
INTRODUCTION
Genetic alterations underlying carcinogenesis have been characterized with the advance of next-generation sequencing technology. One of the main aims of cancer-genome sequencing studies, e.g., the Cancer Genome Atlas [1] and the International Cancer Genome Consortium [2], is to identify driver genes/mutations underlying tumor initiation, maintenance, progression, and metastasis [3]. Distinguishing cancer drivers from millions of somatic mutations (e.g., passengers) remains a monumental challenge. Significantly mutated genes across multiple samples have been exploited based on the hypothesis that positive selection has operated on recurrent mutations and hence has functional relevance [4, 5]. Although TCGA identified significantly mutated genes based on mutation-frequency or -pattern, it remains difficult to prioritize specific driver genes/mutations in patient-specific manners due to substantial tumor heterogeneity. The identification of patient-specific drivers may serve as candidate drug targets for personalized therapy, as advocated in the precision medicine initiative [6].
It is of vital therapeutic potential in finding mutated peptides, which will provide a significant implication in identifying possible driver mutations [7]. In our previous study, we prioritized five personalized candidate mutation-drivers in a hyper-mutated hepatocellular carcinoma patient, which were patient-specific and not reported, by characterizing the expression of tumor-mutated alleles from genome to mRNA to protein [8]. Our proposed ‘multi-omics’ strategy utilized the mutation profile (genome, transcriptome and proteome) from a given patient rather than a population-based study, combined with the catalogue of known cancer drivers and current cancer knowledgebase. However, the roles of the prioritized driver genes and mutations in hepatocarcinogenesis are unclear.
In the present study, we aimed to provide evidence to support the validity of the multi-omics strategy by investigating the impact of missense mutations on structure-function relationship of these genes and assessing the functional effects of the prioritized genes, especially the patient-specific mutations, on cell behaviors in hepatoma cell lines.
RESULTS
A multi-omics strategy for prioritizing patient-specific mutation-driver genes in hepatocellular carcinoma
In our previous study [8], we employed a multi-omics strategy for prioritizing personalized candidate mutation-driver genes (Figure 1). The strategy built upon the following principles: 1) the near-saturation of the number of significantly mutated cancer drivers [9, 10]. Vogelstein [9] commented that ‘the number of frequently altered Mut-driver genes (mountains) is nearing saturation’. Lawrence et al. [10] estimated that near-saturation of significantly mutated genes may be achieved with 600-5000 samples per tumour type. The near-saturation of significant mutated genes provided a basis for prioritizing personalized mutation-driver genes; 2) the effect of tumor-mutated alleles as inferred by its expression at the mRNA and/or protein levels. A fundamental question in proteogenomics is which protein coding alterations are expressed at the protein level [7, 11]. The mutated cancer genomes produce mutant transcriptome and proteome, therefore, mutant proteins that give the cancer cell its oncogenic properties. The mutant proteins are produced only by tumor cells and have found to be functionally related to cancer driver mutations. 3) the current knowledgebase of cancer genes. A causal network analysis based on the Ingenuity Knowledge Base (IPA®) [12] can be used to characterize causal effect between the genes and diseases, e.g., hepatocellular carcinoma.
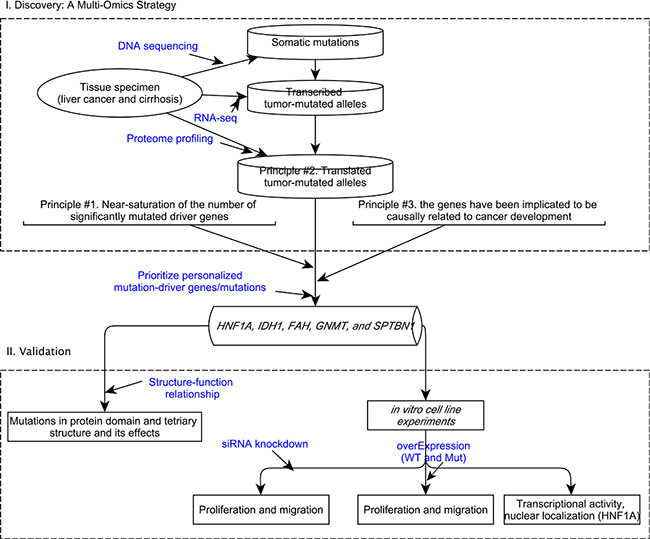
Figure 1: A conceptual framework to prioritize personalized candidate mutation-driver genes based on a multi-omics strategy and its validation.
We obtained high throughput sequencing data by whole-exome sequencing, transcriptomic sequencing (RNA-seq) and proteome profiling in a hyper-mutated (due to MSH2 inactivation) hepatocellular carcinoma patient. We characterized the expression patterns of 4980 tumor-mutated alleles and found that only 42% and 3.5% tumor-mutated alleles were transcribed and translated, respectively. The screening steps in prioritizing personalized mutation-driver genes were illustated in detail previously [8]. Notably, the two most frequently mutated (~20%) genes in HCC (i.e., TP53 and CTNNB1) were not mutated in this patient, and the tumor-mutation allele of a missense mutation in ARID1A was not transcribed. We prioritized five personalized candidate driver genes/mutations in this patient (Table 1), which were patient-specific and were not reported in hepatocellular carcinoma in the Catalog of Somatic Mutation in Cancer database (COSMIC, version 75) [13].
