
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is a clonal lymphoproliferative disorder characterized by the progressive accumulation of mature, typically CD5/CD19-positive B cells, most of which are in the G0/G1 phase of the cell cycle [1, 2]. Despite great advances in the management of CLL with the introduction of targeted drugs, such as ibrutinib and idelalisib [3], chemoimmunotherapy with the purine analog fludarabine in combination with cyclophosphamide and the anti-CD20 monoclonal antibody rituximab (FCR) remains the standard first-line treatment for CLL [4]. However, except for some selected patients with favorable characteristics [5], nearly all patients eventually relapse post-FCR. Therefore, identification of therapies with novel mechanisms of action, particularly agents that could complement existing therapeutic options, remains a priority.
Purine analogs like fludarabine and cladribine are prodrugs that require intracellular conversion into triphosphate derivatives to be active. Triphosphate analogs have several mechanisms of action, which collectively lead to induction of DNA strand breaks, p53 up-regulation and apoptosis [6]. The first and rate-limiting step of purine analog activation is catalyzed by deoxycytidine kinase (dCK), an enzyme that plays a critical role in purine analog efficacy [7]. We previously demonstrated that several genotoxic agents, including UV-irradiation and DNA synthesis inhibitors, such as aphidicolin, enhance dCK activity in leukemic cells by inducing phosphorylation of dCK at Ser-74 [8, 9]. This finding led us to postulate that combination of nucleoside analogs with drugs like aphidicolin might enhance their conversion into nucleoside analog triphosphate and thereby their effectiveness. However, increase of dCK activity in CLL cells through Ser-74 phosphorylation was found to augment the activation of pyrimidine analogs, but not of purine analogs [10]. Yet, we observed in preliminary experiments that aphidicolin potentiated the cytotoxicity of fludarabine and cladribine in primary CLL cells, suggesting a still unidentified mechanism of sensitization to purine analogs.
Aphidicolin is a natural tetracyclic diterpene that inhibits DNA polymerases α, δ, and ε, and therefore DNA replication and certain forms of DNA repair [11]. Especially nucleotide excision repair (NER), which requires DNA polymerases δ and ε for the final step of the repair, is sensitive to aphidicolin [12]. This property of aphidicolin has provided the rationale for combining it with DNA-damaging anticancer drugs whose effectiveness could be limited by activation of NER. In particular, aphidicolin has been shown to enhance the potency of cisplatin in primary ovarian tumors and of chlorambucil in CLL lymphocytes [13–15]. Also, it was found to potentiate the cytotoxicity of the pyrimidine analog cytarabine in blast cells from adult and pediatric patients with acute myeloid leukemia, whereas it did not modify sensitivity of the same cells to fludarabine [16, 17].
In the present study, we aimed to analyze the combination aphidicolin/fludarabine or aphidicolin/cladribine in CLL samples of a larger cohort of patients and determine the mechanism by which aphidicolin could increase purine analog cytotoxicity. Particularly, we assessed whether inhibition of DNA repair could underlie the beneficial effect of this combination in primary CLL cells.
RESULTS
Aphidicolin enhances sensitivity of primary CLL cells to fludarabine and cladribine
Cytotoxicity of aphidicolin and purine analogs, alone or in combination, was assessed using the MTT assay. Aphidicolin was used at 3 μM, a concentration that was found to increase dCK activity by 2- to 3-fold in CLL cells [9, 10]. At this concentration, aphidicolin alone induced negligible cytotoxicity, the loss of CLL cell viability being 5.6 ± 0.9 % after 4 days (n = 47). Overall, we investigated the combination aphidicolin/fludarabine in 47 samples from 29 individual CLL patients and the combination aphidicolin/cladribine in 32 samples from 21 patients. As illustrated in Figure 1A, the DNA polymerase inhibitor significantly increased CLL cell sensitivity to fludarabine and cladribine. On average, aphidicolin decreased the IC50 of fludarabine from 4.5 ± 1.2 to 1.0 ± 0.2 μM (n = 47) and that of cladribine from 2.2 ± 0.7 to 0.8 ± 0.3 μM (n = 32), thus increasing CLL cell sensitivity to fludarabine and cladribine by 4.5- and 2.8-fold, respectively (Figure 1B). Sensitization by aphidicolin (defined as the ratio of the IC50 obtained in the absence or presence of aphidicolin) ranged from 1 to 21.9 for fludarabine and from 1 to 8.2 for cladribine, being ≥ 2 in 39/47 analyses (83% of patients) for fludarabine and in 19/32 analyses (59% of patients) for cladribine. In only one of the 29 CLL samples, aphidicolin had no sensitizing effect for either of the analogs. Interaction of aphidicolin with fludarabine or cladribine, analyzed according to the multiplicative model [18], was found to be synergistic in almost all conditions (Figure 1C).
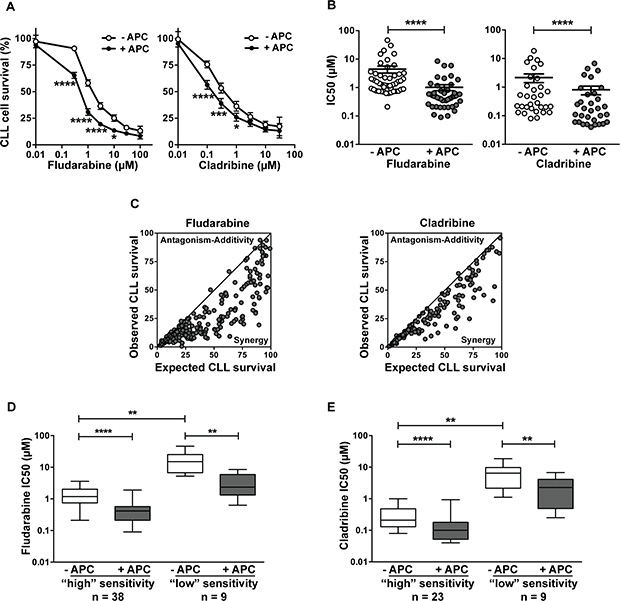
Figure 1: Aphidicolin enhances fludarabine and cladribine cytotoxicity in primary CLL lymphocytes. CLL lymphocytes were incubated in the presence or absence of 3 μM aphidicolin (APC) and various concentrations of fludarabine or cladribine for 96 h. Cell viability was measured using the MTT assay. Forty-seven and thirty-two analyses were performed for fludarabine and cladribine, respectively. (A) Dose-response curves of fludarabine or cladribine in the absence or presence of APC. Values shown are means ± SEM. Significance relative to the absence of APC was analyzed by two-way Anova followed by Bonferroni post-test. (B) IC50 of fludarabine and cladribine in the absence or presence of APC calculated from the individual data of Figure 1A. Individual IC50 and means ± SEM are shown. Significance relative to the absence of APC was analyzed by Wilcoxon matched-pairs signed-rank test. (C) Comparison between observed and expected cell survival for the combination fludarabine/APC and cladribine/APC was performed according to the multiplicative model [18]. The continuous line is x = y. (D–E) Effect of aphidicolin according to the sensitivity of CLL cells to purine analogs. CLL samples were divided into two groups (of “high” or “low” sensitivity) on the base of the IC50 of fludarabine (D) or cladribine (E) obtained in the absence of APC: < or ≥ 5 μM for fludarabine, and < or ≥ 1 μM for cladribine. Significance relative to the absence of APC was analyzed by Wilcoxon matched-pairs signed-rank test and difference between less and more sensitive samples was assessed by Mann-Whitney test. For all panels: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
As the sensitivity to fludarabine or cladribine widely varied among patients, we split the cohort into two subsets, one “more sensitive” and the other “less sensitive” to the drug, as determined by the choice of an arbitrary threshold IC50 value: 5 μM for fludarabine (Figure 1D) and 1 μM for cladribine (Figure 1E). Comparison showed that the sensitizing effect of aphidicolin was present in the two subgroups.
