
INTRODUCTION
The BRCA1 protein is involved in many essential cellular processes that include DNA damage signaling and repair, cell-cycle control, protein ubiquitination, cell differentiation, and gene transcription regulation, all of which are associated with its tumor suppressor function [1, 2]. Women with heterozygous BRCA1 mutations have a greater risk of developing breast and ovarian cancer in their lifetime. BRCA1-related cancers generally occur in younger women, before the age of menopause, and are more aggressive than breast cancers that arise in the general population [3, 4]. Although sex and organ-specific penetrance of BRCA1-related cancers remains poorly understood, ovarian hormones have been implicated in early cell transformation events. Early menarche and late menopause were associated with an increased risk [5, 6]. Prophylactic salpingo-oophorectomy in women with BRCA1 mutations decreases breast cancer risk by 50% [7, 8], or more if the oophorectomy is performed before the age of 40 [9].
BRCA1 has been shown to play a role in the regulation of estrogen receptor (ER) and progesterone receptor (PR) signaling. Rosen et al. demonstrated that BRCA1 inhibited estradiol (E2)-dependent gene transcription [10, 11]. In addition, cross-talk between BRCA1 and ERα was revealed through BRCA1 enhancing transcription of ERα, while ERα in turn increased the transcription of BRCA1 [12, 13]. Similarly, physical interaction between BRCA1 and PR inhibited PR-dependent gene transcription and increased degradation of PR by the proteasome [14, 15]. Normal breast tissues from women with BRCA1 mutations did not have different levels of ERα expression compared to non-mutated BRCA1+/+ breast tissues [16]. However the expression of an ER-inducible gene involved in the migration of human breast cancer cells, the trefoil factor 1 (TTF1 or pS2), was decreased in BRCA1+/mut tissues [16, 17]. A decrease in expression for both isoforms of PR (PR-A and PR-B) was also observed in BRCA1+/mut tissues, with a ratio in favor of PR-A [16]. In addition, p53-/-/brca1f11/f11 mice that were treated with progesterone (P4) alone and in combination with E2 had enhanced mammary gland proliferation and developed mammary tumors [18]. Interestingly, these effects were reversed by mifepristone, a PR antagonist. These data, along with studies that report 80% of BRCA1-related tumors are negative for ER and PR expression [19, 20], suggest that alterations in hormone signaling contribute to early stages of breast cancer development in histologically normal BRCA1mut/+ cells.
Selective hormone receptor modulators are increasingly considered as preventive breast cancer treatments. Five years of selective ER modulator (SERM) therapy reduced the occurrence of breast cancer in high risk women by 50% [21-25]. Although there are strong implications of PR involvement in BRCA1-related breast carcinogenesis, the effect of selective PR modulators (SPRMs) on breast cancer prevention has not yet been evaluated in humans. Among the SPRMs, ulipristal acetate (UPA) was launched as a new generation emergency contraceptive pill and proposed as treatment for uterine fibroids symptoms [26, 27]. Wide use of UPA in the gynecological and clinical fields is due to its ability to efficiently inhibit PR signaling while reducing adverse effects, even with repeated use [28-30].
In this study we analyzed ER and PR expression and responses by immunohistochemistry (IHC) in normal breast tissues from women with heterozygous BRCA1 mutations (BRCA1mut/+ tissues) or from women without BRCA1 mutation (BRCA1+/+ tissues). Fresh tissues were also grafted in hormone-treated mice. We report findings that further support the involvement of ovarian hormones in BRCA1-related tumor development and support the use of SPRM treatment for breast cancer prevention.
RESULTS
Analysis of marker expression in control and BRCA1-mutated breast tissue
Expression of several markers was analyzed by IHC in histologically normal breast tissues from 28 women selected as controls (BRCA1+/+) and 22 women with BRCA1 mutations (BRCA1mut/+). Characteristics of patients bearing BRCA1 mutations are described in Table 1.
Table 1: Clinical features of patients with BRCA1 mutations
Case |
Age |
BRCA1 Mutation |
Salpingo-oophorectomy (age at surgery) |
Pregnancy & parity |
|---|---|---|---|---|
1 |
56 |
NA |
bilateral (32) |
Pr4Pa2 |
2 |
39 |
NA |
bilateral (38) |
NA |
3 |
51 |
NA |
bilateral (45) |
NA |
4 |
26 |
NA |
none |
Pr4 Pa2 |
5 |
37 |
NA |
none |
Pr2 Pa2 |
6 |
45 |
NA |
bilateral (44) |
Pr2 Pa2 |
7 |
43 |
1135insA (X339) |
bilateral (39) |
Pr3 Pa2 |
8 |
37 |
NA |
none |
Pr1 Pa1 |
9a |
42 |
3627dupA (E1210RfsX9) |
bilateral (41) |
Pr1 Pa2 |
9b |
43 |
|||
10 |
48 |
185delAG |
Bilateral (46) |
Pr4 Pa4 |
11 |
28 |
2012insT (X635) |
none |
Pr0 Pa0 |
12 |
45 |
4065del4 |
none |
Pr0 Pa0 |
13 |
28 |
130t>A (C44S) |
none |
Pr0 Pa0 |
14 |
50 |
NA |
Bilateral (16 and 47) |
Pr1 Pa1 |
15 |
36 |
3481del11 |
none |
Pr2 Pa2 |
16 |
33 |
1599C>T (X494) |
none |
Pr3 Pa3 |
17 |
36 |
1731C>T (Q538X) |
none |
NA |
18 |
55 |
5083del19 (X1670) |
none |
Pr0 Pa0 |
19 |
36 |
5382insC |
none |
Pr3 Pa3 |
20 |
39 |
3960C>T (X1281) |
none |
Pr2 Pa2 |
21 |
46 |
917-918delTT (S267fs) |
bilateral (45) |
Pr2 Pa2 |
22 |
57 |
2125-2126insA (G709YfsX3) |
bilateral (57) |
Pr8 Pa8 |
Patients who tissues were included in the xenograft experiments are highlighted in grey.
Pr: number of pregnancy; Pa: parity; NA: not available.
Proliferation marker Ki67 was quantified in luminal epithelial cells of breast tissues. The Ki67-positive cell percentage was similar between the control (4.7 ± 1.3%) and mutated breast tissues (4.6 ± 1.2%) (p=0.974, data not shown). However, when women were sorted according to their menopausal status, we observed that Ki67 expression was significantly reduced in the post-menopause group compared to the pre-menopause group, among patients with BRCA1 mutations (p=0.019) (Figure 1a). Similar results were observed in lobular and ductal structures of breast tissues when analyzed independently (Figure 1b).
