
INTRODUCTION
Long noncoding RNAs (lncRNA) are non-protein coding transcripts of more than 200 nucleotides [1]. In recent years, reports of dysregulated lncRNA expression in numerous tumor types suggest that lncRNAs may act as potential oncogenic or tumor-suppressive RNAs [2]. The X inactivate-specific transcript (Xist), a 19 kb lncRNA (17 kb in mouse) from the inactive X-chromosome (Xi), is required for X-chromosome inactivation (XCI) in female cells [3]. However, the role of Xist as an oncogenic or a tumor-suppressive lncRNA remains largely unclear [4]. Xist loss in hematopoietic stem cells was shown to result in the development of female-specific highly aggressive myelodysplastic syndrome (MDS) and myeloproliferative neoplasm (MPN), suggesting a tumor suppressor role for Xist [5]. In contrast, a recent report indicated an oncogenic role of Xist in glioblastoma stem cells by evidence that Xist expression was elevated in glioma tissues and that Xist knockdown reduced cell proliferation, migration and invasion in glioblastoma stem cells [6]. Interestingly, Xist expression was reduced in some breast cancer cell lines compared to normal cell lines [7]. Very little is known, however, about whether Xist plays an oncogenic or a tumor suppressive role in human breast cancer.
Xist expression is positively and negatively regulated by noncoding Jpx RNA and antisense Tsix RNA, respectively. Jpx RNA could bind CTCF protein and titrate out the repression effect of CTCF on Xist promoter [8]. Conversely, Tsix RNA could facilitate PRDM14 binding to Xist intron 1 to suppress its expression [9]. In addition, pluripotency factors such as OCT4, SOX2, REX1 and NANOG could bind to Xist intron 1 for transcriptional repression, while these factors also activate Tsix expression [10]. Currently, the regulation of decreased Xist levels in breast cancer cells has not been fully elucidated.
Xist plays a crucial role in XCI processes, initially binding to the inactive X-chromosome in cis inducing a cascade of events, including XCI establishment (such as euchromatin mark removal and RNA polymerase II exclusion) and XCI maintenance (such as repressive chromatin modification formation and DNA methylation) [11]. The recruitment of numerous repressive complexes by Xist, such as polycomb repressive complex 2 (PRC2) and DNA-methyltransferase, are required for the XCI maintenance [8]. Two research groups have recently implicated Xist RNA interactors, including SPEN/SHARP, in XCI establishment [12, 13]. The SPEN/SHARP protein directly interacts with the SMRT co-repressor, leading to the recruitment of HDAC3 and further activation of HDAC3 activity in removing euchromatin marks and excluding RNA polymerase II across the X chromosome [12, 14]. While Xist plays an important role in XCI processes, it remains to be elucidated whether decreased Xist-regulated expression of genes, other than in the X chromosome, are associated with breast cancer.
Here, we demonstrate Xist acts as a tumor-suppressor lncRNA in breast cancer cells by decreasing AKT phosphorylation. Expression of Jpx and Xist were downregulated in breast tumor. Knockdown of either Xist or SPEN expression in breast cancer cells suppressed the expression of PHLPP1, a phosphatase in AKT dephosphorylation [15], and was correlated with increased HDAC3 recruitment to the PHLPP1 promoter. Our findings provide a previously undescribed molecular basis of Xist in suppressing the AKT pathway in breast cancer.
RESULTS
Xist expression is significantly reduced in breast cancer cell lines and breast cancer samples
We investigated Xist expression in breast cancer using public data sets. Analysis of microarray data sets (GSE5764, GSE5460 and GSE14017) found Xist was significantly reduced in tumor and metastasis samples, compared to normal tissue, and inversely correlated with a positive control VEGFA expression profile (Figure 1A). Analysis of RNA-sequencing data sets from The Cancer Genome Atlas (TCGA) showed similar results (Figure 1B), suggesting Xist expression was downregulated in breast tumor and its expression in both tumor and metastasis samples was similar. We further examined Xist levels in non-tumorigenic (M10 and MCF10A), tumorigenic (MCF7 and MDA-MB-468), and metastatic (MDA-MB-231 and Hs578T) breast cell lines by quantitative RT-PCR analysis. Interestingly, Xist levels in MDA-MB-468, MDA-MB-231 and Hs578T cells were undetectable, while MCF7 cells expressed very low Xist levels, compared to M10 and MCF10A cells (Figure 1C). These results inversely correlated with a positive control VEGFA (Figure 1C). Our results suggested that Xist expression was decreased in breast tumor samples and breast cancer cell lines.
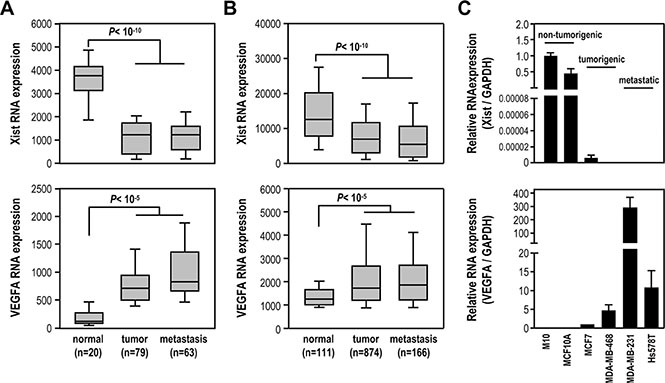
Figure 1: Xist expression is significantly reduced in breast cancer cell lines and breast cancer samples. (A–B) Analysis of Xist and VEGFA expression levels in breast normal and tumor samples using the GEO and the TCGA data sets. (C) Quantification RT-PCR of Xist and VEGFA expression in non-tumorigenic breast cell lines and breast cancer cell lines (n = 3).