Table 1: Prioritized personalized candidate mutation-driver genes
Chr_pos |
Symbol |
Protein |
AA_change |
PDB |
|---|---|---|---|---|
chr12:121431992 |
HNF1A |
NP_000536.5 |
p.S247T |
1IC8 |
chr6:42928545 |
GNMT |
NP_061833.1 |
p.A14T |
1R74 |
chr2:209104698 |
IDH1 |
NP_005887.2 |
p.V294M |
1T0L |
chr15:80473495 |
FAH |
NP_000128.1 |
p.I392V |
NA |
chr2:54882240 |
SPTBN1 |
NP_003119.2 |
p.N1952K |
NA |
Validation of the expression of mutated protein in hepatoma cell lines
Cancer cell lines are commonly used as in vitro models for clinical tumors although cultured cancer cells might have different genetic characteristics from in situ tumors [14]. We assessed both the loss- and gain-of-function of the prioritized genes (Table 1) on hepatoma cell behavior in four hepatoma cell lines (Huh7, SK-Hep1, PLC/PRF/5 and SMMC-7721), which were previously established from human primary hepatocellular carcinoma.
We first assessed the genetic aberration background of the five genes by sequencing the targeted mutation sites in hepatoma cell lines, and we did not note any changes in the sequenced nucleotide (data not shown). The expression of four targeted genes in five hepatoma cell lines and a normal hepatic cell line (LO2) were evaluated, indicating the protein expression of the targeted genes among the hepatoma and normal hepatic cell line were similar (Figure S1).
We used the knockdown approach of multiple siRNAs to reduce the endogenous protein expression in hepatoma cells (Figure S2). We then constructed recombinant vectors exogenously expressing wild-type and mutant allele of HNF1A, GNMT, IDH1 and FAH (SPTBN1 was excluded due to its long coding region (7092 bp)). The expression of both the wild-type and mutant allele of these genes was similar (Figure 2A). These results confirmed our previous findings that both the wild-type and mutant allele of amino acid were identified in the liver cancer tissue by mass spectrometry [8]. A positive correlation of the spectral count of the wild-type and mutant allele of the amino acid (r = 0.31, p = 4.2 × 10−5) from proteomics profiling was noted (Figure 2B).
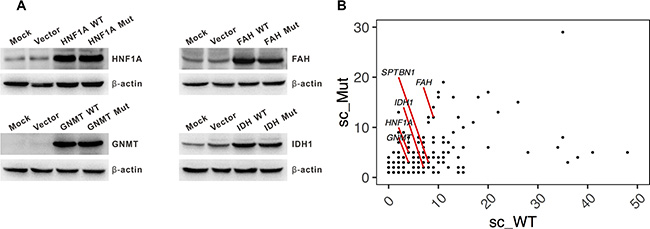
Figure 2: Expression of the wild-type and mutant allele of personalized candidate mutation-driver genes. (A) Western blot showing a similar expression of the wild-type and mutant allele of HNF1A, IDH1, GNMT, and FAH. (B) The spectral count of wild-type (sc_WT) and mutated- (sc_Mut) amino acid identified by proteomic profiling. Spectral count (sc) is defined as the total number of spectra identified for a protein in quantitative proteomics. The sc of the five genes are shown.
HNF1A S247T mutation is a loss-of-function oncogenic event
HNF1A is a transcription factor that is highly expressed in the liver, which is required for the regulation of the expression of several liver-specific genes, including FGA, FGB, SERPINA1, and AFP [15]. It has been suggested that bi-allelic inactivation of HNF1A may be an early step in the development of some hepatocellular carcinomas [16]. Somatic mutations in HNF1A in hepatocellular carcinoma from the COSMIC database [13] indicated that the number of non-silent mutations in the homeobox domain is significantly greater than other domains (p = 8.64 × 10−6) (Figure 3A).
