
INTRODUCTION
SOX9 [sex-determining region Y (SRY)-box 9 protein] is a transcription factor that regulates development under normal circumstances. SOX9 and other members of the SOX family have conserved structural features including a high mobility group (HMG) box DNA-binding domain, as well as a transactivation domain. The range of functions that Sox proteins serve is broad, including maintaining stem cell features, restricting lineage, and directing terminal differentiation. Germline SOX9 heterozygous inactivating missense and nonsense mutations result in campomelic dysplasia, a syndrome resulting in skeletal malformations, central nervous system dysfunction, as well as abnormalities in other organs [1-2].
In the context of cancer, SOX9 has been classified as both tumor suppressor and oncogene depending on the study and type of cancer being investigated. Evidence suggesting its role as a tumor suppressor is derived from several tumor types. SOX9 has decreased expression in cervical carcinoma compared to normal cervical tissue, and in vitro inhibited cell growth and tumor formation when overexpressed [3]. In prostate cancer, SOX9 overexpression has been show to decrease cell proliferation and increase apoptotic activity.[4]. By contrast, both decreased SOX9 activity and oncogenic features of SOX9 have been implicated in colorectal carcinoma (CRC) [5-7].
Recently, exome sequencing has identified SOX9 as a recurrently mutated gene in colorectal carcinoma.[8]. The spectrum of mutations, effect on protein expression, molecular correlates, and clinical features has not been characterized to date. Here, we characterize the spectrum of SOX9 mutations, their molecular and clinical correlates, and their effect on SOX9 protein expression in comparison to SOX9 wild type (WT) carcinoma and normal epithelium within the context of CRC.
RESULTS
Characteristics of SOX9 mutations in CRC
Mutations discovered were either frameshift or nonsense mutations in 31 of 38 (81.6%) of SOX9 mutant CRC. Each of the 38 SOX9 mutations was unique, without recurrent mutations (Figure 1A, Table 1). There were 23 frameshift mutations, 12 nonsense mutations, 8 missense mutations, and 1 in frame deletion. The most frequently mutated exon was the last one (exon 3), which harbored predominantly nonsense and frameshift mutations (92%), while frameshift and truncating mutations only made up 44% of exon 1 mutation and 78% of exon 2 mutations (p = 0.02 for truncating mutations in exon 3 versus exons 1-2). There were 9 mutations in exon 1 (4 missense, 1 in frame deletion, 1 nonsense, and 3 frameshift), 9 mutations in exon 2 (2 missense mutations, 2 nonsense mutations, and 5 frameshift mutations) and 23 mutations in exon 3 (2 missense mutations, 9 nonsense mutations, and 15 frameshift mutations). TCGA data was similar to institutional data in that the majority of SOX9 mutations detected were frameshift or nonsense. Further, the SOX9 mutant CRC in TCGA data displayed transcriptional upregulation of SOX9 (Figure 1B).
Table 1: SOX9 mutations, zygosity, and expression.
