
INTRODUCTION
Great progress has been achieved in AML treatment through past decades, however, some types of AML, especially AML with Fms-like tyrosine kinase-3 internal tandem duplication (FLT3-ITD) mutation, which comprised of 20-30% AML [1], are still incurable using currently approaches except for allogeneic hematopoietic stem cell transplantation [2]. Tyrosine kinase inhibitors (TKIs) emerging as targeted therapy appeared most exciting therapeutics in FLT3-ITD treatment, such as multikinase inhibitor sorafenib [3] and some new compounds [4]. Although some TKIs could achieve high remission rate both in peripheral blood and bone marrow, leukemia cells usually relapse in a short period of time [5]. One reason is that TKIs execute subclone selection in vivo, amplifying drug insensitive clones, such as FLT3 point mutation (D835Y) clone [6]. Another reason is that drug resistant leukemia stem cells (LSCs) were protected from bone marrow niche, leukemia then re-established from LSCs under certain conditions such as TKI withdraw [7].
Leukemia/microenvironment interaction is important for leukemia cells especially leukemia stem cells’ biological behavior [8]. Accumulating evidences indicate that integrins are involved in leukemia/microenvironment interaction by influencing both cell growth and drug sensitivity [9]. For instance, interaction of α4β1 and α5β1 in leukemia cells with fibronection in stromal contributes to chemo-insensitivity and minimal residual disease of AML [10, 11]. Integrin β could directly interact with integrin-linked kinase (ILK), which could trigger downstream signaling pathway, such as PI3K/Akt pathway, resulting in cell survival and proliferation [12]. Recently, an in vivo small hairpin RNA (shRNA) screening approach was used to find out integrin β3 was required for leukemogenesis and LSCs transcriptional programs maintenance in a MLL-AF9 AML mouse model. They also found that integrin αv, which formed a heterodimer with integrin β3, also required for maintaining the leukemic phenotype [13].
In this paper, we demonstrated that the expression level of integrin β3 was associated with the National Comprehensive Cancer Network (NCCN) risk stratification and could be a novel prognostic biomarker in AML especially in cytogenetic-normal FLT3-ITD mutated AML patients. Integrin αvβ3 decreased sorafenib sensitivity when co-culture MV4-11 cells with BMSCs, and it is crucial for OPN induced sorafenib insensitivity in FLT3-ITD mutated AML cells. αvβ3 could enhance β-catenin activation through PI3K/Akt/GSK3β pathway. Our study revealed a novel mechanism in microenvironment influence on sorafenib sensitivity in AML with FLT3-ITD mutation.
RESULTS
Relationship between ITGB3 expression and clinicopathological features of AML patients
To investigate the prognostic significance of ITGB3 gene expression in human AML, we mined The Cancer Genome Atlas (TCGA) database which is available to public. The clinicopathological features of all AML patients are categorized in Table 1. We found that the patients with higher ITGB3 expression were older (median age, 57.8 vs 52.8 years; p<0.05) and had a higher proportion of patients with unfavorable cytogenetics (p=0.002) compared with patients who had lower ITGB3 expression. Moreover, we found patients with higher ITGB3 expression had lower blasts in peripheral and bone marrow, lower WBC counts, higher platelet counts compared with the patients with lower ITGB3 expression (p<0.05). When grouped with prognostic risk stratification of AML according to NCCN, the expression level of ITGB3 was significantly higher in poor group than in favorable and intermediate groups (Figure 1A).
Table 1: Relationship between ITGB3 expression and clinicopathological features of AML patients
Characteristic |
No. |
ITGB3 high |
ITGB3 low |
p value |
|---|---|---|---|---|
No. of patients |
173 |
87 |
86 |
|
Sex |
0.707 |
|||
Male |
93 |
48 |
45 |
|
Female |
80 |
39 |
41 |
|
Median age, y (range) |
57.8 (21-88) |
52.8 (18-81) |
0.043 |
|
FAB classification, no. |
0.252 |
|||
M0 |
16 |
12 |
4 |
|
M1 |
42 |
19 |
23 |
|
M2 |
39 |
18 |
21 |
|
M3 |
16 |
5 |
11 |
|
M4 |
35 |
18 |
17 |
|
M5 |
18 |
10 |
8 |
|
M6 |
2 |
1 |
1 |
|
M7 |
3 |
3 |
0 |
|
Not Classified |
2 |
1 |
1 |
|
Cytogenetic abnormality, no. |
0.002 |
|||
Favorable |
32 |
7 |
25 |
|
Intermediate |
103 |
56 |
47 |
|
Poor |
36 |
23 |
13 |
|
Unknown |
2 |
1 |
1 |
|
FLT3 mutation rate (%) |
24/85 (27.9) |
26/84 (31.3) |
0.699 |
|
Peripheral blast, % (range) |
61.2 (0-97) |
70.4 (0-100) |
0.006 |
|
Bone marrow blast, % (range) |
74.3 (10-100) |
84.9 (10-100) |
0.010 |
|
Median WBC count, ×109 (range) |
26.3 (1-137) |
49.5 (1-297) |
0.001 |
|
Median Hb concentration, g/dL |
9.8 (6-14) |
9.4 (6-12) |
0.295 |
|
Median platelet count, ×109 (range) |
80.5 (9-351) |
47.5 (8-215) |
0.000 |
Cytogenetic abnormality: according to NCCN guideline.