Sensitization by aphidicolin is independent of the clinical parameters of CLL patients
The sensitizing effect of aphidicolin was analyzed in relation with the clinical characteristics of the patients. CLL samples were stratified in different groups based on the IGHV mutational status, the treatment (untreated versus treated) and cytogenetics (17p deletion versus no deletion). Cytotoxicity of fludarabine was significantly lower in CLL cells from unmutated or previously treated than in mutated or untreated patients, but not significantly different between 17p deleted and non-deleted cases (Table 1A). Sensitization by aphidicolin was observed in each subtype, being very similar in samples from patients with good or poor prognostic features. Concerning cladribine, CLL cells from unmutated, pretreated or 17p deleted patients were less sensitive to the drug than their counterparts, but the difference was only significant between untreated and pretreated patients (Table 1B). Sensitization by aphidicolin was also noted in each subgroup. No correlation with CD38 expression or 11q deletion could be performed, due to insufficient number of patients with these characteristics.
Table 1: Correlation between sensitization by aphidicolin and clinical parameters
(A) Fludarabine |
|||
|---|---|---|---|
Characteristics (n) |
IC50 (μM) |
Sensitization ratio (−APC/+APC) |
|
−APC |
+APC |
||
IGHV |
|
|
|
Mutated (15) |
2.8 ± 0.5 |
0.7 ± 0.1 |
4.0**** |
Unmutated (6) |
19.1 ± 7.1b |
3.3 ± 1.3b |
5.8*ns |
Previous treatments |
|
|
|
No (30) |
2.1 ± 0.5 |
0.5 ± 0.1 |
4.2**** |
Yes (14) |
10.3 ± 3.6a |
2.2 ± 0.7a |
4.7***ns |
17p deletion |
|
|
|
Non-deleted (29) |
5.2 ± 1.9 |
1.1 ± 0.3 |
4.7**** |
Deleted (6) |
5.4 ± 2.3ns |
1.6 ± 0.9ns |
3.4*ns |
(B) Cladribine |
|||
|---|---|---|---|
Characteristics (n) |
IC50 (μM) |
Sensitization ratio (−APC/+APC) |
|
−APC |
+ APC |
||
IGHV |
|
|
|
Mutated (12) |
2.2 ± 1.0 |
0.9 ± 0.4 |
2.4*** |
Unmutated (5) |
3.4 ± 2.0ns |
2.1 ± 1.2ns |
1.6*ns |
Previous treatments |
|
|
|
No (21) |
1.0 ± 0.4 |
0.3 ± 0.1 |
3.3**** |
Yes (10) |
4.8 ± 2.0a |
1.9 ± 0.7b |
2.5**ns |
17p deletion |
|
|
|
Non-deleted (21) |
2.0 ± 1.0 |
0.7 ± 0.3 |
2.9**** |
Deleted (5) |
6.3 ± 3.3ns |
0.9 ± 0.4ns |
7.0**a |
When possible, CLL samples were classified according to known clinical prognostic markers and sensitization by aphidicolin (APC) was compared between the matched subgroups. IC50 values obtained for fludarabine (A) and cladribine (B) in the absence or presence of 3 μM APC are means ± SEM. Significance relative to the absence of APC was analyzed by paired t-test or Wilcoxon matched-pairs signed rank test: *P < 0.05; **P <0.01; ***P < 0.001; ****P < 0.0001. Significance relative to IGHV status, previous treatment or 17p deletion was analyzed by unpaired t-test with Welch’s correction: aP <0.05; bP < 0.01; ns: not significant.
Aphidicolin enhances apoptosis induced by purine analogs
Activation of apoptosis through the intrinsic pathway is considered as the major mechanism of cytotoxicity of purine analogs [6]. Analysis of cell death by flow cytometry using FITC-Annexin V and PI staining (Supplementary Figure 1) confirmed that fludarabine induces apoptosis in CLL cells, most of cells being Annexin-V+/PI– after 48 h of treatment, and that aphidicolin could increase fludarabine-induced apoptosis. In accordance with these results, we observed that cleavage of procaspase-9 and procaspase-3, which are crucial events for the initiation of apoptosis by the intrinsic pathway, were enhanced when aphidicolin was combined with fludarabine (Figure 2A). We could not detect the active cleaved form of caspase-3 by Western blot, but a fluorometric assay confirmed that caspase-3 activity was significantly higher when fludarabine was combined with aphidicolin (Figure 2B). Similar results were obtained with cladribine (not shown).
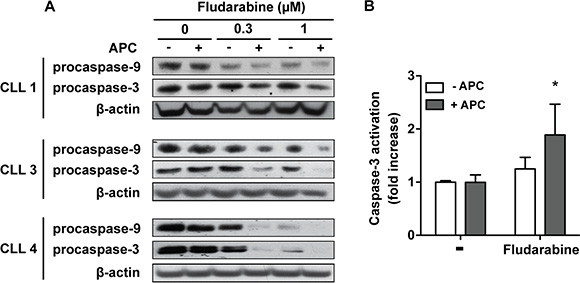
Figure 2: Aphidicolin enhances fludarabine-induced apoptosis. (A) Procaspase-9 and procaspase-3 were analyzed by Western blot in CLL cells from three patients after a 48 h-incubation in the absence or presence of 3 μM aphidicolin (APC) with or without fludarabine at 0.3 and 1 μM. β-Actin served as a loading control. (B) Caspase-3 activity was measured by a fluorometric assay in CLL cells after a 24 h-incubation in the absence or presence of 3 μM APC with or without 1 μM fludarabine (n = 4). Values are means ± SEM. Significance relative to the absence of APC was analyzed by two-way Anova followed by Bonferroni post-test: *P < 0.05.