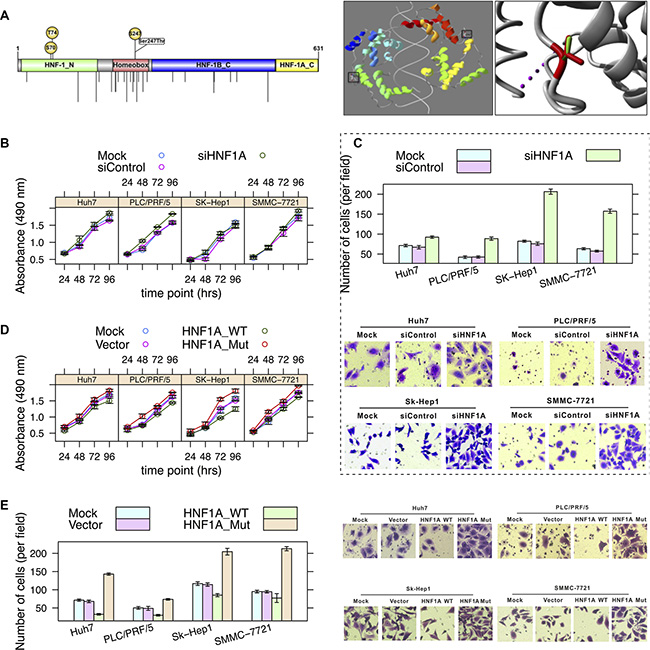
Figure 3: Effects of a loss-of-function mutation (S247T) in HNF1A. (A) Schematic diagram of domains (left panel) and image of crystallographic model (right panel) (PDB: 1IC8) of HNF1A. The Ser247 (phosphoserine) residue is locates on the homeobox domain. Nonsynonymous mutations occurring in hepatocellular carcinoma (from the COSMIC database) are shown by vertical lines. The side chains of both the wild-type and mutant allele of residue are shown in green and red, respectively. (B) Hepatoma cell lines SMMC-7721, SK-Hep1, PLC/PRF/5 and Huh7 cells were infected with HNF1A-targeting siRNA (AdsiHNF1A) or nontargeting control siRNA (AdsiControl). At 12 h after infection, cells were plated into a 24-well plate at 0.5 × 104/ml and were counted every 24 h in triplicate. Data are presented as the mean ± sd. (C) The Transwell assay of cell migration property in hepatoma cells. Cells were treated as described in Figure 3B. Quantitative evaluation of cell migration activity is presented as the means ± sd of five randomly selected microscopic fields from three independent experiments. Magnification:×200. (D) Hepatoma cells were mock-transfected or transfected with HNF1A-WT or HNF1A-Mut (S247T), respectively. Tumor cell growth was measured similar to Figure 3B. (E) Cells were treated as described in Figure 3D. Cell migration was determined by Transwell assay.
We first investigated the impact of S247T on the structure-function relationship of HNF1A based on its crystallographic structure of 83-279 amino acids (PDB: 1IC8) [17] (Figure 3A). The residue is located in the surface of HNF1A, which is involved in a multimer contact according to the PISA-database [18]. The mutation introduces a larger residue (threonine) at this position and this can disturb the multimeric interactions.
Second, we evaluated the effects of knockdown of HNF1A on hepatoma cell growth and migration. We noted that, in comparison with siRNA control, siRNA-mediated depletion of HNF1A significantly promoted hepatoma cell proliferation (ANOVA, p = 0.001, 1.4 × 10−6, 0.091, and 0.025 in Huh7, PLC/PRF/5, SK-Hep1, and SMMC-7721, respectively) (Figure 3B), as well as its migratory capacity (p = 1.2 × 10−5, 1.7 × 10−7, 3.1 × 10−9, and 5.5 × 10−8) (Figure 3C). These results indicated that HNF1A loss-of-function may play an important role in hepatocellular carcinoma tumorigenecity and metastasis.
Third, we tested the effects of overexpression of HNF1A on hepatoma cell growth. Compared with the vector control, overexpression of wild-type HNF1A led to a significantly lower proliferation rate (p = 0.0015, 5.7 × 10−5, 0.0012, and 5 × 10−6) (Figure 3D); the migratory capacity of multiple hepatoma cells was significantly suppressed by exogenously expression of wild-type HNF1A (p = 5.5 × 10−10, 1.6 × 10−4, 1.1 × 10−6, and 5.5 × 10−9) (Figure 3E). When compared with the wild-type HNF1A, overexpression of the mutant allele of HNF1A (S247T) showed a sharply increased cell proliferation (p = 4 × 10−6, 1.4 × 10−8, 2.5 × 10−5, and 7.5 × 10−8) (Figure 3D) and enhanced migratory ability (p = 4.2 × 10−11, 1.5 × 10−9, 3.4 × 10−7, and 8.7 × 10−7) (Figure 3E). Although the protein expression of mutants was clearly detected in hepatoma cells (Figure 2A), functional studies demonstrated the opposite effects of wild-type and mutant allele of HNF1A on hepatoma cell growth and migration.
Mutations in HNF1A may alter protein functions through decreased DNA binding capacity, reduced transactivation, or disrupted nuclear localization [19]. HNF1A was shown to bind to the HNF4A promoter region in mouse [20], and mutation in HNF1A demonstrated reduced transactivation activity of its target (HNF4A) promoter [21], which is essential for the differentiation of the hepatic lineage [22] and loss of HNF4A is a critical event in the progression of hepatocellular carcinoma [23]. Therefore, we first examined the functional consequences of the S247T mutation on HNF1A transcriptional activity by using the HNF4A promoter reporter (Figure 4A). The results demonstrated that the wild-type HNF1A resulted in an enhancement of transcriptional activity of pGL3-HNF4A in HepG2 and SMMC-7721 cells (t-test, p = 0.004 and 0.044, respectively). Compared with the wild-type HNF1A, the reporter activity of S247T mutation was reduced by 40% and 9% (p = 0.006 and 0.058). Next, we examined the cellular distribution of HNF1A in hepatoma cells transduced with wild-type HNF1A as well as S247T-mutated HNF1A (Figure 4B). The nuclear localization of cells infected with wild-type or mutated AdHNF1A did not differ in SMMC-7721 cells. These results indicated that HNF1A has a tumor suppressive effect and a loss-of-function mutation (S247T) in the homeobox domain leads to an oncogenic effect by affecting transcriptional activity.