FAB classification, French-American-British classification; FLT3, Fms-like tyrosine kinase-3; WBC, white blood cell; Hb, hemoglobin.
Table 2: The primer sequences used in the real time PCR experiment
Gene names |
Forward |
Reverse |
|---|---|---|
Ctnnb1 |
5′-AAAGCGGCTGTTAGTCACTGG-3′ |
5′-CGAGTCATTGCATACTGTCCAT-3′ |
beta-actin |
5'-CGTGCGTGACATTAAGGAGAAGC-3' |
5'-CGGACTCGTCATACTCCTGCTTG-3' |
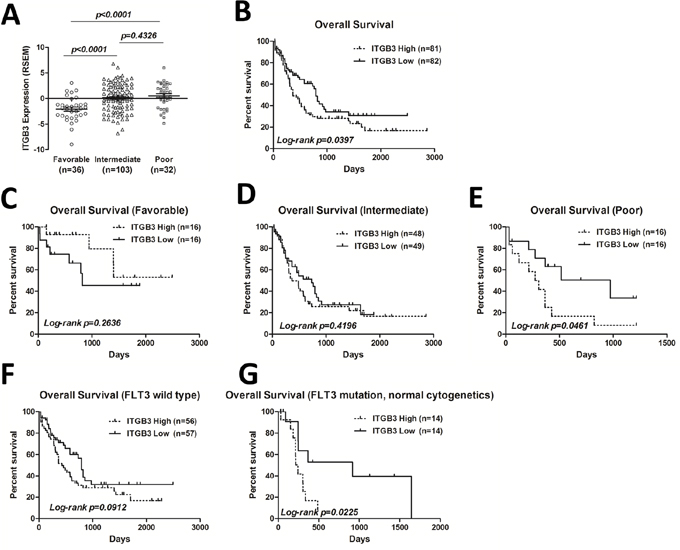
Figure 1: ITGB3 expression and correlation with overall survival (OS) in AML patients. A. ITGB3 expression level in 171 AML patients from TCGA dataset (https://tcga-data.nci.nih.gov/tcga/) was grouped by cytogenetic risk according to NCCN. The expression level is represented by RSEM (RNA-Seq by Expectation Maximization) value. Unpaired Student’s t-test was used to analyze the differences between groups. B. Kaplan-Meier plots of OS of all patients divided by ITGB3 expression. C-E. Kaplan-Meier plots of OS of AML patients segregated by ITGB3 expression in favorable, intermediate and poor groups, respectively. F-G. Kaplan-Meier plots of OS of AML patients segregated by ITGB3 expression in FLT3 wild type and FLT3 mutation groups, respectively.
High level of ITGB3 expression is associated with shorter overall survival of AML patients, especially in patients with FLT3-ITD mutation
To determine the prognostic value of ITGB3 expression in AML patients, we evaluated the overall survival (OS) of all patients by Kaplan-Meier analysis. The median follow-up in this cohort was 557.4 days (0-2861 days). Cut-off value of ITGB3 expression (RNA-Seq by Expectation Maximization, RSEM) is set to -0.10 so that the numbers in two groups are similar [ITGB3 high group (n=81): -0.09 to 6.80; ITGB3 low group (n=82): -6.86 to -0.11]. As shown in Figure 1B, elevated ITGB3 expression was significantly correlated with shorter OS (p<0.05). Then we analyzed the OS depended on ITGB3 expression in each group according to prognostic risk stratification of NCCN. The OS was significantly worse when higher expression level of ITGB3 in poor prognostic group (Figure 1E), however, there was no correlation in favorable and intermediate groups (Figure 1C, 1D), Mutation of FLT3 was frequent and indicated poor prognosis in AML [1]. We thus compared the prognostic significance of ITGB3 on AML patients with or without FLT3 mutation. In FLT3 wild type AML patients, there was no difference of OS between two groups depends on the expression level of ITGB3 (Figure 1F), however, in patients who had normal cytogenetics with FLT3 mutation, the OS of patients with higher ITGB3 expression was significantly worse compared that of patients with lower ITGB3 (Figure 1G).