Exon |
Amino-acid change |
Nucleotide change |
Predicted AA length |
TP (%) |
AF (%) |
H score (IHC) |
Oncoscan result |
Site Tested |
1 |
E57Qfs*50 |
168_179delinsAG |
107 |
22 |
45 |
300 |
liver |
|
1 |
E134* |
400G>T |
134 |
10 |
11 |
0 |
primary |
|
2 |
E159Gfs*25 |
474_475dupGG |
184 |
20 |
21 |
60 |
peritoneum |
|
2 |
E190* |
568G>T |
190 |
46 |
33 |
140 |
primary |
|
2 |
Q208* |
622C>T |
208 |
25 |
50 |
0 |
primary |
|
2 |
S215Pfs*4 |
643delT |
219 |
11 |
7 |
300 |
primary |
|
2 |
E191Rfs*28 |
571delG |
219 |
18 |
24 |
300 |
peritoneum |
|
2 |
Q164*, E226* |
490C>T, 676G>T |
226 |
50 |
10 |
130 |
primary |
|
2 |
D168Efs*84 |
503dupA |
252 |
48 |
39 |
70 |
liver |
|
1 |
W143Lfs*109 |
427dupT |
252 |
70 |
67 |
190 |
Gain, LOH |
recurrence |
3 |
V245Gfs*7 |
733dupG |
252 |
50 |
25 |
300 |
liver |
|
3 |
Q246Cfs*8 |
734_735dupTG |
254 |
53 |
36 |
60 |
primary |
|
3 |
L259* |
776T>A |
259 |
60 |
61 |
0 |
liver |
|
3 |
P267Sfs*9 |
798_807delCCCTATCGAC |
276 |
30 |
14 |
160 |
primary |
|
3 |
G263Afs*16 |
788delG |
279 |
25 |
13 |
1 |
primary |
|
3 |
D274Wfs*6 |
818_819dupTG |
280 |
40 |
29 |
300 |
primary |
|
3 |
E277D, S279Afs*104 |
831G>C, 834delG |
293 |
20 |
40 |
primary |
||
3 |
Q312* |
934C>T |
312 |
30 |
82 |
300 |
Hypotriploid, LOH |
ovary |
3 |
W335* |
1004G>A |
335 |
30 |
76 |
300 |
Gain, no LOH |
peritoneum |
3 |
Q339* |
1015C>T |
339 |
10 |
25 |
300 |
liver |
|
3 |
D290Mfs*93 |
868delG |
383 |
50 |
44 |
300 |
Hypotetraploid, no LOH |
primary |
3 |
P374Rfs*9 |
1212C>A |
383 |
20 |
53 |
300 |
primary, duodenum |
|
1,2,3 |
S23FS, T196A, R394* |
66delC, 586A>G, 1180C>T |
394 |
30 |
15 |
primary |
||
3 |
H396L, E400* in cis |
1187A>T, 1198G>T |
400 |
50 |
55 |
300 |
Gain, LOH |
primary |
3 |
Q393Sfs*10 |
1177delC |
403 |
50 |
25 |
300 |
primary |
|
3 |
H396Rfs*8 |
1185_1186dupGC |
404 |
50 |
33 |
300 |
primary |
|
3 |
Q410* |
1228C>T |
410 |
70 |
85 |
300 |
CN-LOH |
primary |
3 |
Q412* |
1234C>T |
412 |
50 |
73 |
225 |
CN-LOH |
primary |
3 |
H406Sfs*58 |
1215_1234del |
464 |
60 |
23 |
300 |
primary |
|
1 |
A118_A124del |
353_373delCGGCGCGCAGGAAGCTCGCGG |
502 |
54 |
21 |
300 |
primary |
|
2 |
A187V |
560C>T |
509 |
60 |
25 |
5 |
primary |
|
1 |
M113T |
338T>C |
509 |
90 |
28 |
90 |