Aphidicolin increases DNA damage in response to fludarabine and cladribine
To unravel the mechanisms underlying synergy between aphidicolin and purine analogs, we investigated the activation of p53, a key molecular event in chemotherapy using DNA-damaging agents. Whereas aphidicolin alone showed no effect, it increased the phosphorylation of p53 at Ser-15 induced by fludarabine and cladribine (Figure 3A, 3B). Similar results were obtained for p53 protein level, except in the CLL6 sample in which no effect of aphidicolin was noticed.
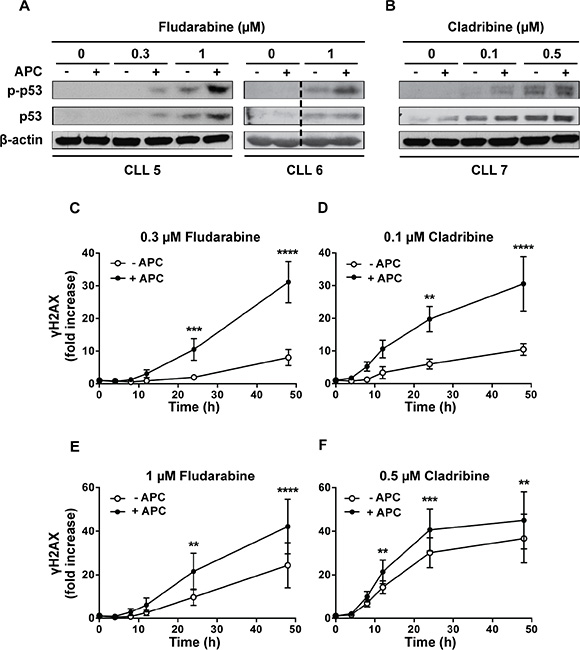
Figure 3: Effect of aphidicolin on p53 phosphorylation and γH2AX accumulation induced by purine analogs. (A–B) CLL cells from 3 different patients were incubated for 24 h with fludarabine (A) or cladribine (B) at the indicated concentrations in the absence or presence of 3 μM aphidicolin (APC). Phosphorylation of p53 at Ser-15 and total p53 were analyzed by Western blot. Data shown for CLL6 were cropped from the same western blot image and combined as indicated by the dotted line. (C–F) Time-course accumulation of γH2AX in CLL cells incubated with 0.3 or 1 μM fludarabine (C, E) or with 0.1 or 0.5 μM cladribine (D, F) in the absence or presence of 3 μM aphidicolin (APC). Results are means ± SEM of 5 independent experiments. Significance relative to the absence of APC was analyzed by two-way Anova followed by Bonferroni post-test: **P < 0.01; ***P < 0.001; ****P < 0.0001. In all panels, γH2AX is expressed as fold increase over untreatred cells.
We also analyzed the phosphorylation of histone H2AX on Ser-139, defined as γH2AX, which occurs shortly after DNA strand breaks and is considered as a surrogate marker of early DNA damage [19, 20]. A dose-dependent increase of γH2AX was clearly detected after incubation with fludarabine or cladribine for 24 h (Supplementary Figure 2). We then examined the influence of aphidicolin on the time-course accumulation of γH2AX at two clinically achievable concentrations of fludarabine (Figure 3C, 3E) or cladribine (Figure 3D, 3F). A latency period (varying between 4 and 12 h depending on the analog concentration) was observed, most probably due to the time required for accumulation of sufficient genotoxic damage. Aphidicolin, which alone did not cause significant γH2AX accumulation (data not shown), reduced this latency and markedly enhanced H2AX phosphorylation induced by fludarabine and cladribine, especially at lower purine analog concentrations (Figure 3C, 3D). These data corroborated that aphidicolin enhances the DNA damage caused by purine analogs.
Using fludarabine at 1 μM and cladribine at 0.1 μM, we verified that a 24 h-incubation in the presence of aphidicolin modified neither their accumulation under triphosphate derivatives, nor their incorporation into nucleic acids (results not shown), confirming our previous results at shorter time points [10]. We thus deduced that increase of purine analog-induced DNA damage by aphidicolin could not be explained by higher activation of these drugs. Finally, we tested the effect of aphidicolin on the cytotoxicity of chemotherapeutic drugs that do not require activation by dCK for their efficacy (Supplementary Table 1). We found that aphidicolin markedly potentiated the cytotoxicity of mafosfamide and doxorubicin in CLL cells, similarly to what was reported previously for chlorambucil [14]. In contrast, aphidicolin did not significantly increase the cytotoxicity of nutlin-3a that acts independently of DNA damage [21].
Aphidicolin impedes the repair of purine analog-induced DNA damage
We investigated whether aphidicolin could increase DNA damage induced by purine analogs through inhibition of DNA repair. CLL cells were incubated for 24 h in the presence of 1 μM fludarabine or 0.1 μM cladribine in order to induce γH2AX accumulation. Then, cells were washed to remove purine analogs, resuspended in fresh culture medium and incubated for several hours in the presence or absence of aphidicolin. In cells pretreated with fludarabine (Figure 4A, left panel) and incubated in the absence of aphidicolin, γH2AX level remained stable for about 12 h before declining gradually, consistent with a progressive repair of the DNA damage induced by fludarabine. In contrast, the presence of aphidicolin provoked additional accumulation of γH2AX, which was maintained over time at significantly higher level than in its absence. In cells pretreated with cladribine (Figure 4A, right panel), γH2AX decrease was also markedly slowed down by aphidicolin. These results indicate that aphidicolin inhibits DNA repair after exposure to purine analogs.
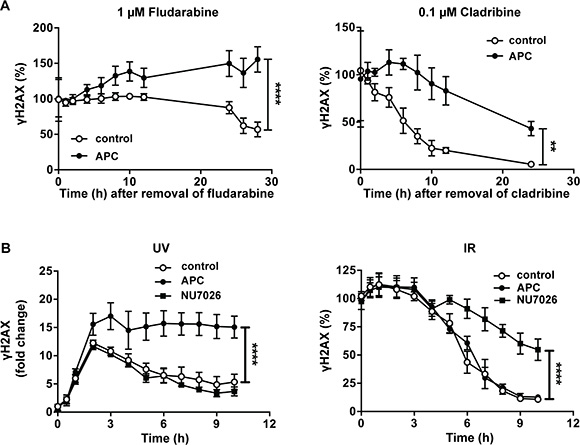
Figure 4: Effect of aphidicolin on DNA repair after various DNA damaging treatments. (A) CLL cells were preincubated for 24 h with 1 μM fludarabine (left panel) or 0.1 μM cladribine (right panel). Cells were then washed, resuspended in fresh medium and incubated in the absence or presence of 3 μM aphidicolin (APC). γH2AX was analyzed by flow cytometry at the indicated times and expressed as % of the value at time 0. (B) CLL cells were subjected to UV-C light (30 J/m2) (left panel) or IR at a dose of 5 Gy (right panel) and immediately incubated in the absence or presence of 3 μM APC or 10 μM NU7026. γH2AX was analyzed by flow cytometry at the indicated times and expressed as fold change (UV) or as % of the value at time 0 (IR). In all panels, results are means ± SEM of 3–5 independent experiments. Significance relative to the absence of APC (shown at the last incubation time) was analyzed by two-way Anova followed by Bonferroni post-test: **P < 0.01; ****P < 0.0001.