Xist knockdown increases cell viability via AKT activation
Our findings of Xist expression in association with breast cancer led us to test the effect of Xist on cell viability, using knockdown and overexpression of Xist in M10 and MCF7 cells, respectively. Depletion of Xist in M10 cells resulted in increased cell viability (Figure 2A, right panel). In contrast, Xist overexpression in MCF7 cells reduced cell viability (Figure 2B, right panel). These results suggested downregulation of Xist expression promoted breast cancer cell viability.
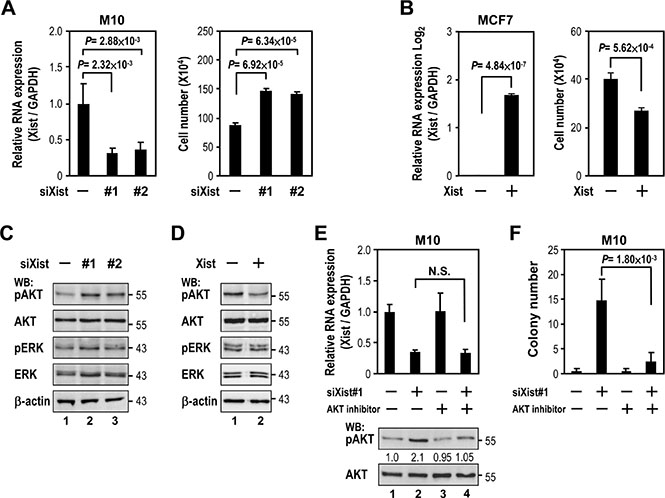
Figure 2: Xist downregulation increases cell viability via AKT activation. (A) M10 cells were transfected with 10 nM specific siRNA oligonucleotides against Xist using RNAiMAX then subjected to quantitative RT-PCR (left panel) and viability assay (right panel). (B) MCF7 cells were transfected with Xist plasmid using Lipofectamine 3000 then subjected to quantitative RT-PCR (left panel) and viability assay (right panel). (C–D) Lysates from transfected M10 and MCF7 cells were immunoblotted with indicated antibodies. (E) Quantification RT-PCR and Western blotting of lysates from M10 cells transfected with Xist siRNA followed by AKT inhibitor treatment (250 nM, 2 days). Phospho- and non-phospho- AKT band intensity was quantified by densitometry. The ratio of phospho- to non-phospho protein is indicated after normalization to mock transfection and treatment samples. (F) Quantitative colony formation analysis of M10 cells transfected with Xist siRNAs and treated with or without 250 nM AKT inhibitor. Error bars: mean ± SD, n = 3.
We next explored the cellular pathway(s) of cell viability regulated by Xist. Since both RAS/ERK and PI3K/AKT pathways are the most frequently dysregulated signaling pathways associated with breast cancer cell growth and viability [16], we tested whether alteration of Xist levels modulated both pathways in breast cells. Xist knockdown by siXist increased phospho-AKT (pAKT) levels in M10 cells (Figure 2C). Conversely, Xist overexpression decreased phospho-AKT in MCF7 cells (Figure 2D). Notably, phospho-ERK was not altered by either Xist knockdown or overexpression (Figure 2C and 2D). These results suggested a role for the AKT pathway in cell viability regulated by Xist.
We reasoned that if AKT activation is involved in the upregulation of cell viability by Xist knockdown, inhibition of AKT activity should attenuate the effect of Xist knockdown on cell viability. Treatment of AKT inhibitor decreased Xist knockdown-elicited AKT phosphorylation in M10 cells (Figure 2E, lanes 2 and 4). Of note, AKT inhibitor treatment did not alter Xist expression levels (left panel, lanes 2 and 4). Under such treatment, cell viability of M10 cells upregulated by Xist knockdown was also attenuated (Figure 2F, lanes 2 and 4). These data suggested that AKT activation mediated Xist knockdown-elicited cell viability regulation.
Negative correlation between Xist and phospho-AKT levels in clinical breast cancer
Our finding that Xist negatively regulated pAKT levels in breast cell lines led us to examine whether an inverse correlation between Xist and pAKT occurred in clinical breast samples. As expected, immunohistochemistry (IHC) and RNA in situ hybridization analyses demonstrated that pAKT levels were higher in Xist-negative breast cancer tissue, while Xist-positive samples yielded lower pAKT levels (Figure 3A), suggesting an inverse correlation between Xist and pAKT levels. These data were supported by analysis of additional breast cancer samples (Figure 3B). These results, in conjunction with the above-mentioned cell line studies, suggested that decreased Xist levels resulted in an increase of pAKT levels in breast tumor.