Blocking integrin αvβ3 enhances leukemia sensitivity to sorafenib in BMSCs and OPN ligation
To figure out the function of ITGB3 in FLT3-ITD AML in bone marrow environment, we used BMSCs to mimic bone marrow environment. We co-cultured MV4-11 cells (a FLT3-ITD mutation AML cell line) with BMSCs and tested the sensitivity of sorafenib, a TKI proved by Food and Drug Administration for hepatocellular carcinoma and renal cell carcinoma. As showed in Figure 2A, MV4-11 was sensitive to sorafenib, by the 50% inhibitory concentration (IC-50) around 10μM (data not shown). Sorafenib induced apoptosis in MV4-11, the apoptotic rate was 38.15% (±1.48). When co-cultured with BMSCs, MV4-11 showed significantly reduced apoptosis rate under sorafenib treatment, the apoptosis rate was 20.54% (±0.44) (Figure 2A, 2B). This phenomenon could be explained by cell adhesion mediated drug resistance. However, when we blocked αvβ3 by pretreating with αvβ3 blocking antibody, this effect was significantly decreased. MV4-11 showed increased apoptosis rate after αvβ3 blocking (Figure 2A, 2B). We also detected lower Bcl-2 and higher Bax level when treated with sorafenib plus αvβ3 antibody in co-culture system (Supplementary Figure S1). These results indicating integrin αvβ3 trigger apoptosis resistance effect. Our previous work showed that bone marrow stromal cells secreted more OPN when co-cultured with MV4-11 cells (Zhaohua Shen, Oncology letters, 2016, in press). OPN was one of the ligand which could bind integrin αvβ3. We also tested the effect of OPN and αvβ3 in MV4-11. OPN could induce sorafenib insensitivity of MV4-11 cells in a dose-dependent manner (Supplementary Figure S2). These effects could be partly abrogated by αvβ3 blocking. These results suggested that integrin αvβ3 induced leukemia insensitivity to sorafenib when co-cultured with BMSCs as well as OPN ligation.
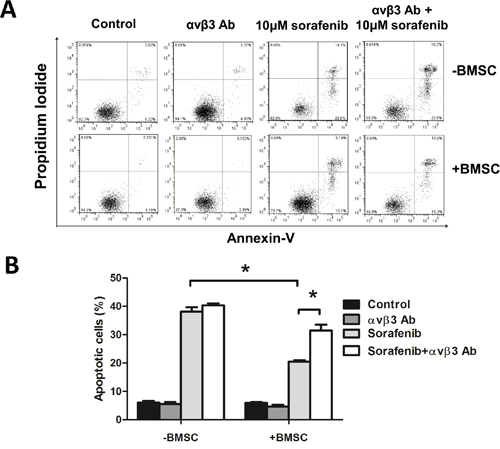
Figure 2: Blocking integrin αvβ3 increased leukemia sensitivity to sorafenib in BMSCs. A. The apoptosis rate measured by flow cytometry by Annexin-V/PI double staining. MV4-11 cells were seeded onto the BMSC monolayer or cultured alone in 6-well plates with or without αvβ3 blocking antibody (1μg/ml) for 2 hours, sorafenib (10μM) was then added and cultured for 24 hours. Suspension cells were used for apoptosis assay. B. The percentage of apoptosis in each group. *p<0.05.
OPN/αvβ3 activated β-catenin via PI3K/Akt/GSK3β signaling pathway
It was reported that OPN/αvβ3 pathway promote a cancer stem cell-like phenotype in hepatocellular carcinoma cells. Because the Wingless-type (Wnt)/β-catenin is the most important cancer stem cell pathway, we hypothesized that αvβ3 could activated β-catenin in MV4-11 cells. We detected higher β-catenin level in OPN treated MV4-11 cells in a dose-dependent manner (Figure 3A). The elevated β-catenin was largely contributed by β-catenin in the nuclear (Supplementary Figure S3). Moreover, we observed higher phosphorylation of Akt, GSK3β and β-catenin in OPN treated MV4-11 (Figure 3B). This effect was largely abrogated when blocking αvβ3 before OPN ligation, indicating β-catenin elevation is αvβ3 dependent and through PI3K/Akt/GSK3β signaling pathway (Figure 3B). We also detected higher phosphorylation of Akt, GSK3β and β-catenin when co-culture MV4-11 with BMSCs, and αvβ3 antibody could partly reduce them (Supplementary Figure S4). To further validate PI3K/Akt/GSK3β contributes to β-catenin elevation and drug sensitivity, we used PI3K specific inhibitor LY294002. As we expected, p-Akt, p-GSK3β and β-catenin were decreased after PI3K inhibition (Figure 3C). Sorafenib sensitivity was increased after PI3K inhibition (Supplementary Figure S2). These results indicated that OPN/αvβ3 activated β-catenin and through PI3K/Akt/GSK3β signaling pathway.
