
INTRODUCTION
Gastric cancer is the fifth most common malignancies and the third leading cause of cancer-related death among all cancers worldwide. According to the global cancer statistics in 2012, an estimated 951,600 new stomach cancer cases and 723,100 deaths occurred [1]. Since it is lack of reliable early diagnostic markers, most patients are diagnosed at the advanced or metastatic stage. Surgery, chemotherapy or radiotherapy plays a minor role to improve the survival rate [2]. Therefore, it is urgent to identify new early diagnostic markers and explore novel therapeutic targets.
H. pylori infection and the resulting gastric inflammation is the strongest identified risk factor for the development of gastric cancer [3]. Persistent infection with H. pylori may cause chronic atrophic gastritis, with development of intestinal metaplasia, dysplasia, and gastric carcinoma [4], and H. pylori has been classified as a class I carcinogenic factor by the International Agency for Research on Cancer since 1994. Recent studies have reported that H. pylori-induced epigenetic modifications, such as DNA methylation and histone modifications, are critical in many oncogenic processes including regulating gene expression, cell cycle and cell proliferation [5, 6]. Dysregulated epigenetic alterations, especially in early neoplastic development, may be just as significant as genetic mutations in driving cancer development and progression [7]. However, the role of H. pylori infection in these processes has not been well defined. JMJD2B, also known as KDM4B, is newly identified and characterized as a member of the histone demethylase JMJD2 family. JMJD2B specifically targets the trimethylated lysine 9 of histone H3 (H3K9) in pericentric heterochromatin and euchromatin [8-10], which has been implicated in inflammation [11], and tumorigenesis [12]. In our previous work, JMJD2B is overexpressed in gastric cancer and plays a role in tumor cell proliferation, survival, invasion and metastasis [13, 14]. However, the precise mechanism of JMJD2B upregulation in gastric cancer has not been completely elucidated. Given the role of H. pylori infection in gastric malignant transformation, we investigate the link between H. pylori infection and JMJD2B expression associated with gastric cancer pathogenesis.
Recent studies suggest that COX-2 is activated in H. pylori-associated gastritis and gastric cancer [15]. COX-2 is reported to be significantly correlated with angiogenesis [16], inhibition of apoptosis [17] and invasiveness [18] in gastric cancer. Although the oncogenic mechanisms of COX-2 and H. pylori-induced inflammation in gastric carcinogenesis have been illuminated, the precise molecular mechanism of its activation is not yet completely defined. Accumulating evidence suggest that COX-2 expression is regulated by epigenetic mechanisms, which associated with the clinical outcome of gastric cancer [19, 20]. Therefore, it arouses our interest to see whether H. pylori-induced COX-2 upregulation may be controlled by JMJD2B-mediated epigenetic mechanisms.
In the present work, we investigated the role of JMJD2B in H. pylori-induced gastric carcinogenesis and discovered a novel intracellular signaling pathway involving JMJD2B and COX-2 in H. pylori-mediated pathogenesis.
RESULTS
Induction of JMJD2B expression by H. pylori in gastric epithelial cells
First, immortalized human gastric epithelial cells (GES-1) and poorly differentiated gastric cancer cells (AGS) were used to study the relationship between H. pylori infection and JMJD2B expression. JMJD2B mRNA and protein levels were significantly increased over time with H. pylori infection in both cell lines (Figure 1A-1C). Similarly, the luciferase assay showed that JMJD2B promoter activity was stimulated by H. pylori infection in both cells (Figure 1D). These findings demonstrated that H. pylori induced JMJD2B upregulation at transcriptional level. To further determine which factor of H. pylori was responsible for the increased JMJD2B expression, CagA plasmid, CagA-depleted H. pylori strain and heat-inactivated H. pylori were used. qRT-PCR and western blot analysis showed that CagA did not promote the mRNA and protein levels of JMJD2B in AGS and GES-1 cells (Figure 1E-1F). Furthermore, when cells were incubated with CagA-depleted H. pylori strain, a comparable increase in JMJD2B expression was detected (Figure 1G-1H). Thus, H. pylori CagA is not the factor that stimulates JMJD2B expression. The mRNA level of JMJD2B was not increased in AGS and GES-1 cells after heat-inactivated H. pylori infection (Figure 1I), suggesting that physical interaction of viable bacteria was required to induce JMJD2B expression. We further examined the biological function of JMJD2B on H. pylori-induced clonogenic potential in both cells. As shown in Figure 1J-1K, H. pylori infection promoted clonogenic ability of both cells, and this promotion was restrained by knockdown of JMJD2B.