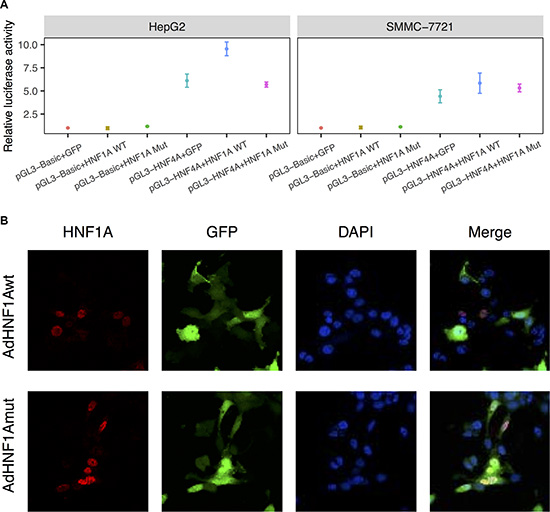
Figure 4: S247T mutation reduced HNF1A transactivity. (A) Luciferase assay of human HNF4A promoter constructs in HepG2 and SMMC-7721 cells. HepG2 and SMMC-7721 cells were co-transfected with pGL3-HNF4A and pAdTrack-HNF1A (HNF1A WT) or pAdTrack-HNF1AS247T (HNF1A Mut). At 36 hours after infection, cells were collected for luciferase assays. Results are presented as the mean relative luciferase activity against the activity of the pGL3-Basic control sample ± SD of three independent experiments. (B) SMMC-7721 cells were infected with adenoviruses expressing wild-type (AdHNF1Awt) or mutated-HNF1A (AdHNF1Amut). Thirty-six hours after adenovirus infection, HNF1A expression was detected by immunofluorescence with an anti-HNF1A antibody and an Alexa Fluor 647-labeled secondary antibody. Cells were counterstained with DAPI to label nuclei. The cellular localization of HNF1A were visualized under a fluorescence microscope. Magnification: 600×.
The V294M mutation in IDH1: loss-of- or gain-of-function?
Isocitrate dehydrogenases (e.g., IDH1) catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. Mutations in IDH1 have been reported in many types of tumors, e.g., gliomas, acute myeloid leukemias, and intrahepatic cholangiocarcinomas [24]. A high mutation frequency of the R132 residue was noted in gliomas and acute myeloid leukemias. Evidence suggested that the IDH1 mutation may be an early event in tumorigenesis with multiple downstream oncogenic consequences [25]. The residue (V294) is located in the isopropylmalate dehydrogenase-like (iso-dh) domain, which is highly conserved across homologous sequences and the mutated-allele was not observed (Figure 5A, left panel). This domain is important for the activity of the protein and is in contact with residues. The mutation (V294M) can affect this interaction and thus protein function (Figure 5A, right panel).
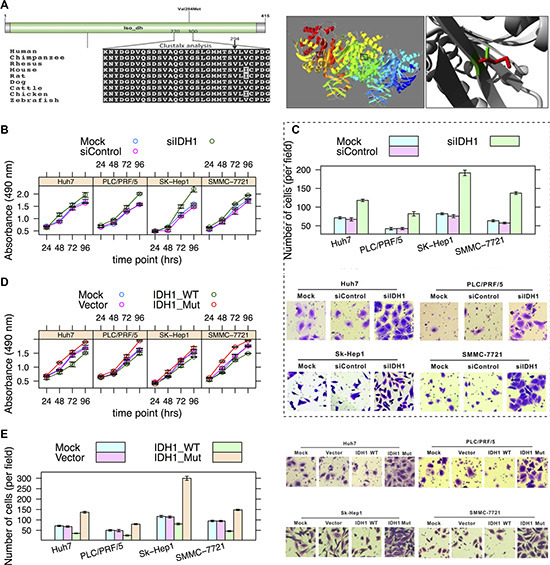
Figure 5: Effects of V294M in IDH1. (A) Schematic diagram of domains and multiple sequence alignment of IDH1 (270-300 AAs) (left) and image of a crystallographic model (right) (PDB: 1T0L) of IDH1 [49]. The V294 residue resides in the iso-dh domain. The side chains of both the wild-type and mutant allele of the residue are shown in green and red, respectively (right). (B) Cell growth curve. Hepatoma cell lines SMMC-7721, SK-Hep1, PLC/PRF/5 and Huh7 cells were infected with AdsiIDH1 or AdsiControl, respectively. Cell growth was determined by MTS assay similar to Figure 3B. (C) The Transwell assay of cell migration property in hepatoma cells depletion of IDH1. Magnification:×200. (D) Hepatoma cells were mock-transfected or transfected with IDH1-WT and IDH1-Mut (V294M), respectively. Tumor cell growth was measured by MTS assay. (E) Cells were treated as described in Figure 3D. Cell migration was determined by Transwell assay.
Whether IDH1 is an oncogene or a tumor suppressor gene is still debated [26]. Our in vitro cell experiment suggested that IDH1 may act as a tumor suppressor gene in hepatocellular carcinoma in that loss-of-function of IDH1 significantly promoted hepatoma cell growth (Figure 5B) and migration (Figure 5C); whereas overexpression of wild-type IDH1 had opposite effects (Figure 5D–5E). In contrast, the V294M mutation in IDH1 resulted in the promotion of cell growth and migration (Figure 5D–5E), suggesting a loss-of-function effect of V294M.