primary |
|
1 |
M113V, R162H |
337A>G, 485 G>A |
509 |
50 |
27 |
170 |
primary |
|
1 |
S39C |
116C>G |
509 |
90 |
45 |
primary |
||
3 |
V486A |
1457T>C |
509 |
25 |
20 |
250 |
recurrence |
|
3 |
A419T |
1255G>A |
509 |
10 |
11 |
250 |
primary |
|
3 |
R508_P509insGGL PRRAKMAEMILK ITEEREDQPEFPL DICVFLFFYFVLF FLLLLL* |
1525_1530+5delCCTTGAGGAGG |
557 |
10 |
18 |
0 |
primary |
|
3 |
M469Ifs*109 |
1406dupT |
578 |
50 |
54 |
3 |
|
liver |
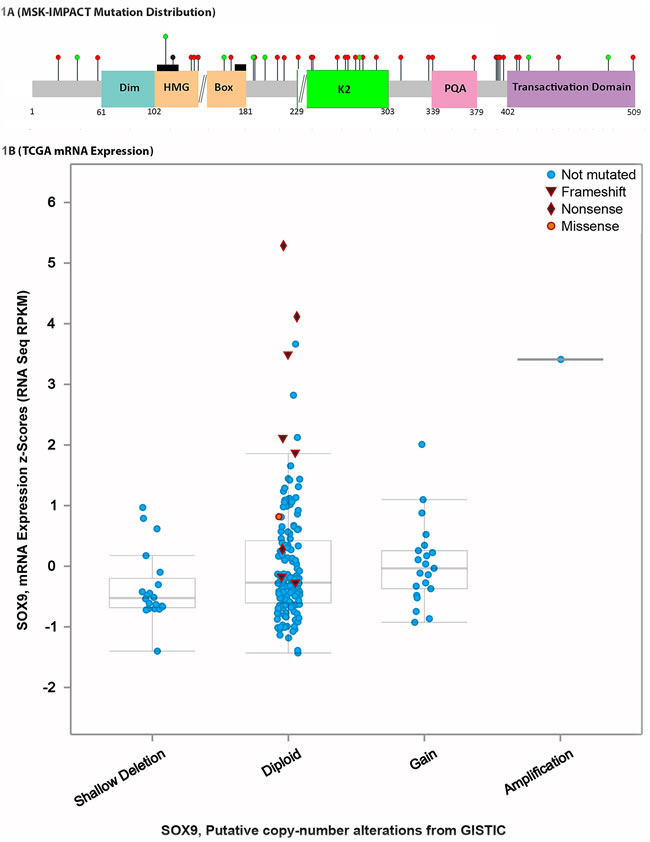
Figure 1: SOX9 mutation distribution and transcription levels. A. SOX9, which only has 3 exons (delineated by black double forward slashes), has a conserved DNA dimerization domain (Dim), a homeobox group binding domain (HMG box) with two nuclear localization signals (black rectangles) that bind DNA, and transactivation domains (K2 and Transactivation Domain) as well as a PQA domain. SOX9 mutations occurred as mostly frameshift and nonsense mutations (red circles) while scattered missense mutations (green circles) and an in frame deletion (black circle) were also detected. The majority of mutations preserved the dimerization and HMG box domains. B. TCGA data shows that, similarly to our institutional study cases, most SOX9 mutations are frameshift or nonsense. The mutations have increased RNA transcription levels on RNA sequencing.
Correlation of SOX9 mutation frequency with known CRC genes
In comparison to SOX9 wild type CRC, SOX9 mutant CRC was strongly associated with the presence of KRAS mutation (p = 0.0001) and the absence of TP53 mutation (p = 0.0004). SOX9 mutant CRC also had higher rates of PIK3CA mutation (p = 0.0451), a trend toward APC mutation (p = 0.0527), and MMR deficiency (p = 0.0472). Interestingly, the majority of SOX9 mutations in the MMR-D CRC were missense or nonsense and did not occur in mononucleotide tracts, while only 2 of the 10 SOX9 mutations in these 6 MMR-D, SOX9 mutant CRC were frameshifts (both in mononucleotide tracts). None of the 38 SOX9 mutant CRC harbored BRAF or NRAS mutations, while 34 (10.8%) and 10 (3.2%) of SOX9 WT CRC harbored BRAF and NRAS hotspot mutations, respectively (Table 2).
Table 2: Clinicopathologic and molecular characteristics of SOX9 mutant versus SOX9 WT CRC.