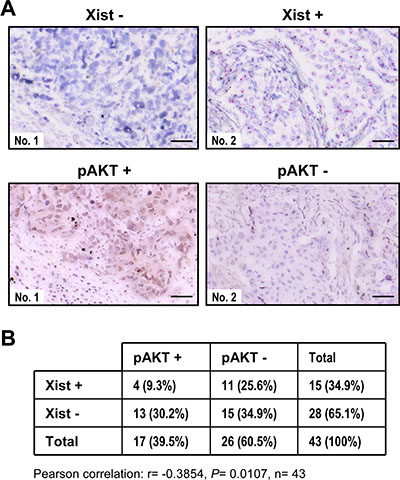
Figure 3: Inverse correlation between Xist and phospho-AKT in clinical breast samples. (A) Immunohistochemistry to detect phospho-AKT (pAKT) (brown) and RNA in situ hybridization to visualize Xist (red) in serial sections of each breast cancer sample. Scale bar = 50 um. (B) Regression analysis of the correlation of Xist and pAKT levels.
Association of Jpx in regulation of Xist expression in breast cancer
We next investigated whether the reduction of Xist expression in breast cancer could be derived from genomic deletion or epigenetic alteration. Xist genomic alteration in breast tumors was analyzed using GEO data sets (GSE26232). We did not observe copy number loss occurring in or near Xist locus, including Jpx and Txis loci (Supplementary Figure S1). These data excluded the possibility of Xist locus deletion in breast cancer.
Previous studies showed that Xist expression was positively controlled by Jpx lncRNA [17] and negatively regulated by Tsix lncRNA and transcription factors, such as PRDM14, OCT4, SOX2, REX1 and NANOG [10]. Bioinformatics analyses was used to test whether these lncRNAs and transcription factors regulated Xist in clinical breast samples. Analysis of GEO data sets revealed that Jpx expression was also reduced in breast tumor and metastatic tissue (Figure 4A, upper panel), suggesting possible Jpx regulation of Xist expression. Tsix expression was also decreased in breast tumor, excluding the possibility of Tsix in regulating Xist expression (Supplementary Figure S2A). Furthermore, the expression of OCT4, but not PRDM14, SOX2, REX1 or NANOG was higher in breast tumor and metastasis samples (Figure 4A, bottom panel, and Supplementary Figure S2B–S2E), suggesting negative regulation of Xist by OCT4.
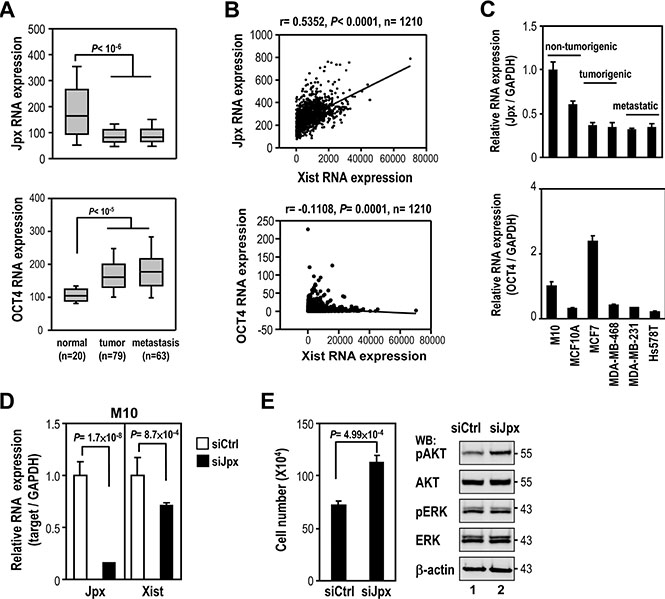
Figure 4: Association of Jpx in regulation of Xist expression in breast cancer. (A) Coexpression analysis of Jpx and OCT4 levels in breast normal and tumor samples using GEO data sets. (B) Coexpression analysis of Xist with Jpx or OCT4 in clinical breast samples using TCGA data sets. (C) Quantitative RT-PCR of Jpx and OCT4 expression in non-tumorigenic breast cell lines and breast cancer cell lines. (D–E) M10 cells were transfected with 10 nM specific siRNA oligonucleotides against Jpx using RNAiMAX then subjected to quantitative RT-PCR, viability assay and Western blot. Error bars: mean ± SD, n = 3.
We further analyzed whether Xist expression was associated with the expression of Jpx or OCT4 in breast cancer by TCGA data set. Interestingly, Xist and Jpx expression showed a robust positive correlation, while a negative correlation between Xist and OCT4 expression was relatively weak (Figure 4B). Accordingly, we found reduced Jpx expression in breast cancer cell lines, compared to non-tumorigenic cells (Figure 4C), correlating with Xist expression in these cell lines. Notably, OCT4 expression was elevated only in MCF7, but not in MDA-MB-468, MDA-MB-231 and Hs578T cell lines. These results implicated Jpx control of Xist expression in breast cancer.
Because Jpx positively modulates Xist expression [18], Jpx knockdown should reduce the negative effect of Xist on cell viability and AKT phosphorylation. As expected, treatment of Jpx siRNA reduced Xist expression in M10 cells (Figure 4D), correlating with increased levels of cell number and phospho-AKT (Figure 4E). These data suggest that reduced Jpx expression enhanced cell viability and AKT phosphorylation through downregulation of Xist in breast cancer.