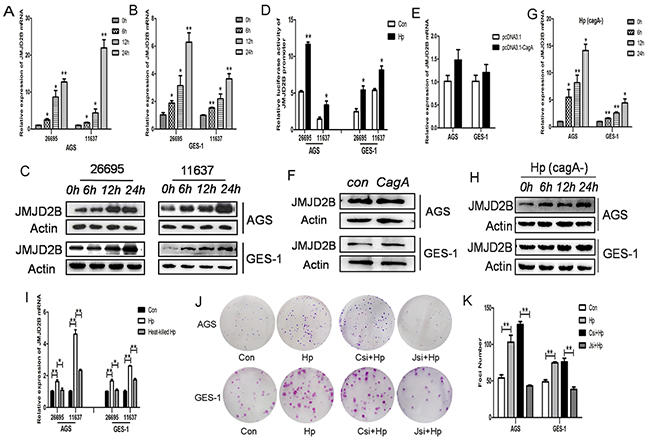
Figure 1: JMJD2B is induced by H. pylori infection independent of CagA. Infection experiments were conducted with MOI (multiplicity of infection) of 100:1. A-C. qRT-PCR and Western blot analysis of JMJD2B mRNA and protein levels in AGS and GES-1 cell lines infected with H. pylori at indicated time points. D. Luciferase activity of JMJD2B promoter in both cells with H. pylori infection for 12h. E-F. qRT-PCR and Western blot analysis of mRNA and protein levels of JMJD2B in AGS and GES-1 cell lines transfected with control and CagA plasmid. G-H. qRT-PCR and Western blot analysis of JMJD2B expression in both cell lines infected with CagA-depleted H. pylori. I. qRT-PCR analysis of JMJD2B mRNA level in H. pylori or heat-killed H. pylori-infected AGS and GES-1 cells. J-K. The colony formation assay was assessed in control cells or JMJD2B knockdown cells with H. pylori infection for 12h. The number of colonies was analyzed. *P <0.05, **P <0.01 compared with control. Data are mean ± SEM of 3 independent experiments.
β-catenin stimulates H. pylori-induced JMJD2B upregulation
Since β-catenin pathway is involved in H. pylori-induced inflammatory response and gastric tumorigenesis, we determined the effect of H. pylori infection on β-catenin activation in our system. Luciferase reporter assay showed significantly increased levels of TOP/FOP ratio compared to control in AGS cells (Figure 2A). The upregulation of β-catenin target genes MMP-7 and c-myc were also detected after H. pylori infection (Figure 2B). The above results demonstrated that H. pylori induced β-catenin activation in our cell lines. Analysis of JMJD2B promoter sequence showed a potential binding site of TCF/LEF and our previous study had shown a physical interaction of JMJD2B and β-catenin in gastric cancer cells, we then asked whether β-catenin regulated JMJD2B expression in these cells. JMJD2B mRNA and protein levels were significantly decreased in AGS and GES-1 cells after knocking down β-catenin expression using specific siRNA (Figure 2C-2D). The luciferase assay revealed that inhibition of β-catenin expression reduced the luciferase activity driven by the JMJD2B promoter (Figure 2E). Finally, we examined the role of β-catenin in H. pylori-induced JMJD2B expression. It can be seen from Figure 2F-2G that β-catenin silencing inhibited H. pylori-induced JMJD2B upregulation. To further determine the regulatory mechanism, we constructed pGL2-JMJD2B promoter vector containing the TCF/LEF binding site CTTTGA which can be bound by β-catenin and pGL2-JMJD2B-mutant promoter vector where β-catenin binding site was mutated to TGGGAC (Figure 2H). The luciferase assay showed that H. pylori-induced JMJD2B promoter activity was dramatically attenuated by knocking down β-catenin (Figure 2I). JMJD2B promoter activity was not increased after mutation of the putative β-catenin binding site, which showed that β-catenin was required for the transcription activation of JMJD2B following H. pylori infection (Figure 2J). Taken together, these results reveal that H. pylori activates β-catenin activity increasing JMJD2B transcription in gastric epithelial cells.
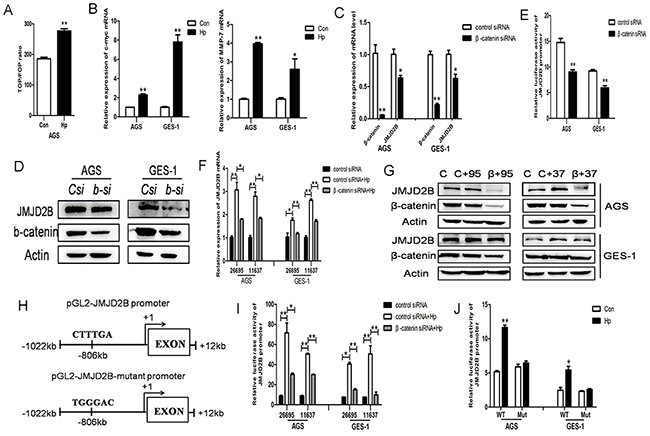
Figure 2: β-catenin stimulates H. pylori-induced JMJD2B upregulation. A. AGS cells were transiently transfected with pTOP-FLASH or pFOP-FLASH reporter plasmids separately and cotransfected with TK plasmid. 48h later, the cells were infected with H. pylori for 12h and luciferase activity was examined. B. qRT-PCR analysis of MMP-7 and c-myc, the target genes of β-catenin, was performed after H. pylori infection in AGS and GES-1 cells. C-D. qRT-PCR and western blot analysis of mRNA and protein levels of β-catenin and JMJD2B in control and β-catenin siRNA-transfected AGS and GES-1 cells. E. Luciferase activity of JMJD2B promoter in control and β-catenin siRNA-transfected AGS and GES-1 cells. F-G. qRT-PCR and Western blot analysis revealed that knocking down β-catenin expression by siRNA suppressed H. pylori-induced upregulation of JMJD2B mRNA and protein levels. H. Schematic structure of pGL2-JMJD2B promoter and pGL2-JMJD2B mutant promoter. I. Luciferase reporter assay revealed that H. pylori-induced JMJD2B promoter activity was significantly inhibited by β-catenin knockdown. J. Luciferase activity was determined with the wild-type or mutant JMJD2B reporter transfection after H. pylori infection. * P <0.05, ** P <0.01 compared with control. Data are mean ± SEM of 3 independent experiments.