GNMT, FAH and SPTBN1
A Gnmt knockout mouse model of hepatocellular carcinoma has shown that genes related to the Wnt pathway (e.g., Ctnnb1, Ccnd1, and Myc) were up-regulated [27], indicating that GNMT is a tumor suppressor gene for liver cancer. GNMT is an enzyme that catalyzes the conversion of S-adenosyl-L-methionine (SAM) to S-adenosyl-L-homocysteine and sarcosine, which is involved in many essential cellular processes including biosynthesis, signal transduction, protein repair, chromatin regulation and gene silencing [28]. The residue (A14) is part of an Interpro domain of SAM-dependent methyltransferase (Figure S3A, left panel), which is important for the activity of the protein and in contact with other residues. The hydrophobicity of the wild-type (Alanine) and mutant (Threonine) residue differs, which may cause loss of hydrophobic interactions with other molecules on the surface of the protein. We noted that loss-of-function of GNMT by siRNA promoted hepatoma cell growth (Figure S3B) and migration (Figure S3C) in comparison with siRNA control. In addition, overexpression of the wild-type and mutant allele of GNMT had opposite effects (Figure S3D–S3E), i.e., inhibition or promotion of cell proliferation and migration, respectively.
FAH was mainly expressed in the liver and kidney [29, 30], which is the last enzyme in the tyrosine catabolism pathway that synthesizes acetoacetate and fumarate from L-phenylalanine [31, 32]. A deficiency of FAH is associated with type 1 hereditary tyrosinemia [33, 34]. The exact crystal-structure of FAH is unknown, and we performed homologous modeling of FAH upon mouse Fah (PDB: 1HYO) (Figure S4A). The residue (I392) is located in the FAA hydrolase domain which functions in hydrolase and catalytic activities. The mutant residue (Valine) is smaller than the wild-type (Isoleucine) residue, which will cause an empty space in the protein core. Therefore, the mutation may affect the domain interaction and protein function. We found that loss-of-function of FAH promoted hepatoma cell growth (Figure S4B) and migration (Figure S4C) in comparison with siRNA controls. Additionally, overexpression of the wild-type and mutant allele of FAH had opposite effects (Figure S4D–S4E), i.e., inhibiting or promoting cell proliferation and migration, respectively.
SPTBN1 plays an important role in the determination of cell shape, the arrangement of transmembrane proteins and the organization of organelles [35]. A recent report showed that by regulating the Wnt inhibitor kallistatin, loss of SPTBN1 activates Wnt signaling and promotes progression of hepatocellular carcinoma and Wnt signaling [36]. The mutation (N1952K) is located in one of the spectrin repeats (Figure S5A). We constructed the protein structure of SPTBN1 by homologous modeling using SWISS-MODEL [37] but with a GMQE (Global Model Quality Estimation) of 0.16, indicating a lower reliability. Loss-of-function of SPTBN1 promoted hepatoma cell proliferation and migration (Figures S5B–S3D).
DISCUSSION
In the present study, we provided supporting evidence for validating the patient-specific mutation-driver genes that was prioritized based on a multi-omics strategy (Figure 1) [8]. We hypothesized that the translated tumor-mutated allele would impact the structure-function relationship of the encoded protein. Therefore, the mutant-type of protein may play an important role in carcinogenesis.
Our results suggested that the prioritized mutation-driver genes act as tumor suppressor genes instead of oncogenes to regulate cell cycle. The in vitro evidence showed that loss- and gain-of-function of these genes had obvious effects on cell proliferation and migration in four hepatoma cell lines. In addition, we confirmed that the mutant-type protein was expressed in vitro (Figure 2), and the patient-specific missense mutations in four genes have similar loss-of-function effects. Of note, we characterized the functional consequence of S247T in HNF1A, in which phosphorylation of HNF1A at Ser247 was involved in HNF1A transcriptional activity [38]. Although both the wild- (serine) and mutant- (threonine) type can be phosphorylated, significantly reduced transcriptional activity was noted in the HNF1A mutant-type for HNF4A reporter (Figure 4A).
IDH1 was thought to be an oncogene whose mutations have stimulated the burgeoning field of tumor metabolism [9]. There is a mutation hotspot (R132H) in IDH1 in glioma [39] and acute myeloid leukemia [40] that is responsible for driving tumor progression. However, the hotspot R132H mutation was significantly under-represented in intrahepatic cholangiocarcinoma [41], suggesting a differential mutation pattern of IDH1 in different cancer types. Mutant IDH1 inhibits HNF4A to block hepatocyte differentiation and promote intrahepatic cholangiocarcinomas [42]. The promotion of cell proliferation and migration by siRNA knockdown (Figure 5B–5C), as well as its effects by overexpression of the wild-type and mutant allele of IDH1 (Figure 5D–5E), indicated that IDH1 may act as a tumor suppressor gene on hepatoma cell lines.
There are several limitations in the present study. First, we adapted tumor cells as model, which may not support the key role of driver genes in tumor cell transformation. However, the prioritized genes/mutations modulate tumor cell growth and aggressiveness (i.e., progression), which features one of malignant characteristics of tumor cells. Second, it is unclear whether there are synergistic effects of these candidate driver genes.
