected with wild type or arginase-deficient Helicobacter pylori. BMC Microbiol. 2012; 12:175.
15. Zhong J, Sharma J, Raju R, Palapetta SM, Prasad TS, Huang TC, Yoda A, Tyner JW, van Bodegom D, Weinstock DM, Ziegler SF, Pandey A. TSLP signaling pathway map: a platform for analysis of TSLP-mediated signaling. Database (Oxford). 2014; 2014:bau007.
16. Dai L, Trillo-Tinoco J, Chen Y, Bonstaff K, Del Valle L, Parsons C, Ochoa AC, Zabaleta J, Toole BP, Qin Z. CD147 and downstream ADAMTSs promote the tumorigenicity of Kaposi’s sarcoma-associated herpesvirus infected endothelial cells. Oncotarget. 2016; 7:3806–3818. doi: 10.18632/oncotarget.6584.
17. Jacobs SR, Damania B. The viral interferon regulatory factors of KSHV: immunosuppressors or oncogenes? Frontiers Immunol. 2011; 2:19.
18. Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, Heise MT, Damania B. The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J Virol. 2013; 87:798–806.
19. Baresova P, Pitha PM, Lubyova B. Distinct roles of Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factors in inflammatory response and cancer. J Virol. 2013; 87:9398–9410.
20. Jacobs SR, Stopford CM, West JA, Bennett CL, Giffin L, Damania B. Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 1 Interacts with a Member of the Interferon-Stimulated Gene 15 Pathway. J Virol. 2015; 89:11572–11583.
21. Lin X, Liang D, He Z, Deng Q, Robertson ES, Lan K. miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS One. 2011; 6:e16224.
22. Bellare P, Ganem D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe. 2009; 6:570–575.
23. Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010; 12:193–199.
24. Lu CC, Li Z, Chu CY, Feng J, Sun R, Rana TM. MicroRNAs encoded by Kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 2010; 11:784–790.
25. Liang D, Gao Y, Lin X, He Z, Zhao Q, Deng Q, Lan K. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell Res. 2011; 21:793–806.
26. Ziegelbauer JM, Sullivan CS, Ganem D. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet. 2009; 41:130–134.
27. Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J Virol. 2010; 84:2697–2706.
28. Dai L, Plaisance-Bonstaff K, Voelkel-Johnson C, Smith CD, Ogretmen B, Qin Z, Parsons C. Sphingosine kinase-2 maintains viral latency and survival for KSHV-infected endothelial cells. PLoS One. 2014; 9:e102314.
29. Qin Z, Dai L, Trillo-Tinoco J, Senkal C, Wang W, Reske T, Bonstaff K, Del Valle L, Rodriguez P, Flemington E, Voelkel-Johnson C, Smith CD, Ogretmen B, et al. Targeting Sphingosine Kinase Induces Apoptosis and Tumor Regression for KSHV-Associated Primary Effusion Lymphoma. Mol Cancer Ther. 2014; 13:154–164.
30. Workman C, Jensen LJ, Jarmer H, Berka R, Gautier L, Nielser HB, Saxild HH, Nielsen C, Brunak S, Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002; 3:research0048.