Aphidicolin inhibits DNA repair induced by UV-C, but not by ionizing radiation
Next step was to identify the DNA repair pathway inhibited by aphidicolin in response to purine analogs. The DNA polymerases δ and ε, which are inhibited by aphidicolin, are implicated in the DNA resynthesis step of various forms of DNA repair, comprising NER, mismatch repair (MMR) and long-patch base excision repair (BER), but not short-patch BER that only employs polymerase β [22]. Long-patch BER and MMR prominently handle repair during S and G2 phase of the cell cycle, whereas NER is active in both dividing and non-dividing cells [23]. Since CLL cells are non-dividing in vitro, we reasoned that NER could be the repair pathway targeted by aphidicolin. To substantiate this hypothesis, we analyzed the effect of aphidicolin on DNA damage repair after UV-C irradiation, a treatment known to activate NER in CLL cells [24, 25]. As illustrated in Figure 4B (left panel), UV-C irradiation triggered γH2AX accumulation that reached a maximal level after 2 h and was higher in the presence of aphidicolin. Thereafter, γH2AX progressively decreased in cells incubated in the absence of aphidicolin, whereas it was maintained at its maximal levels in its presence, indicating that repair of the UV-C-induced DNA damage was inhibited. For comparison, we performed the same type of experiments after ionizing radiation (IR) (Figure 4B, right panel). IR mainly activates the non-homologous end joining (NHEJ) pathway, which does not involve DNA polymerases δ or ε, but polymerases μ and λ [26]. H2AX phosphorylation induced by IR (5 Gy) was almost maximal at the first sampling time and started to decrease after 3 h consistent with progressive DNA repair. As expected, aphidicolin did not influence repair of DNA damage induced by IR in contrast with NU7026 that inhibits DNA-dependent protein kinase, a key actor of NHEJ [27]. We verified that NU7026 did not inhibit DNA repair after UV-C-irradiation (Figure 4B, left panel).
Fludarabine induces nuclear import of XPA in CLL cells
To determine whether purine analogs could activate NER, we analyzed the effect of fludarabine on the DNA repair protein XPA (xeroderma pigmentosum, complementation group A). XPA is an important repair factor in NER, which was shown to be imported from the cytoplasm into the nucleus after DNA damage induced by UV [28] or alkylating drugs [29, 30]. As illustrated in Figure 5A, we found that fludarabine induced dose-dependent translocation of XPA from cytoplasm to nucleus, suggesting that NER can be activated in response to purine analogs.
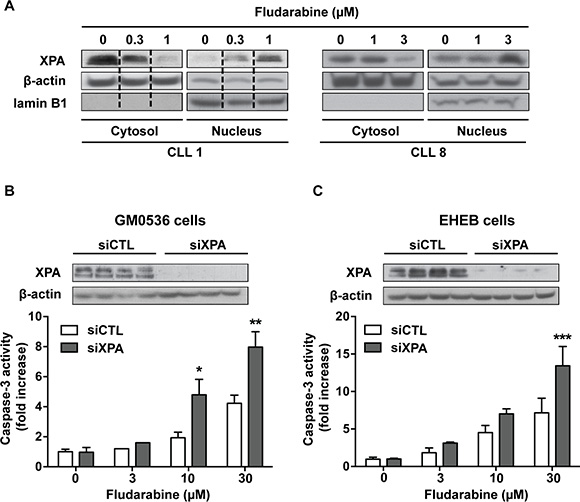
Figure 5: Involvement of XPA in cell sensitivity to fludarabine. (A) CLL cells were incubated for 24 h with fludarabine at the indicated concentrations. Cytoplasmic and nuclear fractions were extracted and analyzed for XPA by Western blot. β-Actin and lamin B1, a specific nuclear protein, served as loading controls. Data shown for CLL1 were cropped from the same western blot image and combined as indicated by the dotted lines. (B) GM0536 and (C) EHEB cells were transfected with scrambled (siCTL) or XPA siRNA (siXPA) for 24 or 48 h, respectively. Then, cells were incubated for additional 24 h with fludarabine at the indicated concentrations. Upper panels show XPA levels analyzed by Western blot in siCTL-treated and siXPA-treated cells and lower panels show caspase-3 activity measured by a fluorometric assay. Results are means ± SEM of 3 independent experiments. Significance relative to siCTL-treated cells was analyzed by two-way Anova followed by Bonferroni post-test: *P < 0.05; **P < 0.01; ***P < 0.001.
Down-regulation of XPA increases purine analog-induced apoptosis in cell lines
To determine if DNA damage repair by NER could limit the sensitivity of CLL cells to purine analogs, we used siRNA to down-regulate XPA. However, the cytotoxicity of electroporation procedure limited usefulness of primary CLL cells for these investigations. Therefore, we analyzed the effect of XPA silencing in lymphoblastoid cells (GM0536) and in the CLL cell line EHEB. Cells were transfected with scramble or XPA siRNA for 24 (GM0536) or 48 h (EHEB) and then incubated with fludarabine at increasing concentrations for additional 24 h. Relatively high concentrations of fludarabine were selected for these experiments due to the lower sensitivity of these cell lines to fludarabine with IC50 of 6.5 and 17.2 μM for GM0536 and EHEB cells, respectively. XPA protein was strongly down-regulated by XPA siRNA (Figure 5B, 5C, upper panels), which was concomitant with significantly higher caspase-3 activation in response to fludarabine (Figure 5B, 5C, lower panels).
DISCUSSION
In this study, we have shown that aphidicolin markedly sensitizes CLL cells to fludarabine and cladribine, decreasing their IC50 by 4.5- and 2.8-fold, respectively. This finding contrasts with previous observations that aphidicolin reduced fludarabine cytotoxicity in leukemic cell lines, in which incorporation into DNA during replication is the key mechanism of cytotoxicity [31, 32]. In quiescent CLL cells, incorporation of purine analogs into DNA occurs through background DNA repair activity, most likely by the aphidicolin-insensitive DNA polymerase β [33], explaining why no inhibition of their incorporation into DNA, and therefore of their cytotoxicity, was recorded in the presence of aphidicolin.