Xist knockdown-elicited AKT phosphorylation is via transrepression of PHLPP1 expression by HDAC3
Although Xist reduction is associated with increased pAKT in breast cancer, the molecular mechanism for such regulation is unknown. A recent report suggested Xist repressed gene expression on X-chromosome in female cells via recruitment of HDAC3 [12]. We questioned whether HDAC3 was involved in Xist-regulated AKT activation. Depletion of HDAC3 in M10 cells decreased the effect of Xist knockdown on pAKT induction (Figure 5A, lane 4 versus lane 2), suggesting HDAC3 mediated Xist knockdown-elicited AKT activation.
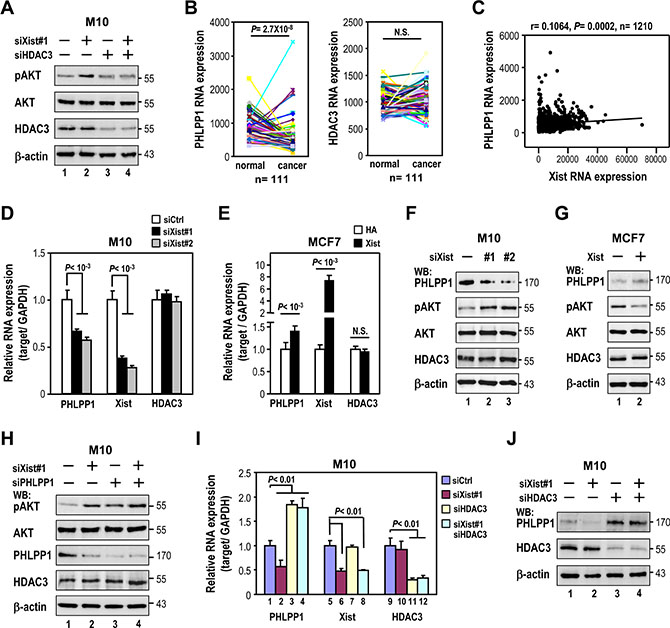
Figure 5: Xist knockdown-elicited AKT phosphorylation is via transrepression of PHLPP1 expression by HDAC3. (A) Western analysis of M10 cells transfected with Xist siRNA and/or HDAC3 siRNA with indicated antibodies. (B) Analysis of PHLPP1 and HDAC3 expression by RNA-seq from 111 pairs of matched normal breast and breast cancer tissues using TCGA data sets. (C) Coexpression analysis Xist and PHLPP1 in clinical breast samples using TCGA data sets. (D and F) M10 cells were transfected with 10 nM specific siRNA oligonucleotides against Xist using RNAiMAX then subjected to quantitative RT-PCR analysis (D) and Western blotting (F). (E and G) MCF7 cells were transfected with Xist plasmid using Lipofectamine 3000 then subjected to quantitative RT-PCR analysis (E) and immunoblotting (G). (H–J). M10 cells were transfected with Xist siRNA and/or HDAC3 or PHLPP1 siRNA using RNAiMAX, then subjected to quantitative RT-PCR and Western analyses. Error bars: mean ± SD, n = 3.
We further investigated the role of HDAC3 in the regulation of AKT activation. Recent research showed HDAC3 knockout increased the expression of the PH domain and leucine-rich repeat phosphatase 1 (PHLPP1) in chondrocytes to suppress AKT phosphorylation [19]. Interestingly, our TCGA data set analysis showed expression levels of PHLPP1, but not HDAC3, were decreased in breast cancer tissues compared to adjacent normal tissues (Figure 5B). Furthermore, PHLPP1 expression was positively associated with Xist expression (Figure 5C). These findings led us to test whether Xist expression could modulate PHLPP1 expression. Depletion of Xist in M10 cells decreased PHLPP1 expression levels (Figure 5D), while overexpression of Xist in MCF7 cells increased it (Figure 5E). Notably, neither knockdown nor overexpression of Xist altered HDAC3 expression (Figure 5D and 5E). We further confirmed the effect of Xist on PHLPP1 expression by Western analysis (Figure 5F and 5G). More importantly, the effect of the Xist knockdown on the increase in pAKT levels was significantly reduced in PHLPP1-depleted cells (Figure 5H, lanes 3 and 4 versus lanes 1 and 2). These results suggested Xist-elicited induction of phospho-AKT levels occurred via downregulation of PHLPP1 expression.
If HDAC3 is involved in the downregulation of PHLPP1 expression by Xist knockdown, depletion of HDAC3 levels should attenuate the negative effect of Xist knockdown on PHLPP1 expression. As expected, silencing of HDAC3 blocked the reduction of the PHLPP1 expression in Xist-depleted cells (Figure 5I and 5J, lanes 3 and 4 versus lanes 1 and 2) These results suggested that Xist knockdown-elicited downregulation of the PHLPP1 expression was HDAC3-dependent.