JMJD2B is required for H. pylori-induced COX-2 activation
Recent studies have reported the induction of COX-2 by H. pylori infection. Here, we also found that COX-2 mRNA level was elevated in GES-1 and AGS cells after H. pylori infection (Figure 3A). Besides, H. pylori activated COX-2 expression independent of CagA (Figure 3B). Since JMJD2B was also increased by H. pylori in a CagA-independent manner, we assessed the role of JMJD2B in H. pylori-induced COX-2 upregulation. As shown in Figure 3C-3D, COX-2 mRNA and protein levels were both decreased by JMJD2B knockdown compared with control in AGS and GES-1 cells. Luciferase reporter assay showed that inhibition of JMJD2B significantly reduced the luciferase activity driven by the COX-2 promoter (Figure 3E). These results suggested that JMJD2B induced COX-2 transcriptional activation in gastric epithelial cells. To further confirm H. pylori-induced COX-2 expression is regulated by JMJD2B, GES-1 and AGS cells were transfected with JMJD2B siRNA and then infected with H. pylori. H. pylori-induced increase in COX-2 mRNA and protein levels were blocked by JMJD2B silencing (Figure 3F-3G). Similarly, H. pylori-induced COX-2 promoter activity was significantly inhibited by JMJD2B knockdown (Figure 3H). Therefore, these findings indicate that JMJD2B is required for H. pylori-induced COX-2 expression.
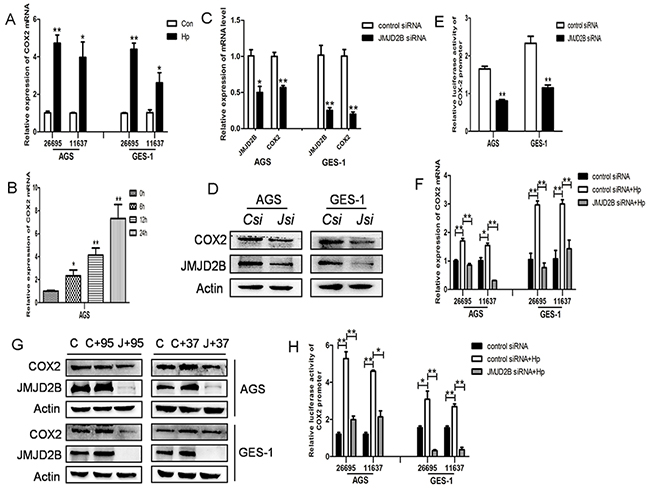
Figure 3: H. pylori-induced COX-2 activation is JMJD2B-dependent. A. qRT-PCR analysis of COX-2 mRNA level in H. pylori-infected AGS and GES-1 cells. B. qRT-PCR analysis of COX-2 mRNA level in CagA-depleted H. pylori-infected AGS cells. C-D. qRT-PCR and Western blot analysis of mRNA and protein levels of JMJD2B and COX-2 in JMJD2B siRNA-transfected AGS and GES-1 cells. E. Luciferase activity of COX-2 promoter in AGS and GES-1 cells by JMJD2B knocking down. F-G. qRT-PCR and Western blot analysis showed that inhibition of JMJD2B by siRNA suppressed H. pylori-induced COX-2 mRNA and protein levels. H. Luciferase reporter assay showed that H. pylori-induced COX-2 promoter activity was inhibited by JMJD2B knockdown. * P <0.05, ** P <0.01 compared with control. Data are mean ± SEM of 3 independent experiments.
JMJD2B regulates COX-2 expression via cooperating with NF-κB in gastric epithelial cells
Next, we determined the molecular mechanism that JMJD2B regulated COX-2 expression. As a histone demethylase, JMJD2B usually cooperates with transcription factors to regulate gene expression by demethylating histone H3K9me3. NF-κB has been demonstrated to be a major transcription factor regulating COX-2 expression, we asked whether JMJD2B modulated COX-2 expression by interaction with NF-κB during H. pylori infection. To this end, we performed Co-IP assay in AGS and GES-1 cells after H. pylori infection and a physical interaction of JMJD2B and NF-κB was detected (Figure 4A). Then, ChIP assay was prepared after H. pylori infection and ChIP-qRT-PCR was performed using specific primers amplifying the COX-2 promoter region (from –327 to +19) containing a NF-κB binding site (Figure 4B). We found that H. pylori significantly increased the binding of JMJD2B and NF-κB (p65) to COX-2 promoter region in AGS cells (Figure 4C). Moreover, a marked reduction of H3K9me3 and sharp accumulation of H3K9me2 at the COX-2 promoter was observed (Figure 4D). Finally, we further investigated the H3K9 methylation pattern in the COX-2 promoter in the presence and absence of JMJD2B as well as in the presence and absence of H. pylori infection. JMJD2B depletion significantly enhanced H3K9me3 level at the COX-2 promoter region, but the increase of H3K9me3 was inhibited by H. pylori infection (Figure 4E). Moreover, a reduction of H3K9me2 was observed after JMJD2B knockdown, while the reduction was restored by H. pylori infection (Figure 4E). Therefore, H. pylori increases binding of JMJD2B to COX-2 promoter where it demethylates H3K9me3 and recruits NF-κB to bind on COX-2 promoter to facilitate COX-2 induction.