At the molecular level, aphidicolin enhanced p53 phosphorylation and accumulation of γH2AX, an early marker of DNA damage [19], indicating that aphidicolin increases DNA damage in response to purine analogs. Since aphidicolin did not trigger H2AX phosphorylation per se, was without effect on the activation of purine analogs, and maximally inhibited DNA repair synthesis in quiescent lymphocytes at the concentration used in our experiments [34], we postulated that aphidicolin enhances purine analog-induced DNA damage by reducing DNA repair. In line with this hypothesis, disappearance of γH2AX, which was found to occur after removal of purine analogs and can be considered as readout of DNA repair, was significantly slowed down by aphidicolin.
The possibility to increase purine analog efficacy by preventing DNA repair has still been little explored. Nevertheless, sensitization of CLL cells to fludarabine has been reported in the presence of NU7441, which impairs the NHEJ pathway through inhibition of DNA-dependent protein kinase [35], and methoxyamine, a BER inhibitor [36]. In addition, cladribine cytotoxicity was increased in NER-deficient lymphoblasts compared to wild-type cells [37]. Our observation that aphidicolin impedes DNA repair after treatment by purine analogs demonstrates that the DNA damage induced by these drugs is repaired, at least partially, by a pathway that involves DNA polymerases δ/ε. Among the DNA repair pathways involving polymerases δ/ε, namely long-patch BER, MMR and NER, only the latter is thought to operate in non-dividing cells [23]. That NER could be the pathway inhibited by aphidicolin in CLL cells in response to purine analogs is supported by several observations: (1) the repair of UV-C-induced DNA damage, known to occur by NER, was inhibited by aphidicolin, (2) XPA, an essential factor for NER, was translocated from the cytosol to the nucleus after fludarabine, as previously observed after UV and alkylating agent treatment [28, 29, 38], and (3) silencing of XPA in cell lines, including a CLL cell line, resulted in higher caspase-3 activation in response to fludarabine. Moreover, using genome-wide expression profiling, we previously observed that four NER-associated genes (PCNA, XPC, GADD45, and DDB2) were among the most up-regulated genes in CLL cells after treatment with fludarabine or cladribine in vitro [39]. Upregulation of XPC was also recorded in response to fludarabine in vivo [40]. All these data strongly indicate that the NER pathway can be activated by purine analogs in CLL cells, resulting in decrease of their efficacy.
Important finding of this study is the ability of aphidicolin to sensitize CLL cells to purine analogs regardless of prognostic factors of patients, including relapse, IGHV status and 17p deletion. Drug resistance represents a major therapeutic challenge in management of CLL [41]. Regarding purine analogs, several factors may be involved such as reduced intracellular accumulation of their active form, due to impaired transport or metabolism, or aberrant expression of certain Bcl-2 family proteins [6]. However 17p deletion, 11q deletion, and/or TP53 gene mutations, which impair the ATM/p53 signaling pathway and apoptosis in response to DNA damage, are among the best-documented factors of clinical resistance to purine analogs as well as to other genotoxic therapies [41, 42]. Strikingly, aphidicolin was able to synergize with purine analogs in all the 17p deleted cases investigated. Although the sample size was limited and p53 dysfunction might not be complete, these data suggest that increase of cell death induced by aphidicolin could occur by p53-dependent and -independent mechanisms. That DNA damage can induce signaling for apoptosis in a p53-independent manner has recently became increasingly evident from literature data in different types of cancer cells including the leukemic cell line HL-60 and primary CLL cells [35, 36, 43].
As inhibitor of DNA replication, aphidicolin possess strong antitumor activity in cancer cell lines in vitro. This property led to Phase I clinical trials with a water-soluble glycinate ester, which showed limited toxicity at concentrations achievable in humans, but also little antitumor effect [44]. Preclinical studies suggested that aphidicolin might be more effective in combination with platinum agents through inhibition of DNA repair elicited by these drugs [13]. However, clinical studies were not finalized, a major obstacle for clinical use being its low solubility and fast clearance from human plasma. Therefore, aphidicolin, as such, could not currently enter clinical trials in combination with fludarabine or cladribine, but studies are in progress to enable the development of novel aphidicolin derivatives with enhanced stability and inhibitory properties [45]. Regarding NER, many efforts have been made in last years to identify specific inhibitors of this pathway, but most so-called NER inhibitors appear to be nonspecific, have weak efficacy and are often toxic [46]. Last attempts pointed to find molecules that block interaction between essential components of the NER pathway, such as the interaction between ERCC1 and XPF, which is obligatory for the endonuclease activity of the complex. Novel inhibitors of this interaction have been recently identified and shown to enhance cytotoxicity of alkylating agents in cancer cell lines [47, 48].
In conclusion, our study indicates that purine analogs elicit a DNA repair pathway that involves the DNA polymerases δ/ε, most likely NER, and provides proof-of-concept that inhibition of this repair pathway could improve their therapeutic efficacy in CLL patients with a good, but also a worse prognosis. These finding should still increase interest in developing aphidicolin-like inhibitors and/or specific inhibitors of the NER pathway, which could be combined with fludarabine that remains a mainstay for CLL treatment.
MATERIALS AND METHODS
Drugs, reagents and antibodies
[8–3H]-Cladribine (7 Ci/mmol) and [8–3H]-fludarabine (4.4 Ci/mmol) were from Moravek Biochemicals (Brea, CA, USA). Fludarabine, 3-[4,5- dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) and propidium iodide were purchased from Sigma–Aldrich (St Louis, MO, USA). Cladribine was synthesized and supplied by Prof. J. Marchand (Laboratory of Organic Chemistry, Université catholique de Louvain, Belgium). Aphidicolin (APC) was from VWR (International Radnor, PA, USA). Ac-DEVD-AMC (Ac-Asp-Glu-Val-Asp-AMC) and AMC (7-amino-4-methyl-coumarin) were purchased from Alexis Biochemicals (San Diego, CA, USA). The annexin V-FITC apoptosis detection kit was from BD Biosciences (Franklin Lakes, NJ, USA). NU7026 was from Selleck (Houston, TX, USA). Antibodies used in this study were: anti-p53 (#sc126) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-p53-pS15 (#9284L), anti-caspase-3 (#9665S) and anti-caspase-9 (#9502S) from Cell Signaling Technologies (Beverly, MA, USA); anti-XPA (ab65963) and anti-lamin B1 (ab16048) from Abcam (Cambridge, UK); Alexa Fluor 488 Mouse anti-H2AX (pS139) from BD Biosciences; anti-β-actin from Sigma-Aldrich. Secondary antibodies (horseradish peroxidase conjugated anti-rabbit and mouse antibodies) were purchased from Sigma–Aldrich. Other chemicals, materials and reagents were from Sigma–Aldrich, Merck Biosciences (Gibbstown, NJ, USA) or Bio-Rad (Hercules, CA, USA) Laboratories.