Depletion of Xist or SPEN increases HDAC3 recruitment to PHLPP1 promoter
Because HDAC3 levels were not altered by Xist expression, we hypothesized that HDAC3 recruitment to repress PHLPP1 promoter activity would be altered by Xist. We examined the effect of Xist on HDAC3 recruitment to the PHLPP1 promoter by chromatin immunoprecipitation (ChIP). Notably, HDAC3 recruitment to the PHLPP1 promoter region was increased in Xist-depleted M10 cells (Figure 6A), and correlated with a decreased level of histone 3 acetylation (H3ac) in this promoter region. These results suggested that Xist negatively regulated HDAC3 targeting to the PHLPP1 promoter.
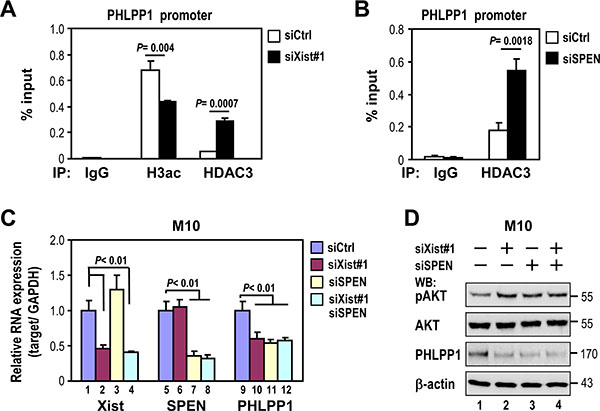
Figure 6: Depletion of Xist or SPEN increases HDAC3 recruitment to the PHLPP1 promoter and decreases PHLPP1 expression. (A and B) M10 cells were transfected with 10 nM specific siRNA oligonucleotides against Xist or SPEN using RNAiMAX then subjected to chromatin immunoprecipitation (ChIP). ChIP analyses were performed with antibodies against HDAC3, acetylated histone H3 (H3-ac) or an IgG control. Subsequent qPCR analysis was carried out by primers specific for PHLPP1 promoter. Input represents 1% of the chromatin used for immunoprecipitation. (C and D) M10 cells were transfected with Xist siRNA and/or SPEN siRNA using RNAiMAX then subjected to quantitative RT-PCR (C) and Western analyses (D). Error bars: mean ± SD, n = 3.
Recent research showed that Xist indirectly recruited HDAC3 via SPEN for XCI establishment [12]. This finding led us investigate whether Xist sequesters HDAC3 from the PHLPP1 promoter via SPEN. CHIP analysis was used to determine if SPEN knockdown also altered HDAC3 recruitment to the PHLPP1 promoter. Similar to Xist, SPEN knockdown increased the level of HDAC3 recruitment to the PHLPP1 promoter region in M10 cells (Figure 6B). These results suggested that both Xist and SPEN retained HDAC3 in X-chromosome, thereby reducing HDAC3 recruitment to the PHLPP1 promoter region.
If SPEN, like Xist, is involved in sequestering HDAC3 from the PHLPP1 promoter region, depletion of SPEN levels should attenuate the expression of PHLPP1. As expected, SPEN knockdown decreased PHLPP1 expression (Figure 6C, lane 11 and Figure 6D, 3rd panel, lane 3), which also correlated with an increase of pAKT (Figure 6D, top panel, lane 3). Notably, SPEN knockdown blocked the effect of Xist knockdown on both PHLPP1 downregulation and pAKT upregulation (Figure 6C, lanes 11 and 12 versus lanes 9 and 10; Figure 6D, lanes 3 and 4 versus lanes 1 and 2). These results provided additional support that Xist positively regulates PHLPP1 expression via sequestering HDAC3 from the PHLPP1 promoter.
DISCUSSION
The molecular basis of Xist as a tumor suppressor lncRNA is largely unclear. In the present study, we demonstrate an inverse relationship of Xist and phospho-AKT levels in breast cancer tissues. More importantly, Xist knockdown-mediated upregulation of AKT phosphorylation occurred via suppression of PHLPP1 expression by HDAC3. Our findings suggest that Xist functions as a tumor suppressor through inhibition of AKT activation in breast cancer. A recent study reported that conditional deletion of Xist from HSCs in female mice lead to the development of highly aggressive MDS/MPN [5]. AKT was also prominently activated in MDS/MPN [20]. Our findings of Xist-enhanced PHLPP1 expression in decreasing AKT phosphorylation may also provide a possible molecular basis for Xist loss-associated MDS/MPN. Indeed, PHLPP1 expression was decreased in the microarray analysis of Xist-deficient HSCs in mice [5]. Furthermore, analysis of GEO data set also revealed decreased levels of PHLPP1 in MDS patients, correlating with reduced Xist levels (data not shown). Our results that Xist negatively modulated AKT activation via PHLPP1 regulation suggest a tumor suppressor role of Xist in breast cancer and MDS/MPN.