SOX9 mutant (n = 38) |
SOX9 WT (n= 315) |
P value |
|
Age (mean; median) |
60; 62 |
55; 59 |
|
Sex (M:F) |
22:16 |
184:131 |
|
Location: cecum to transverse |
18 |
107 |
|
Location: descending to rectum |
20 |
208 |
|
Histology: mucinous, poor |
18%, 0 |
4%, 10% |
|
MMR-D |
26% |
6% |
P = 0.0472 |
KRAS mutant |
66% (25 of 38) |
33% (123 of 315) |
P = 0.0001 |
NRAS mutant |
0 |
11% |
|
BRAF mutant |
0 |
3% |
|
APC mutant |
87% |
72% |
|
TP53 mutant |
42% (16 of 38) |
72% (227 of 315) |
P = 0.0004 |
PIK3CA mutant |
32% |
17% |
P = 0.0451 |
Correlation of SOX9 mutation Type to immunohistochemical expression
All 22 SOX9 WT CRC analyzed showed strong positive expression (mean H-score 296±19.2). (Figure 2A, 2C) while normal epithelium shows weak expression limited to deep crypts (Figure 2A, 2B). Overall, SOX9 mutant CRC showed a wide range of H scores with a mean H-score (188.7±123.3, p value < 0.001). However, closer review indicated that H scores in SOX9 mutant CRC were highly related to both the type of variant and its site within the coding region of the SOX9 gene. Truncating or missense exon 3 mutant CRC had higher H scores in comparison to other mutations (exon 1 or 2 mutant or exon 3 frameshift mutants resulting in loss of stop codon and predicted protein elongation) (p = 0.02): SOX9 exon 1 or 2 mutant mean H score = 147, SOX9 frameshifts resulting in loss of stop codon and predicted protein elongation had mutant mean H score = 2 (Figure 2A, 2D), exon 3 mutant CRC (excluding stop loss mutants) mean H score = 239 (Figure 2A, 2E).
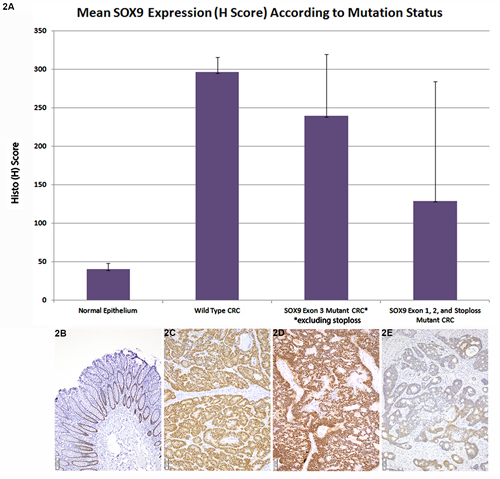
Figure 2: Mean SOX9 Expression (H Score) According to Mutation Status. A. In comparison to normal epithelium (mean H score 40±7.9), which only shows SOX9 expression in deep crypt epithelium, the majority of both SOX9 WT (mean H score = 296±19.2) and mutant CRC overexpressed SOX9. Among SOX9 mutant CRC, truncating and missense exon 3 mutants had a higher mean H score (239±80.3) in comparison to other mutants including exon 1, exon 2, and exon 3 elongating mutants resulting in loss of stop codon (mean H score = 129±155.3), p = 0.02. B. Normal colorectal epithelium has nuclear SOX9 expression restricted to deep crypts. C. Strong, nuclear SOX9 expression is seen in SOX9 wild type CRC. D. A SOX9 p. Q312X mutant with loss of the normal allele displayed strong SOX9 nuclear expression. E. A SOX9 frameshift mutants resulting in loss of the stop codon with paucity of nuclear SOX9 expression.
Overexpression of truncating SOX9 mutations in the absence of a wild type allele
Zygosity analysis was performed on 7 SOX9 expressing (IHC) CRC with SOX9 frameshift or nonsense mutations and high SOX9 mutant allele frequencies in comparison to estimated tumor percent. Five of these 7 cases showed LOH at chromosome 17q24.3 where SOX9 is located. Of these 5 cases, 2 cases displayed copy neutral (CN) LOH while 3 harbored gains of the mutant allele (Table 1) in addition to LOH (Figure 3). The remaining two cases tested showed gain of the mutant allele yet retained the WT allele.