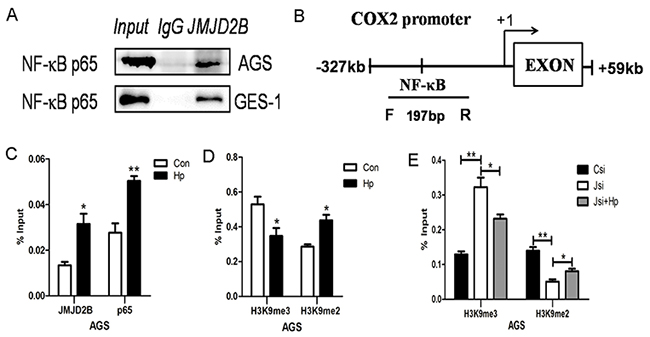
Figure 4: JMJD2B cooperates with NF-κB to regulate COX-2 expression in gastric epithelial cells. A. Interaction between JMJD2B and p65. AGS and GES-1cells infected with H. pylori were immunoprecipitated with anti-JMJD2B antibody and the coimmunoprecipitated p65 was detected using anti-p65 antibody. B. Schematic representation of COX-2 promoter showing the locations of the PCR primer used for ChIP assay. C-D. ChIP-qRT-PCR was performed in AGS cells to evaluate the enrichment of JMJD2B, NF-κB and histone H3K9me3 and H3K9me2 on the COX-2 promoter region. E. The methylation level of H3K9 in the COX-2 promoter region under the presence and absence of JMJD2B as well as H. pylori infection. * P <0.05, ** P <0.01 compared with control. Data are mean ± SEM of 3 independent experiments.
H. pylori regulates JMJD2B and COX-2 expression in vivo
To investigate whether H. pylori induces JMJD2B and COX-2 activation in vivo, C57BL/6J mice were infected with H. pylori (SS1) or PBS using intragastric administration and killed at 8wk after inoculation. qRT-PCR analysis showed that the mRNA levels of JMJD2B and COX-2 were both increased in H. pylori-infected gastric mucosal samples (Figure 5A-5B).
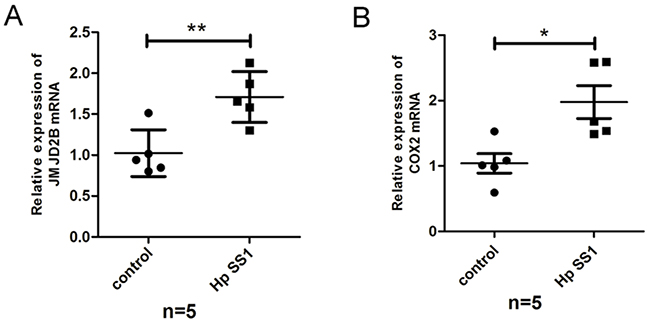
Figure 5: H. pylori regulates JMJD2B and COX-2 expression in vivo. A-B. qRT-PCR analysis of mRNA levels of JMJD2B and COX-2 in gastric mucosal samples of H. pylori-infected mice. * P <0.05, ** P <0.01.
JMJD2B is overexpressed in human gastritis and gastric cancer tissues
Finally, we determined whether the results from the cell lines and animal experiments have any clinical relevance. Immunohistochemical staining showed that JMJD2B was gradually increased in human specimens from gastritis to gastric cancer (Figure 6A). Moreover, the positive staining of JMJD2B were detected in H. pylori positive gastritis samples (61.5%, 8/13), which was higher than that of H. pylori negative samples (15.4%, 6/39) (Table 1). We also found that JMJD2B expressed in 20% (7/35) superficial gastritis tissues and in 66.7% (8/12) atrophic gastritis tissues. These results indicate that JMJD2B is involved in the process of H. pylori-induced inflammation to gastric cancer.
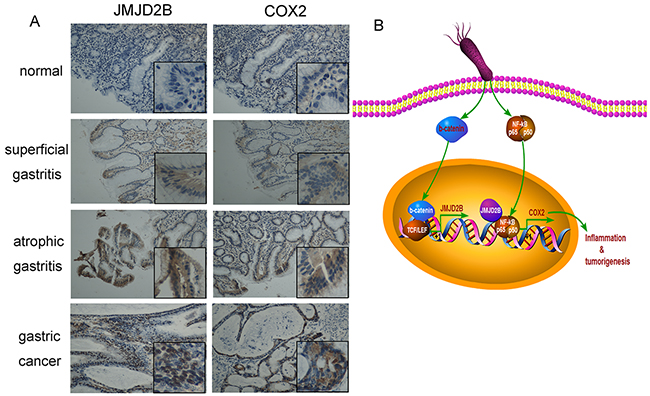
Figure 6: Expression of JMJD2B and COX-2 in human clinical samples from chronic inflammation to gastric cancer. A. Expression of JMJD2B and COX-2 is examined by immunohistochemistry staining in normal, superficial gastritis, atrophic gastritis and gastric cancer samples. Representative images are shown. Original magnification: ×200, magnification in the box: ×400. B. An outline of the main pathways illustrated in our work.
Table 1: Association between JMJD2B expression and H. pylori infection in chronic gastritis patients
Variables |
Total |
JMJD2B expression |
P value |
|
|---|---|---|---|---|
0-1 |
2-3 |
|||
H. pylori |
||||
Positive |
13 |
5 |
8 |
P<0.01 |
Negative |
39 |
33 |
6 |
|
NOTE: 0-1, low expression of genes; 2-3, high expression of genes.
χ2 test
JMJD2B expression positively correlates with COX-2 in chronic gastritis and gastric cancer tissues
We also detected COX-2 expression in gastritis and gastric cancer tissues. COX-2 level was consistent with the expression trends of JMJD2B in gastritis and gastric cancer samples (Figure 6A). Moreover, a strong positive correlation between JMJD2B and COX-2 levels in gastritis (Table 2) and gastric cancer (Table 3) tissues was found.