The loss of Xist expression could induce the reactivation of X-linked genes, which may contribute to breast cancer progression. Recent studies identified X-linked genes in X chromosome reactivation, including HDAC8 and TBL1X [7]. HDAC8 was suggested to activate breast cancer stem cell-like properties and increase cell invasion [21, 22]. Although X-linked gene reactivation might contribute to breast cancer progression [23], our finding of PHLPP1 expression regulated by Xist levels in downregulating AKT phosphorylation provides evidence that non-X-linked gene expression regulated by Xist may also play an important role in breast cancer progression. Currently, the mechanism of Xist regulation of PHLPP1 remains largely unclear. While Xist could indirectly recruit HDAC3 via SPEN/SHARP and SMRT proteins for XCI establishment [12, 13], our findings that knockdown of either Xist or SPEN increased HDAC3 recruitment to the PHLPP1 promoter region for gene suppression led us to hypothesize that Xist reduction may release HDAC3 protein from inactive X-chromosome, thereby increasing the HDAC3 pool for PHLPP1 promoter targeting. In this scenario, Xist could function as an lncRNA decoy to titrate HDAC3 from the PHLPP1 promoter. A similar scenario of lncRNA decoys, such as PANDA, has been well documented [24]. PANDA was shown to inhibit apoptotic gene expression through sequestering transcription factor NF-YA, preventing NF-YA’s transactivation of apoptotic program upon DNA damage [25].
While we demonstrated that Xist expression in breast cancer was associated with Jpx expression, very little is known about Jpx expression downregulation in breast cancer. Two possible scenarios could be considered: A recent report showed that Jpx and Xist promoters were fully hypermethylated in breast cancer cell lines [26]. Thus, one possible scenario is that reduction of both Jpx and Xist expression in breast cancer may resulted from hypermethylation. Research also showed that chromosome conformation at the Tsix topologically associating domain could regulate Tsix transcriptional status [27]. In this scenario, decreased Jpx expression in breast cancer may be due to altered chromosome conformation. These two possibilities are not mutually exclusive. Further studies are required to elucidate Jpx downregulation in breast cancer.
In summary, our data suggest a tumor suppressor role of Xist in inhibiting AKT activation via regulation of non-X-chromosome gene PHLPP1 expression. In addition to the molecular basis of Xist in modulating AKT activation, these findings of decreased Xist levels and increased AKT phosphorylation may potentially be considered as markers for breast cancer diagnosis and therapeutic intervention.
MATERIALS AND METHODS
Analysis of public data sets
Affymetrix HG U133 plus 2.0 arrays data sets (GSE5764, GSE5460 and GSE14017) and Affymetrix Genome-Wide Human SNP 6.0 array data sets (GSE26232) were downloaded from NCBI GEO [28–30]. Expression data derived from downloaded CEL files were normalized using the invariant set and converted into intensity using dChip (www.dchip.org) software [31]. Processed RNA-sequencing data of TCGA breast invasive carcinoma (BRCA) dataset were obtained from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/) [32].
Quantitative RT-PCR (RT-qPCR)
Total RNA was extracted as previously described [33]. Total RNA (5 μg/sample) was reverse-transcribed using Maxima H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, Vilnius, Lithuania). Quantitative real-time PCR was performed using Luminaris Color HiGreen Low ROX qPCR Master Mix (Thermo Scientific, Vilnius, Lithuania) and an ABI 7500 sequence detection system (Life Technologies, California, USA). Purified RNA was subjected to RT-qPCR, using specific primers: Xist (NR_001564): 5′- GCATAACTCGGCTTAGGGCT-3' and 5′-TCCTCTGCCTGACCTGCTAT-3′, VEGFA (NM_001 025366): 5′- CGCAAGAAATCCCGGTATAA -3′and 5′- TCT CCGCTCTGAGCAAGG-3′, GAPDH (NM_002046): 5′- TCTTTTGCGTC GCCAGCCGAG -3′and 5′- TGA CCAGGCGCCCAATACGAC -3′, Jpx (NR_024582): 5′-AG ACTTAAGATGGCGGCGTT-3′and 5′-TGGACTCAT AC TTCGGACGC-3′, and Tsix (NR_003255): 5′-GTGTAGG TGGTTCCCC AAGG-3′ and 5′-AATGACTGACCACTG CTGGG-3′, OCT4 (NM_002701): 5′- TGCAGCAGATCAG CCACAT-3′ and 5′-CTCGGACCACATCCTTCTCG-3′, HDAC3 (NM_003883): 5′- GGCCTATTTCTACGACCCCG -3′ and 5′-TGGTATGGCTTGAAGACGATCA-3′, PHLPP1 (NM_194449): 5′-ACTCCAACTGCATCGAGGTC-3′ and 5′-AGCTCAG GTCCACACACTTG-3′, SPEN (NM_015 001): 5′-GGAGACTTGATGGGGCTTCA-3′ and 5′-GAT CAA ATCCTCCCGTCCCC-3′.
Cell culture, plasmids, RNA interference and transfection
All breast cancer cell lines were cultured in specific medium with 10% fetal bovine serum (FBS). MCF-7, MDA-MB-468, Hs578T cells were cultured in DMEM; MDA-MB-231 and M10 cells in RPMI and MEM medium, respectively, and MCF10A cells in DMEM/F12 with 10 μg/ml insulin, 20 ng/ml EGF, 100 ng/ml choleratoxin, 0.5 μg/ml hydrocortisone and 5% horse serum. Xist plasmid pCMV-Xist-PA was a gift from Rudolf Jaenisch (Addgene plasmid # 26760) [34]. Specific siRNA oligonucleotides against Xist, Jpx, HDAC3 and negative control were synthesized by Life Technologies. The siRNAs sequences were: siXist#1: GUAUCCUAUUUGCACGCUAtt, siXist#2: GCCCUU CUCUUCGAACUGUtt, siJpx: GAAAAGGAAUAAA AUCAAAtt, siHDAC3: CCAAGAGUCUUAAUGCCU Utt, and siSPEN: CACGCAGAGUUACCGAAAAtt. Transfections were performed using RNAiMAX or Lipofectamine 3000 (Life Technologies).