In conclusion, we validated the strategy for prioritizing personalized mutation-driver genes using multi-omics data. Our study provided evidences that these expressed mutations exert potential oncogenic effects, thus may have translational potential in personalized therapy. The proposed strategy provides a new avenue for the identification of personalized cancer driver mutations in a patient-specific manner.
MATERIALS AND METHODS
Homology modeling and mutation analysis
Information about protein domains was based on the Pfam [43] or Interpro [44] database. We used HOPE [45] to analyze the three-dimensional (3D) structural and functional effects of a non-synonymous mutation in the protein. HOPE has been suggested to perform similarly to manual approaches. HOPE collected information from 1) structural calculations on the PDB file base on WHAT IF [46]; if the PDB file was not available, YASARA was used to perform homology modeling, 2) conservation scores estimation by HSSP [47], 3) sequence-based predictions by DAS-servers [48], and 4) sequence annotations by Uniprot. All these data were combined with the known properties of the amino acids in a decision schedule, i.e., the effect of the mutated-allele on the 3D-structure of the protein. We also used the SWISS-model to perform homologous modeling if the crystallographic model was unknown for the interested protein.
Cell lines
Human hepatoma cell lines (SMMC-7721, SK-Hep1, PLC/PRF/5 and Huh7 cells) and a normal hepatic cell line (LO2) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone™, UT). All culture media were supplemented with 10% fetal bovine serum (FBS; Gibco®, Rockville MD), 100 units/mL penicillin, and 100 μg/mL streptomycin (Hyclone™).
Plasmids and adenoviruses
Full length coding sequences of human HNF1A, GNMT, FAH, and IDH1 were amplified by the polymerase chain reaction (PCR) and inserted into the shuttle vector pAdTrack-TO4 (obtained from Dr. T-C He, University of Chicago, Chicago IL). Mutated plasmid of HNF1A (S247T), FAH (I392V), IDH1 (V294M), and GNMT (A14T) (Table 1) were constructed by overlapping extension PCR and subcloned into pAdTrack-TO4 (primer sequences were listed in Table S1). All recombinant vectors were confirmed by Sanger sequencing. Recombinant wild-type HNF1A (AdHNF1Awt) and S247T-mutated HNF1A (AdHNF1Amut) were generated successfully in HEK293 cells using the AdEasy system. All recombinant adenoviruses expressed green fluorescent protein (GFP) as a marker for monitoring of infection efficiency.
Three pairs of oligonucleotides containing siRNA (Table S2) target sites for the coding region of HNF1A, GNMT, FAH, IDH1 and SPTBN1 were designed and subcloned into the Sfi I site of pSES vector (from Dr. T-C He) to generate adenovirus AdR-siHNF1A, AdR-siGNMT, AdR-siFAH, AdR-siIDH1, and AdR-siSPTBN1 using the AdEasy system. A scrambled shRNA control (AdR-siControl) that expresses RFP was used as a control.
The HNF4A promoter-luciferase reporter was generated by cloning an approximately 1 kb PCR fragment into the pGL3-Basic vector (Promega, Madison WI; E1751).
Western blotting
Proteins were extracted from cells with cell lysis buffer (Beyotime Biotechnology, Jiangsu, China) containing 1 mM phenylmethanesulfonyl fluoride (Beyotime). Approximately 50 μg proteins were separated on 10% polyacrylamide gels and electrotransferred to PVDF membranes (Millipore, Billerica MA). The membranes were immunoblotted with the following antibodies: HNF1A (Abcam, Cambridge UK; ab204306), FAH (Bioworld™, Atlanta GA; BS8270), IDH1 (Bioworld™; BS6918), GNMT (Bioworld™; BS8292) and SPTBN1 (Abcam; ab72239). Secondary goat anti-rabbit IgG (H+L)-horseradish peroxidase antibodies were purchased from Bioworld™ (BS13278). Endogenous β-actin (Bioworld™; AP0060) expression was used as the normalization control.
Immunofluorescence staining
Cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% formaldehyde for 30 min. Cells were then permeabilized with 0.5% Triton and incubated with HNF1A antibody (Abcam; ab204306) at 4°C overnight. After washing, cells were incubated with an Alexa Fluor 647-labeled secondary antibody (Invitrogen®, Carlsbad CA; A21244) for 1 h at room temperature and counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Roche, Basel, Switzerland) for 5 min. The expression of HNF1A was visualized under a laser scanning confocal microscope (Nikon, Tokyo, Japan; A1+R).
Cell migration assay
Cell migration was measured using transwell units with a polycarbonate filter (BD, San Jose CA). For knockdown assay, cells were mock-infected or infected with AdR-siHNF1A, AdR-siGNMT, AdR-siFAH, AdR-siIDH1, AdR-siSPTBN1 or AdR-siControl. For overexpression assays, cells were transfected with HNF1A-WT, HNF1A-Mut (S247T), GNMT-WT, GNMT-Mut (A14T), FAH-WT, FAH-Mut (I392V), IDH1-WT, IDH1-Mut (V294M), or vector control. Twenty-four hours after transduction, cells were suspended in 200 μL of serum-free medium and added at 4 × 104 cells/well in the upper chamber. A DMEM medium (600 μL) containing 10% FBS was added into the lower chamber to act as a chemoattractant. Cells were incubated at 37°C under 5% CO2. After 12 h, cells were fixed with 4% formaldehyde and stained with crystal violet. The numbers of migrated cells were counted in five fields (200×) on each membrane and the average per field was calculated.