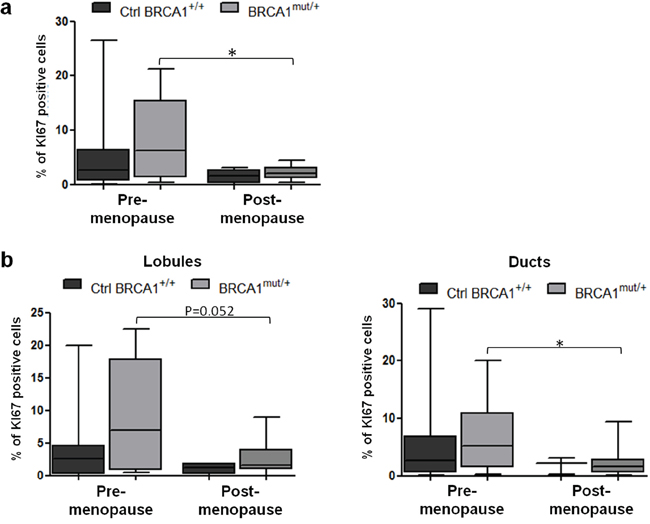
Figure 1: Proliferation status in control and BRCA1mut/+ breast tissues according to menopausal status. Tissue sections were stained for Ki67 by IHC. a. Quantification of Ki67-positive cells in control (Ctrl BRCA1+/+) and BRCA1mut/+ breast tissues, pre- and post-menopause. b. Quantification of Ki67-positive cells in lobules (left panel) and ducts (right panel) from control and BRCA1mut/+ breast tissues, pre- and post-menopause. Each box contains the interquartile range values with the central line indicating the median value and whiskers extending to the minimum and maximum values. * = p<0.05.
As this result suggested a different sensitivity to gonadal hormones in BRCA1 mutated tissues compared to control tissues under different ovarian hormonal stimulation, hormone receptor levels were analyzed. Overall, the percentages of ER-positive epithelial cells were not significantly different between BRCA1mut/+ tissues (41.67 ± 2.9%) and control BRCA1+/+ tissues (33.5 ± 3.3%) (p=0.078, data not shown). When analyzed according to menopause status, ER-positive cells were elevated in post-menopausal BRCA1mut/+ tissues in comparison to control tissues (p=0.0162, Figure 2a). A similar profile of expression was observed in lobular and ductal structures (Supplementary Figure 1a).
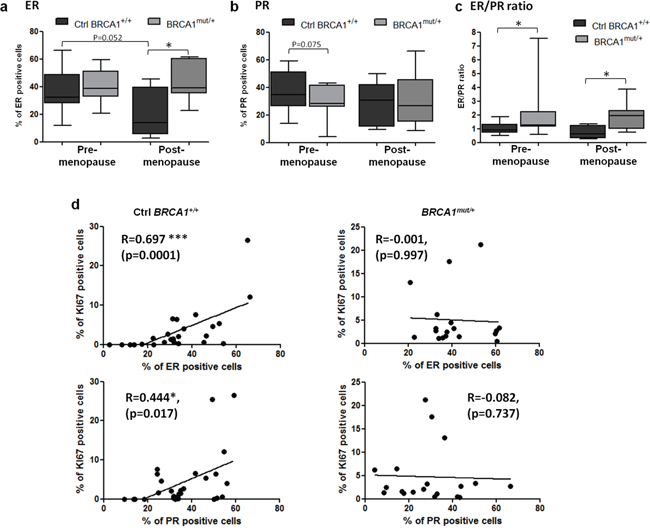
Figure 2: ER and PR expression levels in control and BRCA1mut/+ breast tissues according to menopausal status. Tissue sections were stained by IHC for ER or PR as indicated. a, b. Percentage of ER and PR positive cells scored in control (Ctrl BRCA1+/+) and BRCA1mut/+ breast tissues, pre- and post-menopause. c. ER to PR percentage ratio was calculated in control and BRCA1mut/+ breast tissues, pre- and post-menopause. Each box contains the interquartile range values with the central line indicating the median value and whiskers extending to the minimum and maximum values. d. Correlation curves between Ki67 and hormone receptor expression in control and BRCA1mut/+ breast tissues. Spearman correlation coefficients (R) are indicated. * = p<0.05; ** = p<0.001.
PR levels were also measured. The percentage of PR-positive cells were not significantly different in BRCA1mut/+ breast tissues (29.52 ± 3.3%) compared to the control group (35.8 ± 2.6%) (p=0.13, data not shown). However, pre-menopausal BRCA1mut/+ breast tissues appeared to have a slightly lower percentage of PR-positive cells compared to control tissues (p=0.075, Figure 2b). We also observed that PR expression was significantly reduced in the lobules from pre-menopausal BRCA1mut/+ group (p=0.042) but not in the ducts (Supplementary Figure 1b). PR levels in lobular structures were reduced after menopause in the control group (p=0.017) but not in the BRCA1mut/+ group (Supplementary Figure 1b).
The ER/PR ratio was calculated for each patient breast tissue. This ratio was significantly elevated in the BRCA1 mutated group compared to the control group: 2.27 ± 0.90 vs 1.03 ± 0.09 (p=0.029) for pre-menopause and 1.85 ± 0.30 vs 0.73 ± 0.21 (p=0.028) for post-menopause (Figure 2c). Furthermore, strong correlations were observed between Ki67- and ER-positive cells and between Ki67- and PR-positive cells in control tissues (Figure 2d). In contrast, there were no correlation with BRCA1mut/+ breast tissues (Figure 2d), suggesting that the regulation of epithelial breast cell proliferation by hormone receptor pathways is altered in BRCA1mut/+ breast tissues.
The two isoforms of PR, PR-A and PR-B, are responsible for transcriptional activation of distinct and isoform-specific set of genes. Based on previous findings that showed PR-B as the most active isoform for gene transcription, we analyzed its expression [31, 32]. Interestingly, we observed a significant drop in PR-B levels in BRCA1mut/+ tissues compared to control breast tissues, regardless of menopausal status (Figure 3a-3b). In BRCA1mut/+ tissues, 59.1% of samples displayed loss of PR-B expression whereas PR-B was present in all control tissues. PR-B depletion was observed both in lobular and ductal structures from BRCA1mut/+ tissues (Supplementary Figure 1c).