Cell viability analysis and colony formation assay
Viability of cells transfected with indicated siRNA or plasmid constructs was determined by trypan blue dye exclusion. Colony formation assay was performed with M10 cells transfected with siXist#1 and pTK-hyg, and treated with or without 250 nM AKT inhibitor (A6730, Sigma, St. Louis, USA), further selected with hygromycin (Life Technologies) for 10 days followed by crystal violet staining.
Protein extraction and Western blot analysis
Total protein was extracted as previously described [35]. Briefly, samples were loaded onto SDS-PAGE gels (8~10%, 40 μg/lane), transferred onto Protran Transfer Membrane (NBA085C001EA, PerkinElmer, Boston, USA), probed with antibodies against: β-actin (A5441, Sigma), PHLPP1 (ab71972, Abcam, Cambridge, UK), HDAC3 (17–1 0238, Millipore, Temecula, USA), acetyl-histone H3 (06–5 99, Millipore), phosphorylated-ERK (9101, Cell Signaling Technology), ERK (4695, Cell Signaling Technology), phosphorylated-AKT (4060, Cell Signaling Technology) and AKT (4691, Cell Signaling Technology), and analyzed with a Las-4000 imaging system (Fujifilm, Valhalla, USA).
Immunohistochemistry (IHC) analysis and RNA in-situ hybridization (ISH)
Tissue array slides were purchased from SUPER BIO CHIPS, Seoul, Korea with Institute Review Board approval. Sections were deparaffinized and rehydrated with xylene and a series grade of alcohol. Epitope retrieval was carried out in a 10 mM citrate buffer (pH 6.0, 15 min, 99ºC) using a hot plate, followed by inactivation of endogenous peroxidase with H2O2 and incubation with phosphorylated-AKT (4060, Cell Signaling Technology, 1 h, room temperature). Immunostaining was performed according to standard procedures. Slides were stained with Vectastain Elite ABC kit (Vector Lab, Burlingame, CA), developed using DAB (brown precipitate, Vector Lab), and visualized by light microscopy (Olympus, Allentown, USA). ISH was performed as previously described [36]. Sections were hybridized with custom-designed QuantiGene ViewRNA probes against Xist. Bound probes were amplified using PreAmp and Amp molecules (all reagents from Affymetrix, Santa Clara, USA). Multiple Label Probe oligonucleotides conjugated to alkaline phosphatase were added and Fast Red Substrate was used to produce signal (Cy3). Images of histological samples were digitalized using Mirax Scan. Samples were grouped into four categories based on staining intensity: none (0), weak (+1), medium (+2) and strong (+3); all samples negative (stain intensity 0 and +1) and positive (stain intensity +2 and +3) were included in the analysis.
Chromatin immunoprecipitation quantitative PCR (ChIP-qPCR) assay
ChIP was performed as previously described [33]. ChIP product was analyzed by quantitative real-time PCR using the Applied Biosystem 7500 Real-Time PCR System. Experiments were done in triplicate. A fraction (1%) of the sonicated chromatin was set aside as input control before antibody affinity manipulations. Percent input was calculated by 100 × 2^(Ctadjusted Input - CtIP). Purified DNA was subjected to qPCR, using specific primers for mouse PHLPP1 promoter: 5′-AAGCTGGAGTGCGGGTAAAA-3′ and 5′- CCTCTGCCGTTGAACTCTGT-3′.
Statistical analysis
Analyses were performed with SigmaPlot software. Significance (p ≤ 0.05) was ascertained using a Student’s t-Test.
ACKNOWLEDGMENTS
We thank Dr. Chen-Yang Shen (Institute of Biomedical Sciences, Academia Sinica) for M10 and MDA-MB-231 cells, Dr. Wen-Hwa Lee (Genomics Research Center, Academia Sinica) for MCF10A cells, and Taiwan Mouse Clinic funded by the Ministry of Science and Technology (MOST) of Taiwan for technical support in slide scanning experiment. We are also grateful to Yen-Hsieh Chen and RNA Program in Academia Sinica for comments on the paper.
CONFLICTS OF INTEREST
The authors have declared no conflicts of interest.
GRANT SUPPORT
This work was supported by grant 104-2321-B-001–0 01 from the Ministry of Science and Technology (MOST), Taiwan to HMS.
REFERENCES
1. Pandey GK, Kanduri C. Long noncoding RNAs and neuroblastoma. Oncotarget. 2015; 6:18265–18275. doi: 10.18632/oncotarget.4251.
2. Huarte M, Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet. 2010; 19:R152–161.
3. Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012; 338:1435–1439.
4. Weakley SM, Wang H, Yao Q, Chen C. Expression and function of a large non-coding RNA gene XIST in human cancer. World journal of surgery. 2011; 35:1751–1756.
5. Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, Lee JT. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013; 152:727–742.