Table 2: Association between expression of JMJD2B and COX-2 in human gastritis mucosa
Variables |
Total |
JMJD2B expression |
P value |
|
|---|---|---|---|---|
0-1 |
2-3 |
|||
COX-2 expression |
||||
0-1 |
38 |
32 |
6 |
P<0.01 |
2-3 |
14 |
3 |
11 |
|
NOTE: 0-1, low expression of genes; 2-3, high expression of genes.
χ2 test
Table 3: Association between expression of JMJD2B and COX-2 in human gastric cancer tissues
Variables |
Total |
JMJD2B expression |
P value |
|
|---|---|---|---|---|
0-1 |
2-3 |
|||
COX-2 expression |
||||
0-1 |
18 |
14 |
4 |
P<0.05 |
2-3 |
14 |
6 |
8 |
|
NOTE: 0-1, low expression of genes; 2-3, high expression of genes.
χ2 test
DISCUSSION
Although numerous studies have been conducted in H. pylori pathogenesis and carcinogenesis, the detailed mechanism remains undefined. In this study, we focus on the critical role of histone demethylase JMJD2B in the transformation from chronic inflammation to gastric cancer induced by H. pylori. We demonstrate that JMJD2B is induced by H. pylori in a CagA-independent manner in both gastric epithelial cells and gastric cancer cells. Knockdown of JMJD2B significantly inhibits H. pylori-induced cell proliferation in vitro. H. pylori infection induces β-catenin nuclear translocation and lymphocyte enhancer factor/T cell factor (LEF/TCF) transactivation. β-catenin, together with LEF/TCF binds to JMJD2B promoter and activates its expression. The upregulated JMJD2B then stimulates COX-2 transcription by cooperating with NF-κB, which eventually promotes H. pylori-induced carcinogenic process.
It is well known that H. pylori-induced chronic inflammation is one of the leading factors for the initiation and development of gastric carcinoma [3]. Numbers of studies have been done to elicit the association between H. pylori infection and gastric cancer. H. pylori activates multiple intracellular pathways in epithelial cells, such as NF-κB, β-catenin pathway and PI3K/AKT pathway. These pathways affect various cellular functions, leading to increased inflammatory cytokine production, altered apoptosis rate, deregulated epithelial cell proliferation and differentiation, and finally resulting in gastric oncogenic transformation [21-24]. Moreover, H. pylori-induced epigenetic alterations, especially histone modifications, have been strongly implicated in gene-expression alteration and tumorigenesis [25, 26]. Infection with H. pylori causes acetylation of H4, which is associated with p21 gene expression in gastric epithelial cells [27]. H. pylori induces cag pathogenicity island-dependent H3S10 phosphorylation, which is associated with c-Jun upregulation and hsp70 downregulation and cell cycle premitotic arrest [6]. As an epigenetic molecule, JMJD2B has been reported to possess oncogenic activities in human cancers [12, 28]. Recently, we demonstrated that JMJD2B promoted cell proliferation, survival, invasion and metastasis in gastric cancer [13, 14], indicating that JMJD2B plays a key role in gastric tumorigenesis and progression. However, little is known about the involvement of JMJD2B in H. pylori-induced inflammation to cancer.
In this work, we investigated the early transcription of JMJD2B after H. pylori infection. Both the mRNA and protein levels of JMJD2B were significantly increased in response to H. pylori infection in different gastric epithelial cells, and the induction was not dependent on the CagA status. We also determined that JMJD2B stimulated cell clonogenic ability induced by H. pylori. These results indicate that JMJD2B activation may be an important step in H. pylori pathogenesis.
Among the signaling pathways H. pylori activated, β-catenin plays an important role in inflammation and gastric carcinogenesis. [29]. H. pylori infection induces β-catenin transcriptional activity, LEF/TCF activation, and target genes expression, which are involved in many cellular processes including cell cycle control, differentiation and cell migration [30, 31]. We previously reported that JMJD2B was physically associated with β-catenin and enhanced its nuclear localization and transcription [14], then we determined the role of β-catenin in the regulation of JMJD2B induced by H. pylori. As expected, we found a potential binding site of β-catenin on JMJD2B promoter. Furthermore, our results revealed that β-catenin upregulated JMJD2B expression by increasing JMJD2B promoter transcription activity following H. pylori infection.
Since COX-2 is known to be associated with H. pylori-induced inflammatory response and tumorigenesis [32, 33], illuminating the mechanism that how COX-2 is deregulated would help shed new light on H. pylori pathogenicity. Here we determined that JMJD2B upregulated H. pylori-induced COX-2 transcription. It has been reported that H. pylori induces COX-2 expression by activating transcription factor NF-κB, which is a key regulator of immune and inflammatory responses and regulates many processes in carcinogenesis [34]. Our Co-IP and ChIP assays showed the interaction between JMJD2B and NF-κB in gastric cancer cells and the increased binding of JMJD2B with NF-κB to COX-2 promoter coupled with the decrease of H3K9me3 and increase of H3K9me2 by H. pylori infection.
Consistent with the results from cell lines, JMJD2B and COX-2 expression was increased in H. pylori-infected gastric tissues in vivo. Furthermore, overexpression of JMJD2B and COX-2 was observed in clinical specimens from gastritis to gastric cancer and JMJD2B level was significantly associated with H. pylori infection (P<0.01). We also found that JMJD2B positively correlated with COX-2 expression in both gastritis and gastric cancer tissues. These findings indicate that JMJD2B activation and COX-2 upregulation contribute to gastric inflammation and carcinogenesis.