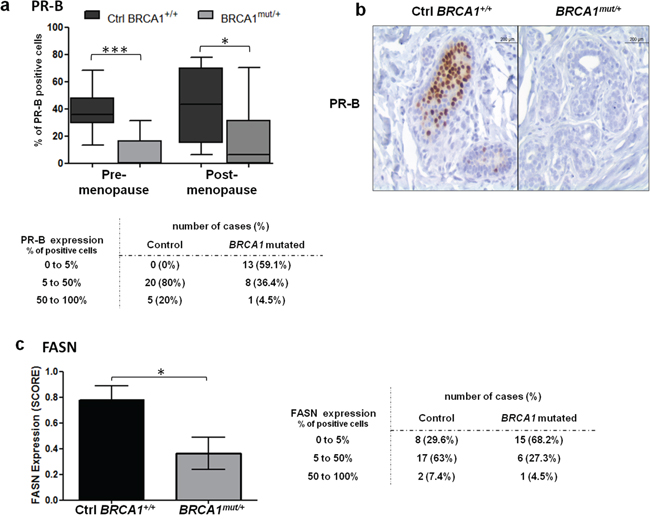
Figure 3: PR-B and FASN expression levels in control and BRCA1mut/+ breast tissues. Tissue sections were stained by IHC for PR-B or FASN as indicated. a. PR-B positive cells quantified in control (Ctrl BRCA1+/+) and BRCA1mut/+ breast tissues, pre- and post-menopause. Each box contains the interquartile range values with the central line indicating the median value and whiskers extending to the minimum and maximum values. Table: PR-B positive cells indicated for control and BRCA1 mutated breast tissues without discrimination of menopausal status. b. PR-B stained IHC sections of control and BRCA1mut/+ breast tissues. c. FASN expression was scored in control and BRCA1mut/+ breast tissues as described in the Materials and Methods. Table: FASN positive cells indicated for control and BRCA1 mutated breast tissues without discrimination of menopausal status. * = p<0.05; ** = p<0.001.
To evaluate the transcriptional activity of PR receptors in BRCA1 mutated tissues, we examined the expression level of fatty acid synthase (FASN), a PR-induced target gene that is associated with tumor growth of breast cancer cells. FASN catalyzes the synthesis of long chain fatty acids, promoting an altered lipogenic metabolism that is beneficial for cancer cell progression [33, 34]. However, FASN is also involved in the promotion of epithelial differentiation in normal breast cells [35]. FASN mRNA expression is activated by PR in breast tissue and was shown to be specifically induced by PR-B isoform [31, 35-37]. We observed significantly higher FASN levels in control tissues compared to BRCA1mut/+ tissues (p=0.0164, Figure 3c). Sixty eight percent of BRCA1mut/+ samples showed loss of FASN expression whereas only 29.6% of control tissues were negative for FASN expression (Figure 3c).
Altogether our results show that hormone receptor expression was impaired in BRCA1+/mut tissues compared to control tissues with a marked loss of the PR-B isoform and a decreased expression of the PR target gene, FASN. In addition, proliferation was increased in pre-menopause tissue compared to post-menopause tissue, among women with BRCA1 mutations. These observations suggest that breast tissues from BRCA1 mutation carriers have differences in proliferation control and in differentiation driven by hormone receptor levels with reduced levels of PR-B, compared to women without BRCA1 mutation.
Response to hormonal treatment in BRCA1 mutated breast tissue xenografts
We studied the cellular responses induced by E2 and P4 in BRCA1mut/+ breast tissues as compared to non-mutated tissues in an NMRInu/nu athymic mouse xenograft model. Four BRCA1+/+ tissue samples and four BRCA1mut/+ tissue samples were grafted subcutaneously onto the backs of mice, on either side of the spine (Figure 4a, see Materials and Methods). Treatments were delivered by pellets inserted under the skin. Time and treatment dose delivery were previously designed to mimic the physiological menstrual cycle in women (Figure 4a and Materials and Methods) [28]. Mice were divided into four groups: Control (C), E2, E2+P4, and E2+P4+UPA.
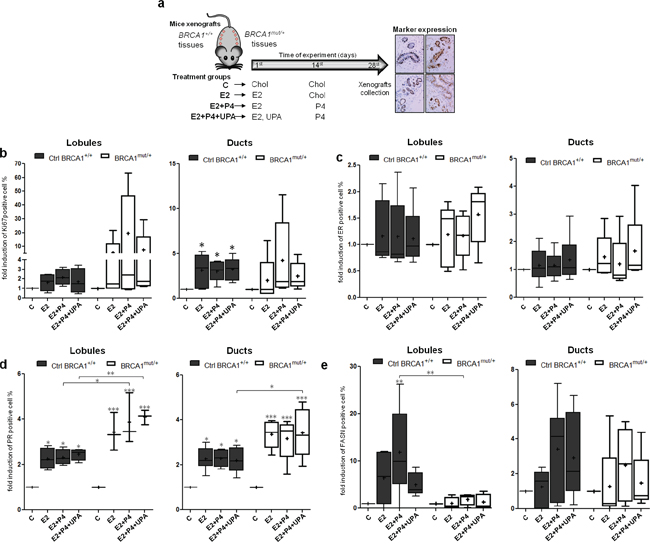
Figure 4: Effects of hormone treatment on BRCA1mut/+ breast tissues xenografted in mice. a. Illustration of breast tissue xenografts and hormonal treatment strategy. Breast tissue samples from 6 patients without mutations (Ctrl BRCA1+/+) and from 5 patients with BRCA1 mutation (BRCA1mut/+) were xenografted in NMRInu/nu athymic mice. Treatment pellets were grafted in two steps to mimic the menstrual cycle: E2 and UPA pellets were grafted on the first day of the experiment while P4 was grafted on the 14th day. Cholesterol (Chol) was used as a placebo for the control condition (C). After 28 days of treatment, mice were sacrificed and breast tissue xenografts were collected for subsequent scoring of marker expression by IHC. Fold change in induction compared to the control (C) were evaluated for proliferation (Ki67) b., ER c., PR d. and FASN e. in lobules (left panel) and ducts (right panel) from Ctrl BRCA1+/+ and BRCA1mut/+ breast tissue xenografts. Each box contains the interquartile range values with the central line indicating the median value, the cross indicating the mean value, and whiskers extending to the minimum and maximum values. * = p<0.05; ** = p<0.01; *** = p<0.001.
We analyzed the effects of ovarian hormone treatments on proliferation in lobular and ductal structures (Figure 4b). In BRCA1mut/+ tissues, proliferative responses were highly heterogeneous compared to normal tissues. In BRCA1mut/+ lobules, E2 treatment increased the Ki67 expression from 1 to 21.7 fold, relative to the control. This range of Ki67 expression was drastically increased, from 1 to 63.4 fold, by the addition of P4. Interestingly, the PR inhibitor UPA, reversed the action of P4 and restored a proliferation profile that was similar to mice treated only with E2 (Figure 4b). In ducts, proliferation was significantly increased in the three treatment groups of BRCA1+/+ tissues whereas addition of P4 was the major enhancer of proliferation in BRCA1mut/+ tissues. As observed in lobules, the P4 effect was reversed by UPA (Figure 4b). These results indicate that the proliferative responses to E2 and P4 are deregulated in BRCA1 mutated breast tissues with a high degree of heterogeneity among patient tissues.