Patients and CLL cell isolation
Peripheral blood mononuclear cells were obtained from 34 different CLL patients, who had provided informed consent following protocol approval by the Ethics Committee of the Cliniques universitaires Saint-Luc (Brussels, Belgium). This study was conducted in accordance with the Declaration of Helsinki. All patients had an established diagnosis by standard morphological and immunological criteria [49], were free of any anticancer treatment for at least 3 months and had lymphocytes ≥ 30 × 109/L. Patient characteristics are summarized in Supplementary Table 2. Conventional karyotype and interphase fluorescence in situ hybridization (FISH) analysis were performed as previously described [50]. Mononuclear cells (> 95% CLL cells, as confirmed by flow cytometry analysis) were isolated and cultured as described before [39]. When successive analyses were performed in samples from the same patient, they were separated by a period of at least one month. In combination studies, CLL cells were incubated with 3 μM aphidicolin for 30 min prior to the addition of fludarabine, cladribine or other chemotherapeutic drugs. Stock solutions of hydrophobic compounds were prepared in DMSO and equal amounts of DMSO were added in untreated and treated cells. The final DMSO concentration was ≤ 0.2%. In some experiments, cells were UV-C irradiated as described before [51] or subjected to IR using a 137Cs source at a dose rate of 2.43 Gy/min at room temperature.
Cell viability and drug interaction analysis
To assess the influence of aphidicolin on the sensitivity of CLL cells to purine analogs, freshly isolated cells (2 × 106 cells/well) were incubated in 96-well plates with increasing concentrations of fludarabine or cladribine in the absence or presence of 3 μM aphidicolin. After 96 h, CLL cell survival was measured using the MTT assay as previously described [52]. Each condition was done in triplicate in the same experiment. The concentrations of fludarabine or cladribine required to kill 50% of cells (IC50) were determined graphically. Interaction between aphidicolin and fludarabine or cladribine, i.e. synergism, additivity or antagonism, was defined according to the multiplicative model as previously reported [18].
Western blot analysis
Cell protein extracts were obtained as reported [8]. Protein concentration was determined using the Bio-Rad protein assay. Equal amounts of denatured protein (usually 50 μg) were subjected to SDS-PAGE in 12% (w/v) polyacrylamide gels and transferred to HybondTM-C Extra membranes (Amersham Biosciences). After blocking with 5% skim milk for 1 h at room temperature, the membranes were probed overnight at 4°C with the primary antibody (diluted 1/1000, or 1/10 000 for the anti-β-actin antibody). After washing, the membranes were incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody (1/10 000). Proteins were visualized using Clarity Western ECL Substrate (Bio-Rad) and ECL chemiluminescence film (Fujifilm). Adobe Photoshop CS6 was used for image processing.
Fludarabine and cladribine metabolism in intact CLL cells
The influence of aphidicolin on the metabolism of fludarabine or cladribine in CLL cells was analyzed as previously described, using 3H-labeled purine analogs [10].
Isolation of nuclear and cytoplasmic fractions
Cell pellets (50 × 106 cells) were resuspended in 80 μl of buffer A (10 mM HEPES, pH 7.9, 0.1 mM EDTA, 10 mM KCl, 1 mM dithiothreitol, 0.1% (v/v) Nonidet-P40 and freshly added protease inhibitors) and incubated for 20 min on ice with gentle shaking. After centrifugation at 7200 × g for 5 min, the supernatant containing cytoplasmic fraction was transferred to a new tube. The remaining insoluble fraction was washed once in Buffer A without NP-40 and then solubilized in 25 μl of buffer B (20 mM HEPES, pH 7.9, 1 mM EDTA, 50 mM KCl, 10 mM dithiothreitol, 1% (v/v) Triton X-100 and protease inhibitors) and incubated 30 min on ice. The supernatant containing nuclear fraction was obtained after centrifugation at 16 000 × g for 10 min.
γH2AX flow cytometry analysis
CLL samples (1 × 106 cells) were fixed in 70% ethanol and maintained at –20°C for at least 1 h. Next, cells were washed twice with cold PBS, and rehydrated for 10 min at 4°C in buffer containing PBS, 4% FBS and 0.1% Triton. After centrifugation, cells were resuspended in 100 μl of the same buffer with the Alexa Fluor 488 Mouse anti-H2AX (pS139) antibody (1/200), incubated for 1 h at room temperature in darkness, washed and resuspended in PBS. Labeled cells were analyzed with FACSVerse flow cytometer (BD Biosciences).
Apoptosis assays
Apoptosis was quantified by Annexin V-FITC/propidium iodide (PI) double staining (Beckton Dickinson) followed by analysis using a FACSVerse flow cytometer. Annexin-V positive and PI negative cells were defined as early apoptotic, while Annexin-V and PI dual positive cells were considered as apoptotic/necrotic. As incubation of CLL cells in fresh medium resulted in spontaneous apoptosis (5 to 20% dead cells after 48 h), we calculated specific drug-induced apoptosis as described elsewhere [53] using the following formula: 100 × [experimental apoptosis (%) – spontaneous apoptosis (%)/[100% - spontaneous apoptosis (%)]. Caspase-3 activity was determined by a fluorometric assay with Ac-DEVD-AMC as a fluorogenic substrate using samples containing 50 × 106 CLL cells, as previously reported [10]. Procaspase-9 and procaspase-3 were analyzed by Western blot.
RNA interference in cell lines
The cell lines used for RNA interference experiments were the EBV-immortalized lymphoblastoid cell line GM0536, obtained from the NIGMS Human Mutant Cell Repository (Camden, NJ, USA), and the CLL cell line EHEB, purchased from DSMZ-German Collection of Microorganisms and Cell Culture (Braunschweig, Germany). Cells were cultured as described previously [54] and transfected using Amaxa™ nucleofector and nucleofection Kit V (Amaxa, Cologne, Germany) according to the Amaxa guidelines. Briefly, 2 × 106 cells were resuspended in 100 μl of the Nucleofector solution V. Targeting or not-targeting small interfering RNAs (siRNAs) were added at a concentration of 900 nM to the cell suspensions and the mixtures were transferred to the Amaxa certified cuvettes. Nucleofection was performed using the M-013 program. Experiments were conducted 24 or 48 h after transfection, for GM0536 or EHEB cells, respectively. siRNA duplexes were from Dharmacon (Lafayette, CO, USA): XPA siRNA (SMARTpool: ON-TARGETplus XPA siRNA) and the non-targeting control (ON-TARGETplus Non-targeting Pool).