In conclusion, we determined the role and regulatory mechanism of JMJD2B in the progress of gastric inflammation to gastric cancer induced by H. pylori infection. The H. pylori-β-catenin-JMJD2B-COX-2 signaling cascade may be a new mechanism for the initiation and development of gastric cancer. JMJD2B may serve as a novel target for the treatment and early intervention of gastric cancer.
MATERIALS AND METHODS
Cell culture, siRNA and plasmid transfection
Human gastric epithelial cell line GES-1 and human gastric cancer cell line AGS were purchased from the Cell Resource Center, Shanghai Institute of Biochemistry and Cell Biology at the Chinese Academy of Sciences (Shanghai, PR China). GES-1 and AGS cells were cultured in RPMI-1640 medium (Gibco, USA) and F12 medium (Gibco, USA) respectively supplemented with 10% fetal bovine serum (Gibco, USA). Both cell lines were incubated at 37°C in a humidified atmosphere containing 5% CO2. Chemical modified Stealth™ JMJD2B siRNA and scramble siRNA were purchased from Invitrogen. β-catenin siRNA was bought from GenePharma company (Shanghai, PR China). The sequences for JMJD2B and control siRNA were: JMJD2B siRNA, 5’-UCU CCA UCA CCU GCC UCA AGC ACA A-3’; control siRNA, 5’- CCU ACA UCC CGA UCG AUG AUG UUG A-3’. Lipofectamine 2000 (Invitrogen, USA) was used to transfect siRNAs into cells according to the protocol. PcDNA3.1-CagA plasmid was kindly provided by Yongliang Zhu (Zhejiang University, China) and it was described previously [35]. PcDNA3.1-CagA plasmid and pcDNA3.1 vector were transfected with X-treme GENE HP Transfection Reagent (Roche Diagnostics, Germany) according to the protocol. TCF/LEF-1 reporter (TOP-FLASH) and mutation vector (FOP-FLASH) were bought from Addgene (Addgene plasmid 12456 and 12457, Cambridge, MA).
H. pylori strains and bacterial culture
H. pylori strains (NCTC 11637, 26695 and SS1) were kindly provided by Dr. Jianzhong Zhang (Chinese Disease Control and Prevention Center; Beijing, China). All strains were inoculated into Brucella broth containing 5% FBS under microaerobic conditions (5% O2, 10% CO2, and 85% N2) at 37°C. After 48h, H. pylori were collected and resuspended in phosphate buffered saline. Heat-killed H. pylori suspensions were prepared by incubating bacteria at 70°C for 10min followed by 95°C for 5 min.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA were extracted using Trizol reagent (Invitrogen, USA). After quantification, RNA was reverse transcribed into cDNA with RevertAid First Strand DNA Synthesis (RT) kit (Fermentas, Canada). Then, quantitative real-time PCR was performed with the SYBR Premix Ex Taq system (TaKaRa) using the Bio-Rad CFX96™ Real-Time PCR System (Bio-Rad). β-actin was used as an endogenous control. The primer sequences were shown in Table 4. Relative expression of the target genes were determined using the 2-ΔΔCt method.
Table 4: primers for PCR
Genes |
Primers |
|---|---|
JMJD2B |
5’-CCAGAGGCTTCCTTGCAGACAA-3’ (forward) |
5’-CCAAACTCCTGCCTCAGCCATT-3’ (reverse) |
|
β-catenin |
5’-GAAGGTGTGGCGACATATGCA-3’ (forward) |
5’-ATCCAAGGGGTTCTCCCTGGGC-3’ (reverse) |
|
COX-2 |
5’-GCCCAGCACTTCACGCATCAG-3’ (forward) |
5’-AGACCAGGCACCAGACCAAAGACC-3’ (reverse) |
|
β-actin |
5’-AGTTGCGTTACACCCTTTCTTG-3’ (forward) |
5’-CACCTTCACCGTTCCAGTTTT-3’ (reverse) |
Western blot analysis
Total proteins were lysed with RIPA lysis buffer containing proteinase inhibitor, PMSF (Solarbio). Protein concentration was determined by the BCA reagent kit (Beyotime). Equal amounts of protein (80ug) were separated by 10% SDS-PAGE and transferred to PVDF membranes, which were blocked in 5% non-fat milk, and then incubated with the primary antibodies against JMJD2B (Bethyl Laboratories, Montgomery, USA), COX-2 (Cayman chemical, USA), β-catenin (Cell Signaling Technology, Billerica, USA) and β-actin (Santa Cruz Biotechnology, Santa Cruz, USA) at 4°C overnight. The membranes were then washed in TBS-T and incubated with anti-mouse or rabbit horseradish peroxidase-conjugated secondary antibodies and developed with the enhanced chemiluminescence method (ECL, Millpore). β-actin was a loading control.
Coimmunoprecipitation (Co-IP)
Cells infected with H. pylori were lysed in RIPA lysis buffer. Extracts were clarified by centrifugation to collect the supernatant. The IP antibody aganist JMJD2B (Bethyl Laboratories, Montgomery, USA), p65 (ab7970, abcam, USA) and IgG (Santa Cruz Biotechnology) was added to the protein, and the mixture was gently shaken and incubated at 4°C for 5h. Then the solution was added with 20μl protein G PLUS-Agarose beads (Santa Cruz Biotechnology) at 4°C overnight. Immunoprecipitates were collected, washed, lysed, and boiled for Western blot analysis.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed in AGS cells using an immunoprecipitation assay kit (Millipore, USA) according to the protocol provided. Immunoprecipitated DNA sample was amplified using primers specific to the COX-2 promoter: 5’-GGCAAAGACTGCGAAGAAGA-3’ (Forward); 5’-ATTGCGTAAGCCCGGTGGG-3’ (Reverse) Quantitative real-time PCR was carried out using the Bio-Rad CFX96™ Real-Time PCR System (Bio-Rad). The amount of DNA coprecipitated with specific antibody was calculated in comparison to the total input DNA.