We then quantified ER and PR levels in response to hormonal treatments. ER levels were not modified by hormone treatments in both BRCA1 mutated and non-mutated tissues (Figure 4c and Supplementary Figure 2). Interestingly PR levels were significantly more elevated by E2 treatment in ductal and lobular structures from BRCA1 mutated tissues compared to BRCA1+/+ tissues (Figure 4d). As expected, FASN expression was induced by E2+P4 treatment in non-mutated lobules and ducts structures. In contrast, induction of FASN was impaired in BRCA1mut/+ tissues, particularly in lobular structures despite having elevated PR levels (Figure 4e and Supplementary Figure 2). These results suggest alterations in P4 responses and in PR-target gene activation in BRCA1 mutated tissues.
The heterogeneity of differences were particularly apparent when marker expressions were analyzed independently in BRCA1mut/+ breast tissues from each of the five patients, and compared with the mean response of the six control BRCA1+/+ tissues (Supplementary Figure 2). Responses were homogeneous in the six non-mutated BRCA1+/+ tissues whereas the five BRCA1mut/+ tissues all displayed a different marker expression pattern. Moreover, none of the five BRCA1mut/+ tissues showed the same profile as the BRCA1+/+ tissues. While Ki67 levels in control BRCA1+/+ tissues were only increased by 2.8 ± 0.5 to 3.6 ± 1.3 fold in the presence of E2+P4 relative to the control, Ki67 expression was dramatically elevated in BRCA1mut/+ tissues #17 and #18 (19.5 and 13.0 fold, respectively). The increase in P4-induced proliferation was generally reversed by UPA in these tissues. Interestingly, breast tissues from both patients #17 and #18 were negative for PR-B expression before engraftment (Supplementary Figure 2).
These results highlight the presence of deregulated hormonal responses in BRCA1mut/+ tissues and the heterogeneity of responses among patients. We observed that P4 combined with E2 had an enhanced proliferation effect but dampened PR-dependent gene induction. Interestingly, the use of UPA reversed the P4 effects on proliferation in BRCA1mut/+ tissues.
DISCUSSION
Hormonal exposure and the overall lifetime number of ovulatory cycles are modifiers of breast cancer risk among BRCA1 mutation carriers [5, 6, 9]. Here we showed that hormone receptor expression and responses are altered in BRCA1 mutated breast tissues compared to BRCA1+/+ breast tissues and that these deregulations occur in histologically normal tissues. Since BRCA1 is involved in DNA repair and cell cycle control, our results suggest that E2 and P4 exposure may enhance proliferation in BRCA1mut/+ breast tissues, and potentially increase the accumulation of unrepaired mutations and DNA lesions. Indeed, previous studies have shown that BRCA1mut/+ epithelial breast cells were haploinsufficient for BRCA1 as they displayed genomic alterations [38, 39], including defects in stalled replication fork repair and a higher frequency in fork collapses [39]. Haploinsufficiency was also involved in impaired differentiation of epithelial luminal cells, leading to an expanded luminal progenitor population in BRCA1mut/+ breast tissues [40]. All these data are consistent with the assumption that BRCA1mut/+ breast cells are likely to cumulate genomic aberrations during mitotic recombination [38]. Higher proliferative rates caused by the hormone signaling in these cells would therefore explain the sex and organ-specific penetrance of BRCA1-related cancers.
Currently, prophylactic mastectomy and/or prophylactic annexectomy are used to decrease breast cancer risk in carriers with BRCA1 mutations; however, there is an urgent need to develop efficient and less aggressive strategies. Here we demonstrated that a PR antagonist inhibited the enhanced proliferative effect of P4 in our BRCA1mut/+ breast tissue xenograft models. PR expression was recently shown to improve the prognosis and treatment response to sporadic ER positive breast cancer by modulating ER functions [41]. However, our results support the use of a PR inhibitor as a potential preventive strategy in women with BRCA1 mutations, and highlight the effect of BRCA1 mutations in the regulation of hormone receptors in normal mammary gland. Although mifepristone prevented the onset and development of mammary tumors in p53-/-/brca1f11/f11 mice [18], this result may only be possible in epithelial cells predominantly expressing the PR-A isoform [42]. In our xenograft experiments, tissues #17 and #18 were associated with the highest P4-induced proliferation levels, and were negative for PR-B before engraftment. The aberrant proliferative effect of P4 was reversed by UPA. Notably, tissue from patient #22 was also negative for PR-B but did not display any hormone-induced proliferation. This may be explained by high levels of breast tissue differentiation [43-45] that the patient most likely experienced through eight full term pregnancies (Table 1). Importantly, UPA did not have any proliferative effect on samples that did not show drastic P4 stimulation. Since E2 also displayed mitogenic action, a combination of an anti-estrogen plus an antiprogestin could be optimal for breast cancer prevention. While tamoxifen has already shown a protective effect against the risk of breast cancer, including contralateral breast cancer, in populations with BRCA1/2 mutations [21-23, 46], the use of tamoxifen as a standard preventive treatment is limited due to its side effects. A recent study reported higher levels of circulating P4 and E2 in BRCA1/2 mutation carriers compared to women without BRCA1/2 mutations [47]. This finding combined with atypical ER and PR profiles in normal breast tissues may also indicate that chemical prevention could be beneficial for these patients. UPA is already used and well tolerated in clinics although further studies are needed to test its potential to decrease cancer risk in women with BRCA1 mutations.
Deregulated responses to estrogen and progesterone were demonstrated in BRCA1mut/+ breast epithelial cells with a higher proliferation before menopause, suggesting an increased sensitivity to ovarian hormone stimulation. This was further supported by our observations in xenografted tissues where hormone treatments were highly mitogenic in some BRCA1mut/+ tissues compared with non-mutated tissues. In breast cancer cells and mice models, BRCA1 limited ER and PR transcriptional activities and mitogenic actions [10, 15, 18, 48]. Our study supports these findings as the BRCA1 heterozygous status was associated with an increase of ER and PR proliferative activity. This could explain the reported association between increased hormone exposure and the risk of breast cancer [5, 6, 49]. These results are also supported by the Anderson group study which highlighted the mitogenic effect of E2 in BRCA1mut/+ tissues xenografted in mice [50]. Women with BRCA1 mutations had an abnormally increased ER/PR ratio that was associated with a striking loss of PR-B receptors. Alternatively, Clarke et al. has shown that PR-B expression was lost or decreased in BRCA1mut/+ tissues whereas ER expression was not altered [16]. This is in line with our BRCA1mut/+ data showing a loss of PR-B in almost 60% of samples and little change in ER expression before menopause. Additionally, we observed higher levels of ER in BRCA1mut/+ compared to control tissues from post-menopausal groups, highlighting the importance of the hormonal status in patients with BRCA1 mutations.