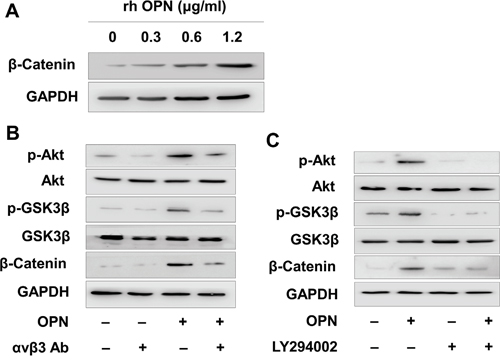
Figure 3: OPN/αvβ3 activated β-catenin via PI3K/Akt/GSK3β signaling pathway. A. MV4-11 was treated by various amount of recombinant human OPN for 24 hours, then β-catenin was detected by Western blot assay. B. MV4-11 was pretreated by αvβ3 blocking antibody (1μg/ml) for 2 hours, then treated by OPN (1.2 μg/ml) for another 24 hours. The phosphorylation level of Akt and GSK3β and the expression of β-catenin were measured by Western blot analysis. C. MV4-11 was pretreated by PI3K inhibitor LY294002 (25μM) for 2 hours, then treated by OPN (1.2 μg/ml) for another 24 hours. The phosphorylation level of Akt and GSK3β and the expression of β-catenin were measured by Western blotting analysis.
β-catenin maintained leukemia cell survival and contributed to sorafenib insensitivity
To determine the effect of β-catenin in leukemia cells, we knocked down β-catenin in MV4-11 using lentiviral delivery of shRNA. To avoided off-target effect, we used two shRNAs which targeted different code sequence of Ctnnb1 and a scramble shRNA as control. β-catenin was dramatically reduced in Ctnnb1 knock down cells (sh-Ctnnb1 cells) by western blotting and real time qPCR assay (Figure 4A, 4B). MV4-11 cell viability was decreased after Ctnnb1 knocked down (Figure 4C). When exposed in different concentration of sorafenib, the cell viability was decreased gradually in each group. However, at the same concentration of sorafenib, the cell viability was higher in control cells compared with sh-Ctnnb1 cells (Figure 4D). Sh-Ctnnb1 cells underwent spontaneous apoptosis. When treated with 10μM sorafenib, the apoptosis rate of sh-Ctnnb1 cells was higher compared with control cells, indicating β-catenin interference could enhance leukemia sensitivity in MV4-11 cells (Figure 4E, 4F).
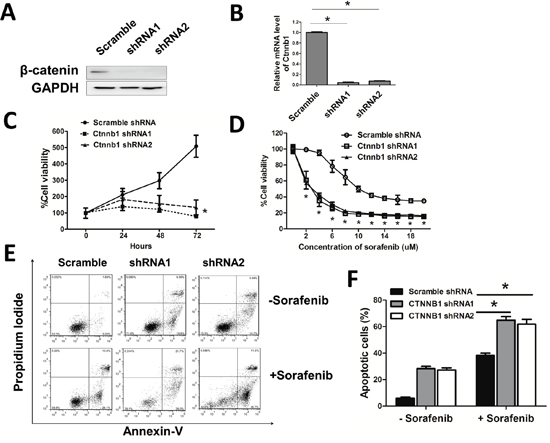
Figure 4: β-catenin maintained leukemia cell survival and contributed to sorafenib insensitivity. A, B. β-catenin protein level and mRNA level were measured by Western blotting and real-time qPCR after Ctnnb1 shRNA vectors infection and puromycin selection for 2 days. *p<0.05. C. Cell viability was measured by CCK-8 assay when cultured for 24, 48, 72 hours, respectively. *p<0.05. D. Cells were treated with different doses of sorafenib for 24 hours. Cell viability was then evaluated by CCK-8 assay. *p<0.05. E. Cells were treated with or without sorafenib (10μM) for 24 hours. F. The percentage of apoptosis in each group. *p<0.05.