Luciferase reporter assay
AGS and GES-1 cells were seeded in 24-well plates and JMJD2B promoter vector (–1022/+12), COX-2 promoter vector (–327/+19), pTOP-FLASH or pFOP-FLASH reporter plasmids (0.5 ug each) were separately transfected into the cells. After 48h, cells were lysed in passive lysis buffer and luciferase activity was measured by use of the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA), and the target promoter-driven firefly luciferase activity was normalized to that of the Renilla control. Each experiment was performed in triplicate and repeated three times.
Colony formation assay
AGS and GES-1 cells were infected with H. pylori or transfected with JMJD2B siRNA or control siRNA for corresponding time, and seeded into 6-well plates (300 cells/well) incubating for 10 days. Then, cells in the plates were fixed with methanol and stained with crystal violet, and the number of colonies with more than 50 cells was counted.
Immunohistochemistry
Paraffin-embedded human gastric tissues underwent deparaffinization and antigen retrieval, then were incubated with primary antibodies against JMJD2B (1:100, Bethyl Laboratories, USA), COX-2 (1:100, Cayman chemical, USA) at 4°C overnight. Then slides were incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies and developed with use of the DAB Kit (Gene Tech, shanghai, China). Slides were finally counterstained with hematoxylin.
Patient specimens
Human gastritis specimens were collected from 52 patients undergoing gastroscopy. None of the patients had taken drugs such as antibiotics or anti-inflammatory drugs before the examination. Resected tissues from 32 patients with gastric cancer were obtained during surgery. None of the patients had received radiotherapy or chemotherapy before surgery. All the tissues from patients were obtained from Qilu Hospital, Shandong University (Jinan, PR China). The study was approved by the local ethics committee.
Animal experiment
Specific-pathogen-free male C57BL/6J mice of 6-week-old (the Experimental Animal Center of Shandong University, China) were used in this study. We randomly divided 10 mice into 2 groups which were control group (n=5) and infection group (n=5). The infection group received 1 × 109 colony-forming units of H. pylori SS1 by intragastric gavage for 3 times after 12h fasting every other day. The control group received 1mL PBS each time in the same way. After eight weeks, mice were sacrificed and gastric mucosal samples were collected for JMJD2B and COX-2 detection. All animal experimental protocols were approved by the local Ethics Committee of Shandong University.
Statistical analysis
Comparisons between different groups were analyzed by Student’s t-test. The correlation between JMJD2B and COX-2 expression was evaluated with the χ2 test. P< 0.05 was considered statistically significant.
ACKNOWLEDGEMENTS
We thank Professor Peter Staller (Biotech Research and Innovation Centre, University of Copenhagen, Copenhagen, Denmark) for providing pGL2-JMJD2B(-1022/+12) plasmid. We also thank Dr Dawei Xu (Karolinska Institute, Stockholm, Sweden) for providing COX-2 promoter vector (originally from Dr Hiroyasu Inoue, Nara Women’s University, Japan). This work was supported by the National Basic Research Program of China (973 Program, No. 2012CB911202) and the National Natural Science Foundation of China (No. 81372680, 81371781, 81402108, 81471991 and 81571960) and the Science Foundation for outstanding young scientists of Shandong Province (No. BS2014SW015) and China Postdoctoral Science Foundation (No. 2014M551915).
CONFLICTS OF INTEREST
No conflicts of interest were declared.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87-108.
2. Crew KD and Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol. 2006; 12:354-362.
3. Peek RM, Jr. and Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006; 208:233-248.
4. Peek RM, Jr. and Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002; 2:28-37.
5. Ding SZ, Fischer W, Kaparakis-Liaskos M, Liechti G, Merrell DS, Grant PA, Ferrero RL, Crowe SE, Haas R, Hatakeyama M and Goldberg JB. Helicobacter pylori-induced histone modification, associated gene expression in gastric epithelial cells, and its implication in pathogenesis. PLoS One. 2010; 5:e9875.
6. Fehri LF, Rechner C, Janssen S, Mak TN, Holland C, Bartfeld S, Bruggemann H and Meyer TF. Helicobacter pylori-induced modification of the histone H3 phosphorylation status in gastric epithelial cells reflects its impact on cell cycle regulation. Epigenetics. 2009; 4:577-586.
7. Kurdistani SK. Histone modifications as markers of cancer prognosis: a cellular view. Br J Cancer. 2007; 97:1-5.
8. Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M and Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006; 125:467-481.
9. Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G and Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006; 20:1557-1562.
10. Martin C and Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005; 6:838-849.
11. Das ND, Choi MR, Jung KH, Park JH, Lee HT, Das A, Kim SH and Chai YG. Functional analysis of histone demethylase Jmjd2b on lipopolysaccharide-treated murine neural stem cells (NSCs). Neurotox Res. 2013; 23:154-165.
12. Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, Yang X, Li Y, Han X, Zhang Y, Xuan C, Yao Z and Shang Y. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc Natl Acad Sci U S A. 2011; 108:7541-7546.