Statistical analysis
Results obtained in at least 3 independent experiments were normalized and expressed as the means ± SEM. Significance was analyzed by different statistical methods as detailed in the figure legend. Changes were considered significantly different when P < 0.05.
ACKNOWLEDGMENTS
We greatly thank all the patients who kindly donated blood samples and made this research possible. We also thank Nicolas Dauguet for help in flow cytometry experiments.
CONFLICTS OF INTEREST
The authors indicate no potential conflicts of interest.
GRANT SUPPORT
This work was supported by grants from the Belgian Fonds National de la Recherche Scientifique (Télévie, FRSM, FRIA), by the PAI Program of the Belgian Science Policy (network P7/43) and by private foundations: Jacques Goor, Salus Sanguinis and Joseph Maisin.
REFERENCES
1. Dameshek W. Chronic lymphocytic leukemia—an accumulative disease of immunolgically incompetent lymphocytes. Blood. 1967; 29:566–584.
2. Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, Murphy EJ, Koduru P, Ferrarini M, Zupo S, Cutrona G, Damle RN, Wasil T, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005; 115:755–764.
3. Woyach JA, Johnson AJ. Targeted therapies in CLL: mechanisms of resistance and strategies for management. Blood. 2015; 126:471–477.
4. Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, Langerbeins P, von Tresckow J, Engelke A, Maurer C, Kovacs G, Herling M, Tausch E, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016; 127:208–215.
5. Rossi D, Terzi-di-Bergamo L, De Paoli L, Cerri M, Ghilardi G, Chiarenza A, Bulian P, Visco C, Mauro FR, Morabito F, Cortelezzi A, Zaja F, Forconi F, et al. Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood. 2015; 126:1921–1924.
6. Van Den Neste E, Cardoen S, Offner F, Bontemps F. Old and new insights into the mechanisms of action of two nucleoside analogs active in lymphoid malignancies: fludarabine and cladribine (review). Int J Oncol. 2005; 27:1113–1124.
7. Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia. 2001; 15:875–890.
8. Smal C, Vertommen D, Bertrand L, Ntamashimikiro S, Rider MH, Van Den Neste E, Bontemps F. Identification of in vivo phosphorylation sites on human deoxycytidine kinase. Role of Ser-74 in the control of enzyme activity. J Biol Chem. 2006; 281:4887–4893.
9. Smal C, Van Den Neste E, Maerevoet M, Poire X, Theate I, Bontemps F. Positive regulation of deoxycytidine kinase activity by phosphorylation of Ser-74 in B-cell chronic lymphocytic leukaemia lymphocytes. Cancer Lett. 2007; 253:68–73.
10. Amsailale R, Van Den Neste E, Arts A, Starczewska E, Bontemps F, Smal C. Phosphorylation of deoxycytidine kinase on Ser-74: impact on kinetic properties and nucleoside analog activation in cancer cells. Biochem Pharmacol. 2012; 84:43–51.
11. Wright GE, Hubscher U, Khan NN, Focher F, Verri A. Inhibitor analysis of calf thymus DNA polymerases alpha, delta and epsilon. FEBS Lett. 1994; 341:128–130.
12. Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, Mullenders LH, Yamashita S, Fousteri MI, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell. 2010; 37:714–727.
13. O’Dwyer PJ, Moyer JD, Suffness M, Harrison SD Jr, Cysyk R, Hamilton TC, Plowman J. Antitumor activity and biochemical effects of aphidicolin glycinate (NSC 303812) alone and in combination with cisplatin in vivo. Cancer Res. 1994; 54:724–729.
14. Bramson J, Panasci L. Potentiation of chlorambucil toxicity in B-CLL lymphocytes using the DNA synthesis inhibitors aphidicolin and 1-beta-D-arabinofuranosylcytosine. Biochem Pharmacol. 1995; 50:131–135.
15. Sargent JM, Elgie AW, Williamson CJ, Taylor CG. Aphidicolin markedly increases the platinum sensitivity of cells from primary ovarian tumours. Br J Cancer. 1996;74:1730–1733.
16. Sargent JM, Elgie AW, Williamson CJ, Lewandowicz GM, Taylor CG. Circumvention of ara-C resistance by aphidicolin in blast cells from patients with AML. Br J Cancer. 2001; 84:680–685.
17. Hubeek I, Peters GJ, Broekhuizen AJ, Sargent J, Gibson BE, Creutzig U, Kaspers GJ. Potentiation of in vitro ara-C cytotoxicity by ribonucleotide reductase inhibitors, cyclin-dependent kinase modulators and the DNA repair inhibitor aphidicolin in paediatric acute myeloid leukaemia. Br J Haematol. 2005; 131:219–222.
18. Kaspers GJ, Veerman AJ, Pieters R, Van Zantwijk I, Hahlen K, Van Wering ER. Drug combination testing in acute lymphoblastic leukemia using the MTT assay. Leuk Res. 1995; 19:175–181.
19. Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010; 24:679–686.
20. Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, Dai W, Darzynkiewicz Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2007; 71:648–661.
21. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844–848.
22. Wood RD, Shivji MK. Which DNA polymerases are used for DNA-repair in eukaryotes? Carcinogenesis. 1997; 18:605–610.
23. Iyama T, Wilson DM, 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 2013; 12:620–636.
24. Ringborg U, Lambert B. Ultraviolet-induced DNA-repair synthesis in lymphocytes from patients with chronic lymphatic leukemia. Cancer Lett. 1977; 3:77–81.
25. Van Den Neste E, Cardoen S, Husson B, Rosier JF, Delacauw A, Ferrant A, Van den Berghe G, Bontemps F. 2-Chloro-2′-deoxyadenosine inhibits DNA repair synthesis and potentiates UVC cytotoxicity in chronic lymphocytic leukemia B lymphocytes. Leukemia. 2002; 16:36–43.
26. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010; 79:181–211.
27. Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, Durkacz BW. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004; 103:4659–4665.
28. Wu X, Shell SM, Liu Y, Zou Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene. 2007; 26:757–764.
29. Park GB, Kim YS, Kim D, Kim S, Lee HK, Cho DH, Lee WJ, Hur DY. Melphalan-induced apoptosis of EBV-transformed B cells through upregulation of TAp73 and XAF1 and nuclear import of XPA. J Immunol. 2013; 191:6281–6291.
30. Li Z, Musich PR, Zou Y. Differential DNA damage responses in p53 proficient and deficient cells: cisplatin-induced nuclear import of XPA is independent of ATR checkpoint in p53-deficient lung cancer cells. Int J Biochem Mol Biol. 2011; 2:138–145.