DISCUSSION
Integrins enhance cell-ECM interaction, intracellular signal transduction, contributing to tumor invasiveness and metastasis [14]. Integrin comprises of two subunits, alpha and beta chain, which form a heterodimer. Integrin beta3 (ITGB3) loss impairs AML survival and homing to endosteum [13]. ITGB3 has prognostic significance in AML especially for unfavorable group and patients with FLT3-ITD mutation according to our data. Higher level of ITGB3 implies older and higher frequency of poor cytogenetics. However, there are significant lower WBC counts, lower blast and higher platelet accompanied with higher level of ITGB3, indicating ITGB3 signaling is not associated with higher leukemia proliferation rate. In myeloid cells, ITGB3 heterodimerizes with ITAV and interact with ECM [15]. We also analyze ITGAV and its clinical relevant, however, there is no significant relevant in TCGA database (data not shown). Whether ITGAV has clinical relevant needs further investigation.
Integrin αvβ3 could bind arginine-glycine-aspartate (RGD)-containing components of matrix, such as OPN, vitronectin and fibronectin, trigger intracellular signal [16]. OPN/αvβ3 was found to drive tumor stemness and resistant to TKI inhibition in solid tumor [17, 18]. In this study, we give evidence that integrin αvβ3 could alternatively enhance β-catenin activation in AML. β-catenin signaling contributes to leukemia stem cell phenotype and influences drug sensitivity in leukemia [19–21]. It is also an independent prognostic indicator in AML [22]. β-catenin could be activated in several ways. First, several Wnt ligands are constitutive expressed in most AML cells and bone marrow stromal cells, leading to Wnt/β-catenin signaling in AML [19, 23]. Second, FLT3-ITD positive AML has ligand independent activated form of FLT3 receptor tyrosine kinase and causes increased activated form of β-catenin [24]. Here, we give evidence that integrin αvβ3 could alternatively enhance β-catenin activation through PI3K/Akt/GSK3β cascade on linking OPN from microenvironment, giving an another interpretation on β-catenin activation in FLT3-ITD mutated AML.
FLT3-ITD positive AML patients might benefit from TKIs combining with integrin αvβ3 blocker and β-catenin inhibitors. Several αvβ3 antibody and β-catenin inhibitors are under clinical trial in several cancer types [25–30]. Based on our study, these compounds might be considered as new pharmaceuticals for therapy of AML with FLT3-ITD mutation combined with TKIs.
Collectively, we demonstrated that integrin β3 has prognostic significance in AML especially in cytogenetic-normal AML with FLT3-ITD mutation. Integrin αvβ3/PI3K/Akt/GSK3β/β-catenin axis is crucial for microenvironment mediated TKI insensitivity in FLT3-ITD AML cells (A schematic drawing is shown in Figure 5). Our data provides a new mechanistic basis for integrin αvβ3 signaling in drug insensitivity in bone marrow microenvironment. Targeting integrin αvβ3/PI3K/Akt/β-catenin signaling might be helpful improving the prognosis of FLT3-ITD mutated AML patients.
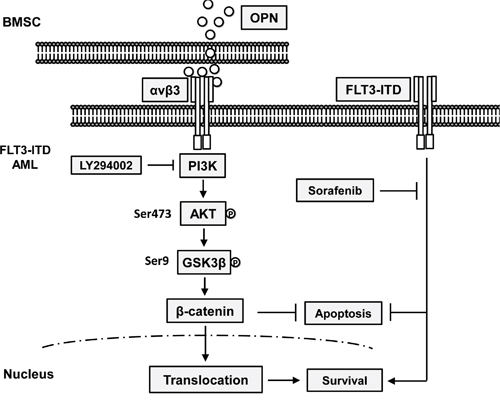
Figure 5: A schematic drawing of Integrin αvβ3/PI3K/Akt/GSK3β/β-catenin axis for microenvironment mediated TKI insensitivity in FLT3-ITD AML cells.
MATERIALS AND METHODS
Clinical data
The Cancer Genome Atlas (TCGA) dataset for AML are available on https://tcga-data.nci.nih.gov/tcga/. High-throughput sequencing data of transcriptome and clinical data were retrieved and analyzed.
Cell line and cell culture
The MV4-11 human acute myeloid leukemia cell line (AML-M5, FLT3-ITD mutation) was obtained from the American Type Culture Collection. Cells were cultured in suspension in IMDM media (Hyclone, USA), supplemented with 10% fetal bovine serum (Gibco, USA) plus 1% penicillin/streptomycin (Hyclone, USA) at 37°C in a humidified 5% CO2 incubator. Cells were passaged every 3 days.