Cell proliferation assay
Cell proliferation was measured using the CellTiter 96 AQ One Solution Cell Proliferation Assay (MTS) (Promega). Cells were treated as mentioned in cell migration assay. Infected cells (2 × 103) were re-plated in 96-well plates, 20 μL of the MTS reagent were added into each well, and the plates were incubated for 2 h at 37°C. The absorbance at 490nm was measured every 24 h until day 4 using a microplate reader (Bio-Tek, Winooski VT).
Luciferase assay
HepG2 and SMMC-7721 cells were cultured in 25 cm2 cell culture flasks and co-transfected with 3 μg of HNF4A responsive luciferase reporter pGL3-HNF4A and pAdTrack-HNF1A wt or pAdTrack-HNF1A mut using Lipofectamine V F2000 (Promega) following the manufacturer’s instructions. The pRL-TK plasmid (Promega) was added as an internal control. Cells were harvested 36 h post-transfection and subjected to the Dual-Luciferase® Reporter Assay (Promega). Each assay was performed in triplicate and repeated three times.
Amplification and sequence analysis of the targeted mutation sites of the five genes
The genome DNA was extracted from hepatoma cell lines using the Wizard Genomic DNA Purification Kit (Promega; A1120) according to the manufacturer’s instructions. The targeted mutation sites of the five genes were amplified by PCR and were sequenced by Sanger sequencing. All primer sequences are listed in Table S3.
Statistical analysis
Pairwise comparison for statistical significance was conducted with Student’s t test, and analysis of variance (ANOVA) was used to test for the effect of overexpression of the wild- and mutant-type gene in cell proliferation. We considered p < 0.05 to be statistically significant.
ACKNOWLEDGMENTS
None.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
The study was supported by the Recruitment Program of Global Youth Experts in China (KD). Research grants from the Major National S&T program (2013ZX10002002-005-003, NT), China National Natural Science Foundation (#81572683 and #81371827, NT), Natural Science Foundation Key Project of CQ CSTC (cstc2015jcyjBX0011, NT), the Outstanding Young talent program of Second Affiliated Hospital of CQMU (NT), and a research grant from CQMU (201401, NT).
REFERENCES
1. The Cancer Genome Atlas Home Page. Available from: http://cancergenome.nih.gov.
2. Chairperson TJH, Anderson W, Aretz A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, Guttmacher A, Guyer M, Hemsley FM, et al. International network of cancer genome projects. Nature. 2010; 464:993–8.
3. Mwenifumbo JC, Marra MA. Cancer genome-sequencingstudy design. Nat Rev Genet. 2013; 14:321–32.
4. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–8.
5. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150:251–63.
6. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015; 372:793–5.
7. Alfaro JA, Sinha A, Kislinger T, Boutros PC. Onco-proteogenomics: cancer proteomics joins forces with genomics. Nature Methods. 2014; 11:1107–13.
8. Ding K, Wu S, Ying W, Pan Q, Li X, Zhao D, Li X, Zhao Q, Zhu Y, Ren H, Qian X. Leveraging a multi-omics strategy for prioritizing personalized candidate mutation-driver genes: A proof-of-concept study. Sci Rep. 2015; 5:17564.
9. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–58.
10. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501.
11. Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014; 11:1114–25.
12. Krämer A, Green J, Pollard J, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014; 30:523–30.
13. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011; 39:D945–50.
14. Borrell B. How accurate are cancer cell lines? Nature. 2010; 463:858–8.
15. Courtois G, Morgan JG, Campbell LA, Fourel G, Crabtree GR. Interaction of a liver-specific nuclear factor with the fibrinogen and alpha 1-antitrypsin promoters. Science. 1987; 238:688–92.
16. Bluteau O, Jeannot E, Bioulac-Sage P, Marqués JM, Blanc J-F, Bui H, Beaudoin J-C, Franco D, Balabaud C, Laurent-Puig P, Zucman-Rossi J. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002; 32:312–5.
17. Chi Y-I, Frantz JD, Oh B-C, Hansen L, Dhe-Paganon S, Shoelson SE. Diabetes mutations delineate an atypical POU domain in HNF-1alpha. Mol Cell. 2002; 10:1129–37.
18. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Bio. 2007; 372:774–97.
19. Bjørkhaug L, Sagen JV, Thorsby P, Søvik O, Molven A, Njølstad PR. Hepatocyte nuclear factor-1 alpha gene mutations and diabetes in Norway. Journal of Clinical Endocrinology & Metabolism. 2003; 88:920–31.
20. Boj SF, Parrizas M, Maestro MA, Ferrer J. A transcription factor regulatory circuit in differentiated pancreatic cells. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:14481–6.
21. Estrada K, Aukrust I, Bjørkhaug L, Burtt NP, Mercader JM, García-Ortiz H, Huerta-Chagoya A, Moreno-Macías H, Walford G, Flannick J, Williams AL, Gómez-Vázquez MJ, Fernandez-Lopez JC, et al. Association of a low-frequency variant in HNF1A with type 2 liabetes in a Latino population. JAMA. 2014; 311:2305–10.