31. Huang P, Plunkett W. Fludarabine- and gemcitabine-induced apoptosis: incorporation of analogs into DNA is a critical event. Cancer Chemother Pharmacol. 1995; 36:181–188.
32. Sampath D, Plunkett W. The role of c-Jun kinase in the apoptotic response to nucleoside analogue-induced DNA damage. Cancer Res. 2000; 60:6408–6415.
33. Huang P, Sandoval A, Van Den Neste E, Keating MJ, Plunkett W. Inhibition of RNA transcription: a biochemical mechanism of action against chronic lymphocytic leukemia cells by fludarabine. Leukemia. 2000; 14:1405–1413.
34. Rao VA, Plunkett W. Activation of a p53-mediated apoptotic pathway in quiescent lymphocytes after the inhibition of DNA repair by fludarabine. Clin Cancer Res. 2003; 9:3204–3212.
35. Willmore E, Elliott SL, Mainou-Fowler T, Summerfield GP, Jackson GH, O’Neill F, Lowe C, Carter A, Harris R, Pettitt AR, Cano-Soumillac C, Griffin RJ, Cowell IG, et al. DNA-dependent protein kinase is a therapeutic target and an indicator of poor prognosis in B-cell chronic lymphocytic leukemia. Clin Cancer Res. 2008; 14:3984–3992.
36. Bulgar AD, Snell M, Donze JR, Kirkland EB, Li L, Yang S, Xu Y, Gerson SL, Liu L. Targeting base excision repair suggests a new therapeutic strategy of fludarabine for the treatment of chronic lymphocytic leukemia. Leukemia. 2010; 24:1795–1799.
37. Lawrenzi J, Bunnell A, Hentosh P. Nucleotide excision repair-deficient human cells in culture exhibit decreased survival after 2-chlorodeoxyadenosine treatment. Anticancer Res. 2003; 23:3141–3145.
38. Li Z, Musich PR, Serrano MA, Dong Z, Zou Y. XPA-mediated regulation of global nucleotide excision repair by ATR Is p53-dependent and occurs primarily in S-phase. PLoS One. 2011; 6:e28326.
39. de Viron E, Knoops L, Connerotte T, Smal C, Michaux L, Saussoy P, Vannuffel P, Beert E, Vekemans MC, Hermans C, Bontemps F, Van Den Neste E. Impaired up- regulation of polo-like kinase 2 in B-cell chronic lymphocytic leukaemia lymphocytes resistant to fludarabine and 2-chlorodeoxyadenosine: a potential marker of defective damage response. Br J Haematol. 2009; 147:641–652.
40. Moussay E, Palissot V, Vallar L, Poirel HA, Wenner T, El Khoury V, Aouali N, Van Moer K, Leners B, Bernardin F, Muller A, Cornillet-Lefebvre P, Delmer A, et al. Determination of genes and microRNAs involved in the resistance to fludarabine in vivo in chronic lymphocytic leukemia. Mol Cancer. 2010; 9:115.
41. Zenz T, Mohr J, Edelmann J, Sarno A, Hoth P, Heuberger M, Helfrich H, Mertens D, Dohner H, Stilgenbauer S. Treatment resistance in chronic lymphocytic leukemia: the role of the p53 pathway. Leuk Lymphoma. 2009; 50:510–513.
42. Zenz T, Habe S, Denzel T, Mohr J, Winkler D, Buhler A, Sarno A, Groner S, Mertens D, Busch R, Hallek M, Dohner H, Stilgenbauer S. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009; 114:2589–2597.
43. Perez-Chacon G, Martinez-Laperche C, Rebolleda N, Somovilla-Crespo B, Munoz-Calleja C, Buno I, Zapata JM. Indole-3-Carbinol Synergizes with and Restores Fludarabine Sensitivity in Chronic Lymphocytic Leukemia Cells Irrespective of p53 Activity and Treatment Resistances. Clin Cancer Res. 2016; 22:134–145.
44. Sessa C, Zucchetti M, Davoli E, Califano R, Cavalli F, Frustaci S, Gumbrell L, Sulkes A, Winograd B, D’Incalci M. Phase I and clinical pharmacological evaluation of aphidicolin glycinate. J Natl Cancer Inst. 1991; 83:1160–1164.
45. Baranovskiy AG, Babayeva ND, Suwa Y, Gu J, Pavlov YI, Tahirov TH. Structural basis for inhibition of DNA replication by aphidicolin. Nucleic Acids Res. 2014; 42:14013–14021.
46. Kelley MR, Logsdon D, Fishel ML. Targeting DNA repair pathways for cancer treatment: what’s new? Future Oncol. 2014; 10:1215–1237.
47. Jordheim LP, Barakat KH, Heinrich-Balard L, Matera EL, Cros-Perrial E, Bouledrak K, El Sabeh R, Perez-Pineiro R, Wishart DS, Cohen R, Tuszynski J, Dumontet C. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol Pharmacol. 2013; 84:12–24.
48. Gentile F, Tuszynski JA, Barakat KH. New design of nucleotide excision repair (NER) inhibitors for combination cancer therapy. J Mol Graph Model. 2016; 65:71–82.
49. Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O’Brien S, Rai KR. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996; 87:4990–4997.
50. Van Den Neste E, Robin V, Francart J, Hagemeijer A, Stul M, Vandenberghe P, Delannoy A, Sonet A, Deneys V, Costantini S, Ferrant A, Robert A, Michaux L. Chromosomal translocations independently predict treatment failure, treatment-free survival and overall survival in B-cell chronic lymphocytic leukemia patients treated with cladribine. Leukemia. 2007; 21:1715–1722.
51. Van Den Neste E, Smal C, Cardoen S, Delacauw A, Frankard J, Ferrant A, Van den Berghe G, Bontemps F. Activation of deoxycytidine kinase by UV-C-irradiation in chronic lymphocytic leukemia B-lymphocytes. Biochem Pharmacol. 2003; 65:573–580.
52. Bontemps F, Delacauw A, Cardoen S, Van Den Neste E, Van Den Berghe G. Metabolism and cytotoxic effects of 2-chloroadenine, the major catabolite of 2-chloro-2’-deoxyadenosine. Biochem Pharmacol. 2000; 59:1237–1243.
53. Debatin KM, Krammer PH. Resistance to APO-1 (CD95) induced apoptosis in T-ALL is determined by a BCL-2 independent anti-apoptotic program. Leukemia. 1995; 9:815–820.
54. Beyaert M, Starczewska E, Van Den Neste E, Bontemps F. A crucial role for ATR in the regulation of deoxycytidine kinase activity. Biochem Pharmacol. 2016; 100:40–50.