Isolation and culture of bone marrow stromal cells
Bone marrow aspirates were obtained from healthy volunteers in Department of Hematology, Xinqiao Hospital after written informed consent was obtained. This study was approved by the Ethical Committee of Xinqiao Hospital and carried out in accordance with the Declaration of Helsinki. BMSCs were isolated and cultured as previously described [31]. Briefly, Mononuclear cells from healthy donors after informed consent were isolated with a density gradient centrifugation (Ficoll-Paque, GE Healthcare, NJ) and cultured in Dulbecco's minimal essential medium (DMEM) with low glucose, supplemented with 10% fetal bovine serum plus antibiotics as mentioned above, and maintained at 37°C in a humidified 5% CO2 incubator. The nonadherent cells were removed 24 hours later. When 80–90% confluency, the adherent cells were trypsinized and passaged. BMSCs were used in the following experiments at passage 3 or 4.
Drugs, chemicals and antibodies
Sorafenib was obtained from Bayer Pharmaceutical Corporation and was dissolved in dimethylsulfoxide (DMSO) (Sigma, St.Louis, Mo), stored in -20°C. For apoptosis assay, the final concentration of sorafenib was 10μM, and DMSO concentration was 0.01%. The αvβ3 blocking antibody was purchased from NOVUS Biologicals (Littleton, CO). Recombinant OPN was purchased from PeproTech (Rocky Hill, NJ). PI3K inhibitor LY294002 was purchased from Selleck Chemicals (Houston, TX). Primary rabbit anti-human antibodies against β-catenin, phospho-Akt (Ser473), Akt, phospho-GSK3β (Ser9), GSK3β were purchased from Cell Signaling Technology, Inc (Beverley, MA). Primary rabbit anti-human antibodies against Bcl-2 and Bax were purchased from Abcam (Cambridge, MA). Rabbit anti-human antibody against GAPDH and Histone H3, horseradish peroxidase-linked anti-rabbit secondary antibody were purchased from Bioworld Technology (Minneapolis, MN).
Assessment of apoptosis by flow cytometry
Apoptosis was measured based on flow cytometry by annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) double staining using the FITC Annexin V Apoptosis Detection Kit (BestBio, Shanghai, China), according to the manufacturer’s instructions. Flow cytometry was carried out using MoFlo XDP system (Beckman Coulter, Pasadena, CA). Data were analyzed by FlowJo software (Treestar Inc). In co-culture experiments, MV4-11 cells were seed onto the BMSCs monolayer in 6-well plates, then cells were treated with or without αvβ3 blocking antibody (1μg/ml) for 2 hours, sorafenib was then added and cultured for 24 hours. After that, suspension cells were removed by gently shaking and used for apoptosis assay.
Lentiviral shRNA production and infection
Scramble shRNA (Sequence: TTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAA) or two Ctnnb1 shRNAs (Sequence 1: TTGGAATGAGACTGCTGATCTCGAGATCAGCAGTCTCATTCCAAGC; sequence 2: GTTATCAGAGGACTAAATACTCGAGTATTTAGTCCTCTGATAAC) were cloned to GV112 vector (Genechem, Shanghai) carrying puromycin resistance gene. GV112-shRNA vector, VSV-G (Addgene plasmid 8454, MA), and psPAX2 (Addgene plasmid 12260, MA) were co-transfected at a 3:1:2 ratio into 293T packaging cells using polyethylenimine (PEI) linear (Polysciences Inc). The medium was replaced 8 hours post-transfection and lentiviral supernatants were harvested at 48 hours. MV4-11 cells were plated at 2 × 105 cells/well on 6-well plates overnight and added with lentiviral supernatants in the presence of 5 μg/mL polybrene (Sigma-Aldrich) with spin infection at 2,500 rpm and 32°C for 90 minutes. Cells were incubated for another 4 hours at 37°C incubator. After changing media, infected MV4-11 cells were cultured 48 hours and selected in 2 μg/mL puromycin for at least 2 days. Then cells were ready for the following experiments.
Determination of cell viability
Infected MV4-11 were seeded in 96-well plates at a density of 5×103 cells per well, cells were treated with various concentration of sorafenib for 24 hours. Cell viability was assessed using the Cell-Counting Kit-8 (CCK-8) (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, after treatment, 10 μl CCK-8 was added to each well and incubated at 37°C for 1 hour. The optical density was read at a wavelength of 450 nm with a microplate reader (BioTek, USA). Cell viability was calculated as the following formula: optical density of treated group/control group×100%.