22. Santangelo L, Marchetti A, Cicchini C, Conigliaro A, Conti B, Mancone C, Bonzo JA, Gonzalez FJ, Alonzi T, Amicone L, Tripodi M. The stable repression of mesenchymal program is required for hepatocyte identity: A novel role for hepatocyte nuclear factor 4. Hepatology 2011; 53:2063–74.
23. Tanaka T, Jiang S, Hotta H, Takano K, Iwanari H, Sumi K, Daigo K, Ohashi R, Sugai M, Ikegame C, Umezu H, Hirayama Y, Midorikawa Y, et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4 in the pathogenesis of human cancer. J. Pathol. 2006; 208:662–72.
24. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010; 465:966–6.
25. Dimitrov L, Hong CS, Yang C, Zhuang Z, Heiss JD. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci. 2015; 12:201–13.
26. Reitman ZJ, Parsons DW, Yan H. IDH1 and IDH2: not your typical oncogenes. Cancer Cell. 2010; 17:215–6.
27. Liao Y-J, Liu S-P, Lee C-M, Yen C-H, Chuang P-C, Chen C-Y, Tsai T-F, Huang S-F, Lee Y-HW, Chen Y-MA. Characterization of a glycine N-methyltransferase gene knockout mouse model for hepatocellular carcinoma: Implications of the gender disparity in liver cancer susceptibility. Int J Cancer. 2009; 124:816–26.
28. Luka Z, Mudd SH, Wagner C. Glycine N-Methyltransferase and regulation of S-Adenosylmethionine levels. J Biol Chem. 2009; 284:22507–11.
29. Klebig ML, Russell LB, Rinchik EM. Murine fumarylacetoacetate hydrolase (Fah) gene is disrupted by a neonatally lethal albino deletion that defines the hepatocyte-specific developmental regulation 1 (hsdr-1) locus. P Nat Acad Sci USA. 1992; 89:1363–7.
30. Jacobs SMM, Beurden DHA van, Klomp LWJ, Berger R, Berg IET van den. Kidneys of mice with hereditary tyrosinemia type I are extremely sensitive to cytotoxicity. Pediatr. Res. 2006; 59:365–70.
31. Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG, Soriano P. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Gene Dev. 1993; 7:2298–307.
32. Endo F, Kubo S, Awata H, Kiwaki K, Katoh H, Kanegae Y, Saito I, Miyazaki J-i, Yamamoto T, Jakobs C, Hattori S, Matsuda I. Complete rescue of lethal Albino c 14CoS mice by null mutation of 4-hydroxyphenylpyruvate dioxygenase and induction of apoptosis of hepatocytes in these mice by in vivo retrieval of the tyrosine catabolic pathway. J Biol Chem. 1997; 272:24426–32.
33. Aponte JL, Sega GA, Hauser LJ, Dhar MS, Withrow CM, Carpenter DA, Rinchik EM, Culiat CT, Johnson DK. Point mutations in the murine fumarylacetoacetate hydrolase gene: Animal models for the human genetic disorder hereditary tyrosinemia type 1. Proc Natl Acad Sci USA. 2001; 98:641–5.
34. Phaneuf D, Lambert M, Laframboise R, Mitchell G, Lettre F, Tanguay RM. Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient. J Clin Invest. 1992; 90:1185–92.
35. Zhang R, Zhang C, Zhao Q, Li D. Spectrin: Structure, function and disease. Science China. Life sciences 2013; 56:1076–85.
36. Zhi X, Lin L, Yang S, Bhuvaneshwar K, Wang H, Gusev Y, Lee M-H, Kallakury B, Shivapurkar N, Cahn K, Tian X, Marshall JL, Byers SW, et al. II-Spectrin (SPTBN1) suppresses progression of hepatocellular carcinoma and Wnt signaling by regulation of Wnt inhibitor kallistatin. Hepatology. 2015; 61:598–612.
37. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014; 42:W252–8.
38. Lim S, Jin K, Friedman E. Mirk protein kinase is activated by MKK3 and functions as a transcriptional activator of HNF1alpha. J Biol Chem. 2002; 277:25040–6.
39. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–73.
40. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009; 361:1058–66.
41. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discovery. 2013; 3:730–41.
42. Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, Gurumurthy S, Akbay EA, Sia D, Cornella H, Miltiadous O, Walesky C, Deshpande V, et al. Mutant IDH inhibits HNF-4 to block hepatocyte differentiation and promote biliary cancer. Nature. 2014; 513:110–4.
43. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014; 42:D222–30.
44. Mitchell A, Chang H-Y, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, Sangrador-Vegas A, Scheremetjew M, Rato C, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015; 43:D213–21.
45. Venselaar H, Te Beek TAH, Kuipers RKP, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010; 11:548.
46. Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990; 8:52–6–29.
47. Glaser F, Rosenberg Y, Kessel A, Pupko T, Ben-Tal N. The ConSurf-HSSP database: the mapping of evolutionary conservation among homologs onto PDB structures. Proteins. 2005; 58:610–7.
48. Cserzö M, Wallin E, Simon I, Heijne G von, Elofsson A. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng. 1997; 10:673–6.
49. Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004; 279:33946–57.