In breast cancer tissue, the typical 1:1 ratio of PR-A and PR-B isoforms in normal epithelial cells is frequently altered due to the apparent loss of PR-B [51, 52]. In T47D breast cancer cells, PR-A overexpression resulted in loss of adherent properties and insufficient FASN mRNA transcription, supporting our observation of decreased FASN protein levels in BRCA1mut/+ tissues [31]. Loss of PR-B expression could be due to increased PR-B degradation or decreased PR-B transcription. Lange et al. has shown that ligand-induced transcriptional activity of PR-B was associated with PR-B rapid ubiquitin-dependent degradation, resulting in PR-B loss [53]. However in our study, PR-B was lost without a gain in transcriptional activity as shown by decreased FASN expression. Moreover, BRCA1 is responsible for PR-ubiquitination and its subsequent degradation which is more likely impaired in BRCA1mut/+ tissues, suggesting that another mechanism is responsible for PR-B loss. E2 is the main regulator of transcription of both PR isoforms, and may also contribute to the silencing of PR-B by selective methylation of the promoter under certain conditions [54]. Other possible mechanisms of PR-B loss may include MAPKs which are involved in the control of phosphorylation and turnover of PR-A and PR-B [55]. Alteration of MAPK activities may result in the loss of BRCA1 function [56, 57]. Notably, we showed that BRCA1 loss of expression was associated with impaired MAPK p38 phosphorylation leading to decreased levels of the activated (S211 phosphorylated) glucocorticoid receptor [56]. Further studies are required to understand the exact mechanisms underlying PR-B loss in BRCA1mut/+ tissues.
The results of our study may also impact the administration of exogenous hormones used for contraception or menopausal hormonal therapy (MHT). The most recent studies reported an elevated risk for breast cancer in women with BRCA1 mutations if oral contraception was used before the age of 20 years or before the first full term pregnancy [58, 59]. Unlike natural progesterone, contraceptives include synthetic progestins with different affinities for other steroid receptors such as androgen, glucocorticoid or mineralocorticoid receptors, leading to differences in risk for breast cancer. Although further studies are required, the use of MHT after surgical or spontaneous menopause did not appear to increase the risk of subsequent cancers among a small and heterogeneous patient cohort [60-62].
Our study is the first to investigate the effect of BRCA1 mutations in lobules and ducts separately. Hormone treatment has a greater impact on the terminal ductal lobular unit (TDLU), which is consistent with the observed increase of E2+P4-dependent proliferation in lobules compared to ducts. In women with BRCA1 mutations, triple negative tumors are predominant but their cellular origin most likely arises from the luminal progenitor [40, 63]. Our results support the idea that these cancers could originate specifically from the TDLU. Analysis of breast tissues according to the menopausal status allowed for differentiation according to hormonal stimulation. However, the number of samples included in our study was low, limiting the strengths of our conclusions. Additional studies are needed to delineate the use of chemoprevention in women with BRCA1 mutation according to their breast tissues phenotypes.
In conclusion, our findings indicate that BRCA1 mutation status is associated with alterations in proliferation and in hormone receptor expression and activities in histologically normal breast tissues. These deregulations could participate in the early events of breast cancer development in BRCA1 mutation carriers. Importantly, for the development of new strategies to prevent the onset of BRCA1-related breast cancer, this study suggests that a subset of women with BRCA1 mutations could be candidates for a UPA treatment as a preventive breast cancer strategy.
MATERIALS AND METHODS
Patient recruitment
Normal breast tissues from healthy volunteers were collected between years 2007 and 2012 from various hospital centers in France as part of the BRACAPS consortium cooperation. Breast tissue samples were obtained from women who had signed an informed consent according to the French law on clinical experimentation (L. 1243-3 and L. 1243-4), as part of a biomedical study that included the collection and conservation of cell cultures and xenografts of breast tissues. The authorization number filed for this project is 11826, from the French ethical committee “Comité de Protection des Personnes”.
The cohort included 22 BRCA1 mutation carriers (BRCA1mut/+) undergoing prophylactic mastectomies, and 28 women as controls without BRCA1 mutation (BRCA1+/+), undergoing breast reductions, and without any reported history of breast disease. The absence of breast malignancy was ensured before and after surgery by breast imaging and anatomopathological review of collected samples, respectively. Hematoxylin–phloxine–saffron staining was used to detect healthy breast tissue.
Women with BRCA1 mutations had genetic testing that revealed a pathogenic germ-line mutation in the BRCA1 gene. Among the 22 BRCA1 mutation carriers, one patient underwent two prophylactic mastectomy surgeries one year apart. For this patient, breast tissues were collected at each surgery and considered as independent samples resulting in n=23 women with BRCA1 mutation. Clinical characteristics of women bearing a BRCA1 mutation are described in Table 1.
There was no significant difference between ages at time of surgery for women with or without BRCA1 mutations. Control women were between 21 to 56 years of age: 37 ± 2.2 years (mean ± SEM). Women with BRCA1 mutation were between 26 to 57 years of age: 41.6 ± 1.8 years. When hormonal status was uncertain, women above 50 were considered as post-menopausal. Oophorectomized women were included in the post-menopausal group. Premenopausal status was assigned to 23 of 28 women in the control BRCA1+/+ group and to 11 of 22 patients in the BRCA1 mutation carrier group.
Mice xenograft experiments
Breast tissue samples were taken from 6 women of the control cohort and from 5 women with BRCA1 mutations (patients 17, 18, 19, 21 and 22, Table 1) and were xenografted in four week old ovariectomized female NMRInu/nu athymic mice (Janvier laboratory, Le Genest Saint Isle, France). Mean age of the control cohort was 36.0 ± 2.1 years (range: 29-42) and was not significantly different from the mean age of women with BRCA1 mutation: 46.0 ± 4.5 years (range: 36-57).