6. Yao Y, Ma J, Xue Y, Wang P, Li Z, Liu J, Chen L, Xi Z, Teng H, Wang Z, Li Z, Liu Y. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer letters. 2015; 359:75–86.
7. Chaligne R, Popova T, Mendoza-Parra MA, Saleem MA, Gentien D, Ban K, Piolot T, Leroy O, Mariani O, Gronemeyer H, Vincent-Salomon A, Stern MH, Heard E, et al. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res. 2015; 25:488–503.
8. Galupa R, Heard E. X-chromosome inactivation: new insights into cis and trans regulation. Curr Opin Genet Dev. 2015; 31:57–66.
9. Payer B, Rosenberg M, Yamaji M, Yabuta Y, Koyanagi-Aoi M, Hayashi K, Yamanaka S, Saitou M, Lee JT. Tsix RNA and the germline factor, PRDM14, link X reactivation and stem cell reprogramming. Mol Cell. 2013; 52:805–818.
10. Navarro P, Oldfield A, Legoupi J, Festuccia N, Dubois A, Attia M, Schoorlemmer J, Rougeulle C, Chambers I, Avner P. Molecular coupling of Tsix regulation and pluripotency. Nature. 2010; 468:457–460.
11. Escamilla-Del-Arenal M, da Rocha ST, Heard E. Evolutionary diversity and developmental regulation of X-chromosome inactivation. Human genetics. 2011; 130:307–327.
12. McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015; 521:232–236.
13. Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. Systematic discovery of Xist RNA binding proteins. Cell. 2015; 161:404–416.
14. You SH, Lim HW, Sun Z, Broache M, Won KJ, Lazar MA. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat Struct Mol Biol. 2013; 20:182–187.
15. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005; 18:13–24.
16. Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, Dancey JE, Piccart-Gebhart MJ. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer treatment reviews. 2013; 39:935–946.
17. Froberg JE, Yang L, Lee JT. Guided by RNAs: X-inactivation as a model for lncRNA function. J Mol Biol. 2013; 425:3698–3706.
18. Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, Lee JT. Jpx RNA activates Xist by evicting CTCF. Cell. 2013; 153:1537–1551.
19. Bradley EW, Carpio LR, Westendorf JJ. Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J Biol Chem. 2013; 288:9572–9582.
20. Khan I, Huang Z, Wen Q, Stankiewicz MJ, Gilles L, Goldenson B, Schultz R, Diebold L, Gurbuxani S, Finke CM, Lasho TL, Koppikar P, Pardanani A, et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia. 2013; 27:1882–1890.
21. Chao MW, Chu PC, Chuang HC, Shen FH, Chou CC, Hsu EC, Himmel LE, Huang HL, Tu HJ, Kulp SK, Teng CM, Chen CS. Non-epigenetic function of HDAC8 in regulating breast cancer stem cells by maintaining Notch1 protein stability. Oncotarget. 2016; 7:1796–1807. doi: 10.18632/oncotarget.6427.
22. Park SY, Jun JA, Jeong KJ, Heo HJ, Sohn JS, Lee HY, Park CG, Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncology reports. 2011; 25:1677–1681.
23. Chaligne R, Heard E. X-chromosome inactivation in development and cancer. FEBS Lett. 2014; 588:2514–2522.
24. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011; 43:904–914.
25. Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Wang Y, Kong B, Langerod A, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011; 43:621–629.
26. Lin IH, Chen DT, Chang YF, Lee YL, Su CH, Cheng C, Tsai YC, Ng SC, Chen HT, Lee MC, Chen HW, Suen SH, Chen YC, et al. Hierarchical clustering of breast cancer methylomes revealed differentially methylated and expressed breast cancer genes. PLoS One. 2015; 10:e0118453.
27. Giorgetti L, Galupa R, Nora EP, Piolot T, Lam F, Dekker J, Tiana G, Heard E. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell. 2014; 157:950–963.
28. Lu X, Lu X, Wang ZC, Iglehart JD, Zhang X, Richardson AL. Predicting features of breast cancer with gene expression patterns. Breast cancer research and treatment. 2008; 108:191–201.
29. Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009; 16:67–78.
30. Turashvili G, Bouchal J, Baumforth K, Wei W, Dziechciarkova M, Ehrmann J, Klein J, Fridman E, Skarda J, Srovnal J, Hajduch M, Murray P, Kolar Z. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer. 2007; 7:55.
31. Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001; 98:31–36.
32. Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
33. Huang YS, Chang CC, Huang TC, Hsieh YL, Shih HM. Daxx interacts with and modulates the activity of CREB. Cell Cycle. 2012; 11:99–108.
34. Wutz A, Jaenisch R. A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell. 2000; 5:695–705.
35. Chang CC, Naik MT, Huang YS, Jeng JC, Liao PH, Kuo HY, Ho CC, Hsieh YL, Lin CH, Huang NJ, Naik NM, Kung CC, Lin SY, et al. Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Mol Cell. 2011; 42:62–74.
36. Huang YS, Hsieh HY, Shih HM, Sytwu HK, Wu CC. Urinary Xist is a potential biomarker for membranous nephropathy. Biochem Biophys Res Commun. 2014; 452:415–421.