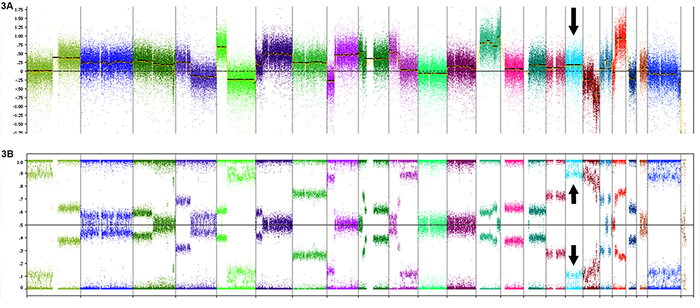
Figure 3: Copy number and heterozygosity of SOX9 mutants. A. Oncoscan analysis of the SOX9 p. Q312X mutant displayed in Figure 2D shows gain of SOX9 (arrow shows increase in log2 copy number over chromosome 17 including SOX9) and B. loss of normal allele (arrows show splitting of beta allele frequency).
Frequency of SOX9 mutations in CRC and relationship to clinical features
From January 1st 2014 to December 31st, 2015; 353 CRC patients underwent MSK-IMPACT testing. Of these, 38 (10.7%) harbored SOX9 mutations. SOX9 mutant CRC did not differ from SOX9 WT CRC in male to female ratio, patient age distribution, WHO morphologic subtype distribution, or primary tumor location (proximal versus distal) (Table 2). Patients with metastatic SOX9 mutant CRC (n = 317) had longer overall survival in comparison to those with SOX9 WT on univariate analysis (log rank p value = 0.049) (Figure 4A). To adjust for microsatellite status because MSI-H was more frequent in SOX9 mutant CRC, we compared overall survival in MSS CRC with versus without SOX9 mutation. In this microsatellite stable analysis, 11 metastatic SOX9 WT and 2 metastatic SOX9 mutant CRC were MSI-H and excluded. Metastatic SOX9 mutant CRC tended to have a longer overall survival of borderline statistical significance (log rank p value = 0.058) (Figure 4B).
Five patients with SOX9 mutant, RAS/ RAF WT CRC received anti-EGFR therapy (cetuximab or panitumumab) in combination with other treatment modalities (FOLFIRI, irinotecan). Of these, 3 (60%) showed progression of disease, 1 patient (unknown whether irinotecan naïve or not) received cetuximab and irinotecan and showed decreased tumor volume of hepatic metastasis, and 1 patient received adjuvant panitumumab with folfiri, floxuridine and a hepatic pump after metastatectomy and has not recurred.
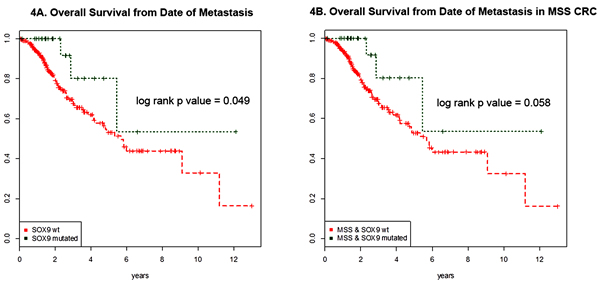
Figure 4: Survival curves of metastatic SOX9 mutant and wild type CRC. A. Overall survival from date of metastasis of SOX9 mutant (green) and SOX9 WT (red) are shown for the 317 CRC patients with metastatic disease (285 WT and 32 mutants) are shown. SOX9 mutation was associated with better survival (log rank p value = 0.049) on univariate analysis. B. Adjusting for microsatellite instability, the overall survival of microsatellite stable (MSS) CRC are shown. This analysis excluded 2 microsatellite instability-high (MSI-H) SOX9 mutant CRC and 30 MSI-H SOX9 wild type CRC. MSS SOX9 mutant CRC tended to have longer overall survival than SOX9 wild type CRC from date of metastasis with borderline significance log rank p value of p = 0.058.
DISCUSSION
In this study, we demonstrate several findings including the recurrent nature of truncating SOX9 mutations in CRC, the overexpression of SOX9 in the majority of both SOX9 mutant (including those with SOX9 LOH) and WT CRC, and the strong association of SOX9 mutation with mutant KRAS and WT TP53.