Six independent tissue xenograft experiments were conducted as described previously [28]. In four experiments, breast tissues from one control woman and one woman with BRCA1 mutations were concomitantly xenografted in mice since the dates of patient surgeries were concurrent. One experiment included breast tissue xenografts from two BRCA1+/+ control patients and one other experiment was performed with tissues from only one patient with BRCA1 mutation. Four tissue fragments per patient were used for subcutaneous xenografts placed on one side of the back of each mouse. Four treatment groups were used per experiment which included the control (C), E2, E2+P4 and E2+P4+UPA (n=4 mice and 16 patient tissue fragments per group). Treatments were delivered by pellets, administered subcutaneously (Figure 4a). Mice were sacrificed 28 days after xenografting, and blood and tissue xenografts were collected. Tissues were immediately fixed in paraformaldehyde solution for IHC analysis. All study protocols and environmental conditions were approved by the French Ethic Charles Darwin committee for the care and use of laboratory animals.
Immunohistochemistry
IHC analyses were performed using the BOND-MAX workstation (Leica, Nanterre, France) as previously described [28]. Tissue sections were stained with antibodies against Ki67 at 1:100 dilution, ERα at 1:300 (NCL-L-Ki67-MM1 and NCL-L-ER-6F11, Novocastra, Leica, Nanterre, France), PR at 1:80 (MU-328-UC, Biogenex, Fremont, CA, USA) and FASN at 1:400 (sc-20140, Santa Cruz, Dallas, TX, USA). For signal detection, the Bond Polymer Refine Detection kit (Leica) was used. Reagents were purchased from Menarini-Diagnostic (Rungis, France). A negative control (no primary antibody) was included in each set. Marker expression was analyzed as previously described [28].
Marker analyses
For each marker, the number of positive cells was counted among a total of 1000 lobular and 1000 ductal luminal epithelial cells. The mean percentage of expression was calculated either for all counted cells per section or only for lobular or ductal cells. Breast tissues showing less than 100 lobular and 100 ductal cells were excluded from the analysis. A scoring system was established for FASN quantification of positive cell percentages: 0 (0<5%), 1 (5-50%), 2 (>50%). In xenograft experiments, the final percentage of marker expression was the mean of percentages in tissues from the four mice per treatment group.
Hormone concentration analyses
Methods and results for measuring serum concentration were described previously [28]. E2 concentration was 36.88 ± 4.25 pg/ml. P4 concentration was 13.05 ± 1.14 ng/ml. UPA concentration was 63.49 ± 10.46 ng/ml which was the same range observed in clinical use [64]. Hormone levels were undetectable in control mice (E2 < 0.8 pg/ml; P4 < 0.4 ng/ml; UPA < 0.5 ng/ml).
Statistical analysis
Results were expressed as mean ± SEM. Missing values were not considered. The Kolmogorov–Smirnov test and Shapiro–Wilk test were used to test for normality of the group distributions (GraphPad Prism 5, USA). One-way analysis of variance or non-parametric Kruskal-Wallis test followed by Tukey’s or Dunn’s multiple comparison post-hoc tests were performed according to the normality of the group distributions. When two groups were compared, an unpaired t-test or a non-parametric Mann Whitney test was performed. The Spearman test was used for correlation analysis. A P-value < 0.05 was considered significant and n represented the number of independent experiments.
ACKNOWLEDGMENTS
We thank Drs Michele Resche-Rigon, Andre Ulmann and Erin Gainer (HRA Pharma, Paris) for helpful discussions, provision of the antiprogestin UPA, and analytical contributions on mice serum UPA concentrations. We are very grateful to Sylvie Dumont and Fatiha Merabtène (Plateforme d’Histomorphologie UMS30 – LUMIC (UPMC) Hôpital Saint Antoine Paris France) for their technical assistance in immunohistochemistry, and to Jacqueline Chung and Kishanda Vyboh for their work editing the publication.
CONFLICTS OF INTEREST
The authors have no conflict of interest to declare.
GRANT SUPPORT
This research was supported by grants from INSERM-UPMC, HRA Pharma and the Institut National du Cancer (INCa, France). Laudine Communal was the recipient of a CIFRE grant from HRA Pharma and the government. Myriam Vilasco was the recipient of a post-doc fellowship (INCa). Aurélie Courtin was a recipient of a grant from the Association pour la Recherche sur le Cancer (ARC).
REFERENCES
1. Silver DP, Livingston DM. Mechanisms of BRCA1 tumor suppression. Cancer discovery. 2012; 2:679-684.
2. Boulton SJ. Cellular functions of the BRCA tumour-suppressor proteins. Biochemical Society transactions. 2006; 34:633-645.
3. Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, Davidson R, Eccles D, Cole T, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. Journal of the National Cancer Institute. 2013; 105:812-822.
4. King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003; 302:643-646.
5. Lecarpentier J, Nogues C, Mouret-Fourme E, Buecher B, Gauthier-Villars M, Stoppa-Lyonnet D, Bonadona V, Fricker JP, Berthet P, Caron O, Coupier I, Pujol P, Faivre L, et al. Breast cancer risk associated with oestrogen exposure and truncating mutation location in BRCA1/2 carriers. Cancer epidemiology, biomarkers & prevention. 2015; 24:698-707.
6. Pan H, He Z, Ling L, Ding Q, Chen L, Zha X, Zhou W, Liu X, Wang S. Reproductive factors and breast cancer risk among BRCA1 or BRCA2 mutation carriers: results from ten studies. Cancer epidemiology. 2014; 38:1-8.
7. Rebbeck TR, Kauff ND, Domchek SM. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. Journal of the National Cancer Institute. 2009; 101:80-87.
8. Noruzinia M, Coupier I, Pujol P. Is BRCA1/BRCA2-related breast carcinogenesis estrogen dependent? Cancer. 2005; 104:1567-1574.
9. Eisen A, Lubinski J, Klijn J, Moller P, Lynch HT, Offit K, Weber B, Rebbeck T, Neuhausen SL, Ghadirian P, Foulkes WD, Gershoni-Baruch R, Friedman E, et al. Breast cancer risk following bilateral oophorectomy in BRCA1 and BRCA2 mutation carriers: an international case-control study. Journal of clinical oncology. 2005; 23:7491-7496.
10. Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, Erdos M, Goldberg ID, Webb P, Kushner PJ, Pestell RG, Rosen EM. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001; 20:77-87.
11. Wang C, Fan S, Li Z, Fu M, Rao M, Ma Y, Lisanti MP, Albanese C, Katzenellenbogen BS, Kushner PJ, Weber B, Rosen EM, Pestell RG. Cyclin D1 antagonizes BRCA1 repression of estrogen receptor alpha activity. Cancer research. 2005; 65:6557-6567.
12. Hosey AM, Gorski JJ, Murray MM, Quinn JE, Chung WY, Stewart GE, James CR, Farragher SM, Mulligan JM, Scott AN, Dervan PA, Johnston PG, Couch FJ, et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. Journal of the National Cancer Institute. 2007; 99:1683-1694.