13. Li W, Zhao L, Zang W, Liu Z, Chen L, Liu T, Xu D and Jia J. Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is overexpressed in gastric cancer. Biochem Biophys Res Commun. 2011; 416(3-4):372-378.
14. Zhao L, Li W, Zang W, Liu Z, Xu X, Yu H, Yang Q and Jia J. JMJD2B promotes epithelial-mesenchymal transition by cooperating with beta-catenin and enhances gastric cancer metastasis. Clin Cancer Res. 2013; 19:6419-6429.
15. Jackson LM, Wu KC, Mahida YR, Jenkins D and Hawkey CJ. Cyclooxygenase (COX) 1 and 2 in normal, inflamed, and ulcerated human gastric mucosa. Gut. 2000; 47:762-770.
16. Leung WK, To KF, Go MY, Chan KK, Chan FK, Ng EK, Chung SC and Sung JJ. Cyclooxygenase-2 upregulates vascular endothelial growth factor expression and angiogenesis in human gastric carcinoma. Int J Oncol. 2003; 23:1317-1322.
17. Tatsuguchi A, Matsui K, Shinji Y, Gudis K, Tsukui T, Kishida T, Fukuda Y, Sugisaki Y, Tokunaga A, Tajiri T and Sakamoto C. Cyclooxygenase-2 expression correlates with angiogenesis and apoptosis in gastric cancer tissue. Hum Pathol. 2004; 35:488-495.
18. Saukkonen K, Rintahaka J, Sivula A, Buskens CJ, Van Rees BP, Rio MC, Haglund C, Van Lanschot JJ, Offerhaus GJ and Ristimaki A. Cyclooxygenase-2 and gastric carcinogenesis. APMIS. 2003; 111:915-925.
19. de Maat MF, van de Velde CJ, Umetani N, de Heer P, Putter H, van Hoesel AQ, Meijer GA, van Grieken NC, Kuppen PJ, Bilchik AJ, Tollenaar RA and Hoon DS. Epigenetic silencing of cyclooxygenase-2 affects clinical outcome in gastric cancer. J Clin Oncol. 2007; 25:4887-4894.
20. Park GY, Joo M, Pedchenko T, Blackwell TS and Christman JW. Regulation of macrophage cyclooxygenase-2 gene expression by modifications of histone H3. Am J Physiol Lung Cell Mol Physiol. 2004; 286:L956-962.
21. Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, Shiratori Y and Omata M. Distinct mechanism of Helicobacter pylori-mediated NF-kappa B activation between gastric cancer cells and monocytic cells. J Biol Chem. 2001; 276:44856-44864.
22. Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, Whitehead R, Gaus K, O’Brien DP, Romero-Gallo J and Peek RM, Jr. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005; 102:10646-10651.
23. Nagy TA, Frey MR, Yan F, Israel DA, Polk DB and Peek RM, Jr. Helicobacter pylori regulates cellular migration and apoptosis by activation of phosphatidylinositol 3-kinase signaling. J Infect Dis. 2009; 199:641-651.
24. Cho SO, Lim JW, Kim KH and Kim H. Involvement of Ras and AP-1 in Helicobacter pylori-induced expression of COX-2 and iNOS in gastric epithelial AGS cells. Dig Dis Sci. 2010; 55:988-996.
25. Nardone G, Compare D, De Colibus P, de Nucci G and Rocco A. Helicobacter pylori and epigenetic mechanisms underlying gastric carcinogenesis. Dig Dis. 2007; 25:225-229.
26. Ding SZ, Goldberg JB and Hatakeyama M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010; 6:851-862.
27. Xia G, Schneider-Stock R, Diestel A, Habold C, Krueger S, Roessner A, Naumann M and Lendeckel U. Helicobacter pylori regulates p21(WAF1) by histone H4 acetylation. Biochem Biophys Res Commun. 2008; 369:526-531.
28. Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D, Leek R, Gatter KC, Ragoussis J and Harris AL. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010; 70:6456-6466.
29. Polakis P. Wnt signaling and cancer. Genes Dev. 2000; 14:1837-1851.
30. Gnad T, Feoktistova M, Leverkus M, Lendeckel U and Naumann M. Helicobacter pylori-induced activation of beta-catenin involves low density lipoprotein receptor-related protein 6 and Dishevelled. Mol Cancer. 2010; 9:31.
31. Brabletz T, Jung A, Dag S, Hlubek F and Kirchner T. beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol. 1999; 155:1033-1038.
32. Chang YJ, Wu MS, Lin JT and Chen CC. Helicobacter pylori-Induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J Immunol. 2005; 175:8242-8252.
33. Rodriguez C, Lopez P, Pozo M, Duce AM, Lopez-Pelaez M, Fernandez M and Alemany S. COX2 expression and Erk1/Erk2 activity mediate Cot-induced cell migration. Cell Signal. 2008; 20:1625-1631.
34. Lin MT, Zuon CY, Chang CC, Chen ST, Chen CP, Lin BR, Wang MY, Jeng YM, Chang KJ, Lee PH, Chen WJ and Kuo ML. Cyr61 induces gastric cancer cell motility/invasion via activation of the integrin/nuclear factor-kappaB/cyclooxygenase-2 signaling pathway. Clin Cancer Res. 2005; 11:5809-5820.
35. Zhu YL, Zheng S, Qian KD and Fang PC. Biological activity of the virulence factor cagA of Helicobacter pylori. Chin Med J (Engl). 2004; 117:1330-1333.