The distribution and nature of SOX9 mutations in CRC is similar to that seen in classic tumor suppressors, namely mostly truncating mutations across the length of the gene without hotspots. However, the fact that we found gain of the mutant SOX9 allele as well as overexpression of the mutant allele in the absence of the WT allele argues that truncated SOX9 may serve as an oncogene in CRC, or as a gain of function tumor suppressor gene as well described in the case of TP53. However, data across the Cancer Genome Atlas (TCGA) show that SOX9 is altered in a pattern typically seen in oncogenes: it is mutated and amplified in multiple cancer types, and rarely is the whole gene deleted [11].The higher expression of SOX9 for exon 3 truncating mutations is potentially due to the fact that exon 3 is the last exon in SOX9, and truncating mutations within the last exon do not activate nonsense-mediated decay.[13].This, however, may not be true for SOX9 mutations that extend the protein via distal frameshift mutations, removing the WT stop codon and activating nonstop mediated decay [14] both of our cases with distal SOX9 exon 3 frameshift mutations with stop loss mutations had very low H scores for expression (0 and 3 out of 300).
Interestingly, studies have shown overexpression of an isoform of the SOX9 protein in CRC that is truncated due to retention of intron 2 (termed ‘miniSOX9’), missing the transactivation domain yet still conserving the N terminal dimerization and DNA binding domains [15].
MiniSOX9 upregulates Wnt/β-catenin-dependent transcriptional activity and is associated with nuclear β catenin accumulation [16-18]. While one proposed explanation for this has been splice site mutations affecting intron 2, many CRC with miniSOX9 expression lack a mutational basis for the truncated and overexpressed SOX9 product. As the majority of SOX9 mutations in CRC are truncating and result in deletion of the C-terminal protein including the transactivation domain, the protein product is predicted to be functionally similar to the MiniSOX9 isoform of SOX9.
The fact that truncated SOX9 is highly associated with mutant KRAS and WT TP53 has not been previously demonstrated. Wild type SOX9 has been shown to cooperate with RAS mutants in several in vivo experiments [6] These experiments showed higher mRNA transcription levels of SOX9 when an HRAS codon 12 mutation was knocked in. These experiments also showed 5 times more tumor foci in a CRC cell line with HRAS codon 12 mutation when SOX9 was overexpressed in comparison to when SOX9 was not overexpressed [6] In regards to TP53 status and correlation with SOX9 mutation status, it has been found that overexpression of SOX9 results in lower levels of TP53 [6].This indirect relationship may be due to SOX9 overexpression having decreased p14 levels, and p14 stabilizes TP53. Therefore, even though truncating SOX9 mutations that result in SOX9 overexpression are associated with WT TP53, they may indirectly downregulate TP53 activity.
Finally, the correlation of SOX9 mutation with longer survival has not been previously investigated. While we find that SOX9 mutation is associated with increased survival in patients with metastatic CRC on univariate analysis (p = 0.049), we also know that SOX9 mutation is associated with MMR deficiency in a non-random way as the majority of SOX9 truncating mutations in MMR deficient CRC were not in mono- or di- nucleotide repeat areas. MMR deficiency has been shown to have a positive impact on prognosis [19]. Even after adjustment for microsatellite status, SOX9 mutant MSS CRC tended to have longer overall survival than SOX9 WT MSS CRC with borderline statistical significance (p = 0.058), suggesting that the SOX9 mutation may positively affect prognosis independent of MSI status. A significant limitation within the survival data is that each patient’s treatment cannot be compared or normalized for as there is a significant amount of variation.