13. Jeffy BD, Hockings JK, Kemp MQ, Morgan SS, Hager JA, Beliakoff J, Whitesell LJ, Bowden GT, Romagnolo DF. An estrogen receptor-alpha/p300 complex activates the BRCA-1 promoter at an AP-1 site that binds Jun/Fos transcription factors: repressive effects of p53 on BRCA-1 transcription. Neoplasia. 2005; 7:873-882.
14. Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA, Rosen EM. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006; 20:14-34.
15. Katiyar P, Ma Y, Riegel A, Fan S, Rosen EM. Mechanism of BRCA1-mediated inhibition of progesterone receptor transcriptional activity. Mol Endocrinol. 2009; 23:1135-1146.
16. Mote PA, Leary JA, Avery KA, Sandelin K, Chenevix-Trench G, Kirk JA, Clarke CL. Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes, chromosomes & cancer. 2004; 39:236-248.
17. Prest SJ, May FE, Westley BR. The estrogen-regulated protein, TFF1, stimulates migration of human breast cancer cells. FASEB journal. 2002; 16:592-594.
18. Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006; 314:1467-1470.
19. King MC, Wieand S, Hale K, Lee M, Walsh T, Owens K, Tait J, Ford L, Dunn BK, Costantino J, Wickerham L, Wolmark N, Fisher B. Tamoxifen and breast cancer incidence among women with inherited mutations in BRCA1 and BRCA2: National Surgical Adjuvant Breast and Bowel Project (NSABP-P1) Breast Cancer Prevention Trial. JAMA. 2001; 286:2251-2256.
20. Eisinger F, Nogues C, Guinebretiere JM, Peyrat JP, Bardou VJ, Noguchi T, Vennin P, Sauvan R, Lidereau R, Birnbaum D, Jacquemier J, Sobol H. Novel indications for BRCA1 screening using individual clinical and morphological features. International journal of cancer. 1999; 84:263-267.
21. Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, Daly M, Wieand S, Tan-Chiu E, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. Journal of the National Cancer Institute. 1998; 90:1371-1388.
22. Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Jr., Wade JL, 3rd, Robidoux A, Margolese RG, James J, et al. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA : the journal of the American Medical Association. 2006; 295:2727-2741.
23. Vogel VG, Costantino JP, Wickerham DL, Cronin WM, Cecchini RS, Atkins JN, Bevers TB, Fehrenbacher L, Pajon ER, Wade JL, 3rd, Robidoux A, Margolese RG, James J, et al. Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: Preventing breast cancer. Cancer Prev Res (Phila). 2010; 3:696-706.
24. Phillips KA, Milne RL, Rookus MA, Daly MB, Antoniou AC, Peock S, Frost D, Easton DF, Ellis S, Friedlander ML, Buys SS, Andrieu N, Nogues C, et al. Tamoxifen and risk of contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. Journal of clinical oncology. 2013; 31:3091-3099.
25. Goss PE, Ingle JN, Ales-Martinez JE, Cheung AM, Chlebowski RT, Wactawski-Wende J, McTiernan A, Robbins J, Johnson KC, Martin LW, Winquist E, Sarto GE, Garber JE, et al. Exemestane for breast-cancer prevention in postmenopausal women. The New England journal of medicine. 2011; 364:2381-2391.
26. Trefoux Bourdet A, Luton D, Koskas M. Clinical utility of ulipristal acetate for the treatment of uterine fibroids: current evidence. International journal of women's health. 2015; 7:321-330.
27. Glasier A. The rationale for use of Ulipristal Acetate as first line in emergency contraception: biological and clinical evidence. Gynecological endocrinology. 2014; 30:688-690.
28. Communal L, Vilasco M, Hugon-Rodin J, Courtin A, Mourra N, Lahlou N, Dumont S, Chaouat M, Forgez P, Gompel A. Ulipristal acetate does not impact human normal breast tissue. Hum Reprod. 2012; 27:2785-2798.
29. Nieman LK, Blocker W, Nansel T, Mahoney S, Reynolds J, Blithe D, Wesley R, Armstrong A. Efficacy and tolerability of CDB-2914 treatment for symptomatic uterine fibroids: a randomized, double-blind, placebo-controlled, phase IIb study. Fertility and sterility. 2011; 95:767-772 e761-762.
30. Donnez J, Hudecek R, Donnez O, Matule D, Arhendt H, Zatik J, Kasilovskiene Z, Dumitrascu MC, Fernandez H, Barlow DH, Bouchard P, Fauser BC, Bestel E, et al. Efficacy and safety of repeated use of ulipristal acetate in uterine fibroids. Fertility and sterility. 2014.
31. McGowan EM, Clarke CL. Effect of overexpression of progesterone receptor A on endogenous progestin-sensitive endpoints in breast cancer cells. Mol Endocrinol. 1999; 13:1657-1671.
32. Giangrande PH, Kimbrel EA, Edwards DP, McDonnell DP. The opposing transcriptional activities of the two isoforms of the human progesterone receptor are due to differential cofactor binding. Molecular and cellular biology. 2000; 20:3102-3115.
33. Rochefort H, Chalbos D. The role of sex steroid receptors on lipogenesis in breast and prostate carcinogenesis: a viewpoint. Hormones & cancer. 2010; 1:63-70.
34. Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer research. 2006; 66:5977-5980.
35. Joyeux C, Chalbos D, Rochefort H. Effects of progestins and menstrual cycle on fatty acid synthetase and progesterone receptor in human mammary glands. The Journal of clinical endocrinology and metabolism. 1990; 70:1438-1444.
36. Chalbos D, Joyeux C, Galtier F, Escot C, Chambon M, Maudelonde T, Rochefort H. Regulation of fatty acid synthetase by progesterone in normal and tumoral human mammary glands. Revista espanola de fisiologia. 1990; 46:43-46.
37. Courtin A, Communal L, Vilasco M, Cimino D, Mourra N, de Bortoli M, Taverna D, Faussat AM, Chaouat M, Forgez P, Gompel A. Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast cancer research and treatment. 2012; 131:49-63.
38. Rennstam K, Ringberg A, Cunliffe HE, Olsson H, Landberg G, Hedenfalk I. Genomic alterations in histopathologically normal breast tissue from BRCA1 mutation carriers may be caused by BRCA1 haploinsufficiency. Genes, chromosomes & cancer. 2010; 49:78-90.
39. Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, Feunteun J, Garber JE, Livingston DM. BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nature communications. 2014; 5:5496.
40. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, Feleppa F, Huschtscha LI, Thorne HJ, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nature medicine. 2009; 15:907-913.
41. Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, Serandour AA, Birrell SN, Bruna A, Saadi A, Menon S, Hadfield J, Pugh M, et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature. 2015; 523:313-317.
42. Wargon V, Riggio M, Giulianelli S, Sequeira GR, Rojas P, May M, Polo ML, Gorostiaga MA, Jacobsen B, Molinolo A, Novaro V, Lanari C. Progestin and antiprogestin responsiveness in breast cancer is driven by the PRA/PRB ratio via AIB1 or SMRT recruitment to the CCND1 and MYC promoters. International journal of cancer. 2015; 136:2680-2692.
43. Layde PM, Webster LA, Baughman AL, Wingo PA, Rubin GL, Ory HW. The independent associations of parity, age at first full term pregnancy, and duration of breastfeeding with the risk of breast cancer. Cancer and Steroid Hormone Study Group. Journal of clinical epidemiology. 1989; 42:963-973.
44. Russo J, Moral R, Balogh GA, Mailo D, Russo IH. The protective role of pregnancy in breast cancer. Breast cancer research. 2005; 7:131-142.
45. Hinkula M, Pukkala E, Kyyronen P, Kauppila A. Grand multiparity and the risk of breast cancer: population-based study in Finland. Cancer causes & control. 2001; 12:491-500.
46. Xu L, Zhao Y, Chen Z, Wang Y, Chen L, Wang S. Tamoxifen and risk of contralateral breast cancer among women with inherited mutations in BRCA1 and BRCA2: a meta-analysis. Breast Cancer. 2015.
47. Widschwendter M, Rosenthal AN, Philpott S, Rizzuto I, Fraser L, Hayward J, Intermaggio MP, Edlund CK, Ramus SJ, Gayther SA, Dubeau L, Fourkala EO, Zaikin A, et al. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. The Lancet Oncology. 2013; 14:1226-1232.
48. Jones LP, Tilli MT, Assefnia S, Torre K, Halama ED, Parrish A, Rosen EM, Furth PA. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERalpha-negative and ERalpha-positive mammary preneoplasia and cancer. Oncogene. 2008; 27:794-802.
49. Lee EY. Promotion of BRCA1-associated triple-negative breast cancer by ovarian hormones. Current opinion in obstetrics & gynecology. 2008; 20:68-73.
50. Bramley M, Clarke RB, Howell A, Evans DG, Armer T, Baildam AD, Anderson E. Effects of oestrogens and anti-oestrogens on normal breast tissue from women bearing BRCA1 and BRCA2 mutations. British journal of cancer. 2006; 94:1021-1028.
51. Mote PA, Bartow S, Tran N, Clarke CL. Loss of co-ordinate expression of progesterone receptors A and B is an early event in breast carcinogenesis. Breast cancer research and treatment. 2002; 72:163-172.
52. Mote PA, Graham JD, Clarke CL. Progesterone receptor isoforms in normal and malignant breast. Ernst Schering Foundation symposium proceedings. 2007; 77-107.
53. Hagan CR, Lange CA. Molecular determinants of context-dependent progesterone receptor action in breast cancer. BMC medicine. 2014; 12:32.
54. Vazquez-Martinez ER, Mendoza-Garces L, Vergara-Castaneda E, Cerbon M. Epigenetic regulation of Progesterone Receptor isoforms: from classical models to the sexual brain. Molecular and cellular endocrinology. 2014; 392:115-124.
55. Khan JA, Amazit L, Bellance C, Guiochon-Mantel A, Lombes M, Loosfelt H. p38 and p42/44 MAPKs differentially regulate progesterone receptor A and B isoform stabilization. Mol Endocrinol. 2011; 25:1710-1724.
56. Vilasco M, Communal L, Hugon-Rodin J, Penault-Llorca F, Mourra N, Wu Z, Forgez P, Gompel A. Loss of glucocorticoid receptor activation is a hallmark of BRCA1-mutated breast tissue. Breast cancer research and treatment. 2013; 142:283-296.
57. Yan Y, Haas JP, Kim M, Sgagias MK, Cowan KH. BRCA1-induced apoptosis involves inactivation of ERK1/2 activities. The Journal of biological chemistry. 2002; 277:33422-33430.
58. Kotsopoulos J, Lubinski J, Moller P, Lynch HT, Singer CF, Eng C, Neuhausen SL, Karlan B, Kim-Sing C, Huzarski T, Gronwald J, McCuaig J, Senter L, et al. Timing of oral contraceptive use and the risk of breast cancer in BRCA1 mutation carriers. Breast cancer research and treatment. 2014; 143:579-586.
59. Brohet RM, Goldgar DE, Easton DF, Antoniou AC, Andrieu N, Chang-Claude J, Peock S, Eeles RA, Cook M, Chu C, Nogues C, Lasset C, Berthet P, et al. Oral contraceptives and breast cancer risk in the international BRCA1/2 carrier cohort study: a report from EMBRACE, GENEPSO, GEO-HEBON, and the IBCCS Collaborating Group. Journal of clinical oncology. 2007; 25:3831-3836.
60. Rebbeck TR, Friebel T, Wagner T, Lynch HT, Garber JE, Daly MB, Isaacs C, Olopade OI, Neuhausen SL, van 't Veer L, Eeles R, Evans DG, Tomlinson G, et al. Effect of short-term hormone replacement therapy on breast cancer risk reduction after bilateral prophylactic oophorectomy in BRCA1 and BRCA2 mutation carriers: the PROSE Study Group. Journal of clinical oncology. 2005; 23:7804-7810.
61. Eisen A, Lubinski J, Gronwald J, Moller P, Lynch HT, Klijn J, Kim-Sing C, Neuhausen SL, Gilbert L, Ghadirian P, Manoukian S, Rennert G, Friedman E, et al. Hormone therapy and the risk of breast cancer in BRCA1 mutation carriers. Journal of the National Cancer Institute. 2008; 100:1361-1367.
62. Kotsopoulos J, Huzarski T, Gronwald J, Moller P, Lynch HT, Neuhausen SL, Senter L, Demsky R, Foulkes WD, Eng C, Karlan B, Tung N, Singer CF, et al. Hormone replacement therapy after menopause and risk of breast cancer in BRCA1 mutation carriers: a case-control study. Breast cancer research and treatment. 2016; 155:365-373.
63. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, Mackay A, Grigoriadis A, Tutt A, Ashworth A, Reis-Filho JS, Smalley MJ. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell stem cell. 2010; 7:403-417.
64. Pohl O, Zobrist RH, Gotteland JP. The clinical pharmacology and pharmacokinetics of ulipristal acetate for the treatment of uterine fibroids. Reprod Sci. 2015; 22:476-483.