Immunoblotting
MV4-11 cells were collected and total protein was lysed in cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH8], 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS), containing protease and phosphatase inhibitors (Bayotime, Shanghai, China). In certain experiment, nuclear and cytoplasmic protein was extracted using Nuclear and Cytoplasmic Protein Extraction Kit (Bayotime, Shanghai, China). Protein samples were resolved by 10% SDS-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (PVDF, Millipore Corporation, MA). PVDF membranes were blocked in Tris-buffered saline (TBS; 25 mM Tris, 0.15 M NaCl; pH 7.2) containing 5% nonfat dry milk and 0.1% Tween-20 for 1 hour and then incubated overnight in 4°C with various primary antibodies. After washing with TBST (TBS with 0.1% Tween-20), blots were incubated with horseradish peroxidase-linked secondary antibody at room temperature for 1 hour. Blots were developed using WesternBright ECL western blotting detection kit (Advansta Inc., Menlo Park, CA).
Real-time quantitative PCR (qRT-PCR)
Total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. QRT-PCR was performed using SYBR Premix ExTaq (Takara, Japan) and an Applied Biosystems 7500 Fast real-time PCR system according to the manufacturer’s instructions. The sequences of primers used in this study were listed in Table 2. The PCR cycling conditions were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15s and 60°C for 35s. Beta-actin (Actb) was used as an internal normalization control. Data analyses were performed using the comparative CT (ΔΔCT) method for calculating relative gene expression.
Statistical analysis
Statistical analysis was conducted by SPSS software, version 18.0 (SPSS Inc., Chicago, IL, USA). The normality distribution test was performed with the Kolmogorov-Smirnov test. Clinical features of sex and FLT3 mutation rate were compared between ITGB3 high and low groups by the Pearson χ2 test. Clinical features of FAB classification and Cytogenetic abnormality were compared between two groups using Fisher’s exact test. Other clinical features of age, peripheral blast, bone marrow blast, WBC count, Hb concentration and Platelet count between two groups were compared using the unpaired two-tailed Student's t-test. Overall survival curves were plotted according to the Kaplan-Meier methods with the log-rank test applied for comparison. Unpaired two-tailed Student’s t-test for two groups was applied in the in-vitro experiments. All values are presented as mean±SEM. A P value<0.05 was considered statistically significant (assigned as *).
ACKNOWLEDGMENTS
We thank Ms. Jing Qiu (Central Laboratory, Xinqiao Hospital, Chongqing, China) for assistance in Flow Cytometry and Dr. Jun Rao for helpful discussion.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
GRANT SUPPORT
This work was supported by National Natural Science Foundation of China (Grant No. 81500132, No. 81000195).
REFERENCES
1. Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. The New England journal of medicine. 2013; 368:2059-2074.
2. Ma Y, Wu Y, Shen Z, Zhang X, Zeng D, Kong P. Is allogeneic transplantation really the best treatment for FLT3/ITD-positive acute myeloid leukemia? A systematic review. Clin Transplant. 2015; 29:149-160.
3. Wolleschak D, Schalk E, Krogel C, Schnoeder TM, Luehr H, Jentsch-Ullrich K, Fischer T, Heidel FH. Rapid induction of complete molecular remission by sequential therapy with LDAC and sorafenib in FLT3-ITD-positive patients unfit for intensive treatment: two cases and review of the literature. Journal of hematology & oncology. 2013; 6:39.
4. Guo Y, Chen Y, Xu X, Fu X, Zhao ZJ. SU11652 Inhibits tyrosine kinase activity of FLT3 and growth of MV-4-11 cells. Journal of hematology & oncology. 2012; 5:72.
5. Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010; 116:5089-5102.
6. Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, Choi WW, Lok S, Cheung AM, Eaves C, Kwong YL, Leung AY. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012; 119:5133-5143.
7. Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. Journal of clinical oncology. 2011; 29:591-599.
8. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009; 23:2233-2241.
9. Tabe Y, Jin L, Tsutsumi-Ishii Y, Xu Y, McQueen T, Priebe W, Mills GB, Ohsaka A, Nagaoka I, Andreeff M, Konopleva M. Activation of integrin-linked kinase is a critical prosurvival pathway induced in leukemic cells by bone marrow-derived stromal cells. Cancer research. 2007; 67:684-694.
10. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A, Akiyama T, Kuroda H, Kawano Y, Kobune M, Kato J, Hirayama Y, Sakamaki S, Kohda K, Miyake K, Niitsu Y. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nature medicine. 2003; 9:1158-1165.
11. De Toni-Costes F, Despeaux M, Bertrand J, Bourogaa E, Ysebaert L, Payrastre B, Racaud-Sultan C. A New alpha5beta1 integrin-dependent survival pathway through GSK3beta activation in leukemic cells. PloS one. 2010; 5:e9807.
12. Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nature reviews Cancer. 2005; 5:51-63.