In summary, we have provided evidence that truncating mutations in SOX9 (particularly exon 3 truncating mutations) are recurrent in CRC and result in a truncated, overexpressed (in comparison to normal epithelium) protein that is likely oncogenic. These mutations are enriched in KRAS mutant, TP53 WT cases; and are associated with better overall survival.
materials and METHODS
Molecular testing
After approval by the local institutional review board, data from all patients with CRC being treated at Memorial Sloan Kettering Cancer Center between January 1, 2014 and December 31, 2015 who had molecular testing with Memorial Sloan Kettering- Integrated Molecular Profiling of Actionable Targets (MSK-IMPACT) was analyzed. MSK-IMPACT molecular testing is performed on MSKCC patients with CRC harboring distant metastases for KRAS, NRAS, and BRAF status analysis. This panel uses patient’s matched normal and tumor DNA to interrogate somatic mutations, structural variants, and copy number alterations in all coding regions and select introns of 410 cancer-related genes as previously described in full detail (see reference) [9]. Mutations in the following genes were recorded: KRAS (c. G12, G13, Q61, A59, K117, A146), NRAS (c. G12, G13, A59, Q61, K117, A146), BRAF (c. V600E, fusions), APC (all coding mutations), TP53 (all coding mutations), PIK3CA (all coding mutations) and SOX9 (all coding mutations). Microsatellite instability (MSI) was assessed with MSIsensor [10] on next generation sequencing (NGS) data for cases that did not have mismatch repair (MMR) immunohistochemical (IHC) analysis for MLH1, PMS2, MSH2, and MSH6 expression retention (MMR-P) or loss (MMR-D). Tumor purity was estimated by a combination of histologic assessment and mutation allele frequencies. Possible candidates with SOX9 loss of heterozygosity (LOH) included cases with mutant SOX9 allele frequencies that were higher than half the estimated tumor purity. Cases with suspected SOX9 LOH were studied for allele-specific copy number by Affymetrix Oncoscan arrays, provided they had sufficient remaining DNA.
Additionally, RNA transcription level data on SOX9 mutant and WT CRC from The Cancer Genome Atlas (TCGA) was reviewed via cbioportal [11].
Immunohistochemistry (IHC)
A cohort of 22 SOX9 WT and all available (n = 35) SOX9 mutant CRC underwent SOX9 IHC staining on whole sections of formalin fixed, paraffin embedded tissue. Immunohistochemical analysis for SOX9 expression was performed with the rabbit monoclonal SOX9 antibody, EPR14335-78 (Abcam, Cambridge, MA). Both the intensity of staining (0-3) and the percent of tumor cells staining (0-100%) were recorded for nuclear SOX9 IHC expression and used to generate a Histo (H) score that ranged from 0-300 [12].Immunohistochemical overexpression of SOX9 was interpreted relative to normal colorectal epithelium.
Clinical assessment
Electronic medical records were used to obtain data points including sex, age, tumor histology according to World Health Organization classification, primary site classified as either ‘proximal’ extending from cecum to distal transverse colon or ‘distal’ extending from splenic flexure to rectum, response to anti-EGFR therapy, time from metastasis to death were recorded.
Statistical analysis
Statistical analysis was performed using Fisher’s exact tests with two-tailed p values for categorical data, and unpaired Student’s t tests for comparison of continuous data (IHC H scores). P values less than 0.05 are mentioned. P values less than 0.01 were considered statistically significant to approximately account for multiple hypothesis testing. Kaplan-Meier curves were drawn and a log-rank p-value was computed to compare survival from time of metastasis based on SOX9 status for 317 patients who developed metastatic disease, including 285 SOX9 WT and 32 SOX9 mutant patients.
Acknowledgments
This study was funded by the Functional Genomics Initiative at Memorial Sloan Kettering Cancer Center, and by the National Cancer Institute (NCI) under the MSK Cancer Center Support Grant/Core Grant (P30 CA008748).
ConflictS of Interest
There is no conflict of interest.
REFERENCES
1. Matsushita M, Kitoh H, Kaneko H, Mishima K, Kadono I, Ishiguro N, Nishimura G. A novel SOX9 H169Q mutation in a family with overlapping phenotype of mild campomelic dysplasia and small patella syndrome. Am J Med Genet. 2013; 161A: 2528-34. doi: 10.1002/ajmg.a.36134.
2. Mattos EP, Sanseverino MT, Magalhães JA, Leite JC, Félix TM, Todeschini LA, Cavalcanti DP, Schuler-Faccini L. Clinical and molecular characterization of a Brazilian cohort of campomelic dysplasia patients, and identification of seven new SOX9 mutations. Genet Mol Biol. 2015; 38: 14-20. doi: 10.1590/S1415-475738120140147.
3. Wang HY, Lian P, Zheng PS. SOX9, a potential tumor suppressor in cervical cancer, transactivates p21WAF1/CIP1 and suppresses cervical tumor growth. Oncotarget. 2015; 6: 20711-22. doi: 10.18632/oncotarget.4133.
4. Drivdahl R, Haugk KH, Sprenger CC, Nelson PS, Tennant MK, Plymate SR. Suppression of growth and tumorigenicity in the prostate tumor cell line M12 by overexpression of the transcription factor SOX9. Oncogene. 2004; 23: 4584-93. doi: 10.1038/sj.onc.1207603.
5. Darido C, Buchert M, Pannequin J, Bastide P, Zalzali H, Mantamadiotis T, Bourgaux JF, Garambois V, Jay P, Blache P, Joubert D, Hollande F. Defective claudin-7 regulation by Tcf-4 and Sox-9 disrupts the polarity and increases the tumorigenicity of colorectal cancer cells. Cancer Res. 2008; 68: 4258-68. doi: 10.1158/0008-5472.CAN-07-5805.
6. Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, Canamero M, Bujanda L, Schedl A, Cheah KS, Skotheim RI, Lothe RA, López de Munain A, et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012; 72: 1301-15. doi: 10.1158/0008-5472.CAN-11-3660.
7. Lü B, Fang Y, Xu J, Wang L, Xu F, Xu E, Huang Q, Lai M. Analysis of SOX9 expression in colorectal cancer. Am J Clin Pathol. 2008; 130: 897-904. doi: 10.1309/AJCPW1W8GJBQGCNI.
8. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487: 330-7. doi: 10.1038/nature11252.
9. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’ Reilly C, Sadowska J, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015; 17: 251-64. doi: 10.1016/j.jmoldx.2014.12.006.
10. Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, Wendl MC, Ding L. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014; 30:1015-6. doi: 10.1093/bioinformatics/btt755.
11. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2: 401-4. doi: 10.1158/2159-8290.CD-12-0095.
12. Thike AA, Chng MJ, Fook-Chong S, Tan PH. Immunohistochemical expression of hormone receptors in invasive breast carcinoma: correlation of results of H-score with pathological parameters. Pathology. 2001; 33: 21-5. doi: 10.1080/00313020123290.
13. Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, Kulozik AE. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA. 2011; 17: 843-54. doi: 10.1261/rna.2401811.
14. Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002; 295: 2258-61. doi: 10.1126/science.1067338.
15. Abdel-Samad R, Zalzali H, Rammah C, Giraud J, Naudin C, Dupasquier S, Poulat F, Boizet-Bonhoure B, Lumbroso S, Mouzat K, Bonnans C, Pignodel C, Raynaud P, et al. MiniSOX9, a dominant-negative variant in colon cancer cells. Oncogene. 2011; 30: 2493-503. doi: 10.1038/onc.2010.621.
16. Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN, Clevers H, Jay P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol. 2004; 166: 37-47. doi: 10.1083/jcb.200311021.
17. Jay P, Berta P, Blache P. Expression of the carcinoembryonic antigen gene is inhibited by SOX9 in human colon carcinoma cells. Cancer Res. 2005; 65: 2193-8. doi: 10.1158/0008-5472.CAN-04-1484.
18. Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, Blache P, Jay P. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J Cell Biol. 2007; 178: 635-48. doi: 10.1083/jcb.200704152.
19. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58: 5248-57.