13. Miller PG, Al-Shahrour F, Hartwell KA, Chu LP, Jaras M, Puram RV, Puissant A, Callahan KP, Ashton J, McConkey ME, Poveromo LP, Cowley GS, Kharas MG, Labelle M, Shterental S, Fujisaki J, et al. In Vivo RNAi screening identifies a leukemia-specific dependence on integrin beta 3 signaling. Cancer cell. 2013; 24:45-58.
14. Guo W, Giancotti FG. Integrin signalling during tumour progression. Nature reviews Molecular cell biology. 2004; 5:816-826.
15. Keskinov AA, Shurin MR. Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology. 2015; 220:236-242.
16. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nature reviews Cancer. 2010; 10:9-22.
17. Cao L, Fan X, Jing W, Liang Y, Chen R, Liu Y, Zhu M, Jia R, Wang H, Zhang X, Zhang Y, Zhou X, Zhao J, Guo Y. Osteopontin promotes a cancer stem cell-like phenotype in hepatocellular carcinoma cells via an integrin-NF-kappaB-HIF-1alpha pathway. Oncotarget. 2015; 6:6627-6640. doi: 10.18632/oncotarget.3113.
18. Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, Cascone T, Diao L, Wang J, Wistuba, II, Heymach JV, Lippman SM, et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nature cell biology. 2014; 16:457-468.
19. Simon M, Grandage VL, Linch DC, Khwaja A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene. 2005; 24:2410-2420.
20. Dandekar S, Romanos-Sirakis E, Pais F, Bhatla T, Jones C, Bourgeois W, Hunger SP, Raetz EA, Hermiston ML, Dasgupta R, Morrison DJ, Carroll WL. Wnt inhibition leads to improved chemosensitivity in paediatric acute lymphoblastic leukaemia. British journal of haematology. 2014; 167:87-99.
21. Hu K, Gu Y, Lou L, Liu L, Hu Y, Wang B, Luo Y, Shi J, Yu X, Huang H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/beta-catenin signaling pathway. Journal of hematology & oncology. 2015; 8:1.
22. Ysebaert L, Chicanne G, Demur C, De Toni F, Prade-Houdellier N, Ruidavets JB, Mansat-De Mas V, Rigal-Huguet F, Laurent G, Payrastre B, Manenti S, Racaud-Sultan C. Expression of beta-catenin by acute myeloid leukemia cells predicts enhanced clonogenic capacities and poor prognosis. Leukemia. 2006; 20:1211-1216.
23. Yang Y, Mallampati S, Sun B, Zhang J, Kim SB, Lee JS, Gong Y, Cai Z, Sun X. Wnt pathway contributes to the protection by bone marrow stromal cells of acute lymphoblastic leukemia cells and is a potential therapeutic target. Cancer letters. 2013; 333:9-17.
24. Kajiguchi T, Chung EJ, Lee S, Stine A, Kiyoi H, Naoe T, Levis MJ, Neckers L, Trepel JB. FLT3 regulates beta-catenin tyrosine phosphorylation, nuclear localization, and transcriptional activity in acute myeloid leukemia cells. Leukemia. 2007; 21:2476-2484.
25. Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, Lozahic S, Marty M, Faivre S. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against alphavbeta3 integrin receptor, in patients with advanced solid tumors. Investigational new drugs. 2008; 26:35-43.
26. Vansteenkiste J, Barlesi F, Waller CF, Bennouna J, Gridelli C, Goekkurt E, Verhoeven D, Szczesna A, Feurer M, Milanowski J, Germonpre P, Lena H, Atanackovic D, Krzakowski M, Hicking C, Straub J, et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: results of an open-label, randomized, controlled phase II study (CERTO). Annals of oncology. 2015; 26:1734-1740.
27. Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, Park E, Naing K, Klemm L, Swaminathan S, Conway EM, Pelus LM, Crispino J, Mullighan CG, McMillan M, Muschen M, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014; 33:2169-2178.
28. Lenz HJ, Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer science. 2014; 105:1087-1092.
29. US National Library of Medicine. ClinicalTrials.gov, http://clinicaltrials.gov/show/NCT01606579 (2012).
30. Griffiths EA, Golding MC, Srivastava P, Povinelli BJ, James SR, Ford LA, Wetzler M, Wang ES, Nemeth MJ. Pharmacological targeting of beta-catenin in normal karyotype acute myeloid leukemia blasts. Haematologica. 2015; 100:e49-52.
31. Zeng D, Hao L, Xu W, Li Z, Li W, Li J, Zhang X, Chen X, Kong P. Pinch-1 was up-regulated in leukemia BMSC and its possible effect. Clinical and experimental medicine. 2013; 13:21-27.