
INTRODUCTION
Urinary bladder cancer is the fourth most common cancer in men of the United States [1]. Known risk factors include increasing age, male sex, certain occupational exposures and cigarette smoking. Cigarette smoking is a major cause for bladder cancer, accounting for approximately 50% of all incident cases [2]. Carcinogenic arylamines present in tobacco smoke, including 4-aminobiphenyl (4-ABP) and 2-naphthylamine are believed to be responsible for the increased risk of bladder cancer among smokers [3].
Many environmental and occupational carcinogens undergo catalysis by N-acetyltransferase 2 (NAT2). The enzyme detoxifies carcinogens such as arylamines in the liver by transferring an acetyl group to an exocyclic amine (N-acetylation). Alternatively, NAT2 may activate carcinogens by transferring an acetyl group to the oxygen group of N-hydroxy-aromatic or heterocyclic amines (O-acetylation) [4] (Figure 1). Hepatic NAT2 N-acetylation capacity can be determined phenotypically by quantifying the urinary caffeine metabolite ratio (CMR) [5]. More recently, the NAT2 acetylation status can be inferred by a panel of seven single nuclear polymorphisms (SNPs) [6] or a tag SNP rs1495741 of the NAT2 gene [7]. The genetic variants may represent both NAT2 N-acetylation status in the liver and O-acetylation activity in the urinary tract. Currently, there is no method available for direct measure of local O-acetylation activity in vivo.
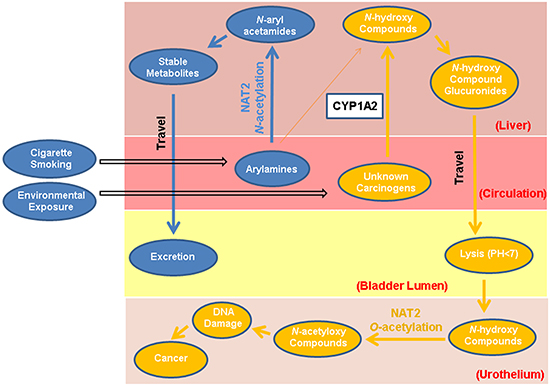
Figure 1: Dual functions of NAT2 in bladder carcinogenesis. Potential dual functions of the NAT2 enzyme involve carcinogenic detoxification (N-acetylation) and activation (O-acetylation). Arylamines from cigarette smoke and other occupational or environmental sources can be N-acetylated by hepatic NAT2 function, or possibly ring-oxidized by CYP1A2. The derivatives can form more stable metabolites through conjugation catalyzed by glutathione S-transferases, as well as UDP-glucuronosyltransferases, and ultimately excreted through urine. Alternatively, arylamines and other unknown bladder carcinogens could be procarcinogens that require metabolic activation to electrophilic intermediates to exert their carcinogenic effect. The first step in this process is N-oxidation catalyzed by cytochrome P450 (CYP) enzymes, such as CYP1A2 in the liver, to form metabolically active N-hydroxy- compounds. The N-hydroxy-compounds are transported through circulation to the bladder lumen, where they can be O-acetylated by local NAT2 or hydrolyzed in the acidic environment to form highly electrophilic derivatives that can covalently bind to urothelial DNA or generate reactive oxygen species (ROS). Certain potential bladder carcinogens such as heterocyclic amines (HCAs) do not go through the detoxifying N-acetylation pathway.
Epidemiological studies in Europe and United States, especially those among smokers, have reported associations between SNP-inferred NAT2 slow acetylation status and increased risk of bladder cancer. It is consistent with the hypothesis that NAT2 plays an important role in detoxifying arylamines present in tobacco smoke through hepatic N-acetylation activities [8–12]. However, in two studies of factory workers in China who were occupationally exposed to benzidine, a group I human bladder carcinogen classified by the International Agency for Research on Cancer (IARC) [13], SNP-inferred NAT2 slow acetylation was associated with a significant decreased risk of bladder cancer [14, 15]. It has been hypothesized that NAT2 enzyme may activate rather than deactivate benzidine at the urinary bladder [15], suggesting a more complex function of NAT2. Tobacco smoking is a major identified risk factor for bladder cancer, but it only accounts for 50-60% of disease burden [2]. Thus exposure to diffused environmental bladder carcinogens as yet to be identified may contribute to the remaining 40-50% of disease burden [16]. Furthermore, the incidence rate of bladder cancer in Chinese men is only about one-third that of their Caucasian counterparts, despite much higher prevalence of cigarette smoking in the former than the latter [17, 18]. On the other hand, the proportion of SNP-inferred NAT2 slow acetylators is considerably lower in Chinese (~19%) than in Caucasians (~55%) [9, 19–21]. Such difference certainly contributes to the difference in the bladder cancer rates between the two races. Thus, the inconsistent results of the NAT2 acetylation status and bladder cancer risk from epidemiological studies in different races could be the results of different prevalence of genetic variants in the NAT2 gene and different levels of exposure to tobacco and other environmental bladder carcinogens.
There are few reports that examined the association between NAT2 acetylation status and bladder cancer risk among Asian populations. Their results were inconclusive [9, 22–27]. Most of these studies were hospital-based and with relatively small sample sizes. None have studied NAT2 acetylation status simultaneously inferred by genotype and phenotype in relation to risk of bladder cancer. The present study was designed to address these limitation and to examine and further clarify the following associations: (1) the concordance of acetylation status inferred by the conventional NAT2 SNP panel and the novel tag SNP of NAT2 gene in a Chinese population, (2) the causal relationship between the SNP-inferred acetylation status and CMR, a functional measure of hepatic N-acetylation, and (3) the association between SNP- and CMR-determined NAT2 acetylation status separately or in combination and the risk of bladder cancer in a relatively large population-based case-control study in Shanghai, China.
RESULTS
Participant characteristics are shown in Table 1. Patients with bladder cancer did not differ significantly from controls in the distributions of sex, age at reference date (2 years before diagnosis for cases or 2 years before interview for controls), body mass index (BMI), and level of education. Cases were more likely to smoke cigarettes than controls. Among smokers, patients with bladder cancer had a significantly younger age at starting to smoke and a greater number of years of smoking than controls.
Table 1: Distributions of baseline characteristics in bladder cancer patients (cases) and control subjects (controls), The Shanghai Bladder Cancer Study
Characteristics |
Cases |
Controls |
Pa |
|---|---|---|---|
Among all subjects |
(n=478) |
(n=473) |
|
Age at Reference (Years), mean ± SD |
61.0±9.9 |
62.1±10.0 |
0.08 |
Body Mass Index (Kg/m2), mean ± SD |
22.5±3.2 |
22.2±3.0 |
0.25 |
Gender, % |
|||
Male |
376 (78.7) |
370 (78.2) |
0.87 |
Female |
102 (21.3) |
103 (21.8) |
|
Education, % |
|||
No Formal Education |
42 (8.8) |
36 (7.6) |
0.76 |
Primary School |
115 (24.1) |
116 (24.5) |
|
High School |
264 (55.2) |
272 (57.5) |
|
College and Graduate School |
57 (11.9) |
49 (10.4) |
|
Cigarette smoking, % |
|||
Never |
165 (34.5) |
208 (44.0) |
0.003 |
Former |
73 (15.3) |
79 (16.7) |
|
Current |
240 (50.2) |
186 (39.3) |
|
Among Current and Former Smokers |
(n=313) |
(n=265) |
|
Age starting to smoke (years), mean ± SD |
22.8±7.6 |
24.7±8.2 |
0.005 |
Number of cigarettes per day, mean ± SD |
15.9±9.6 |
15.8± 9.1 |
0.82 |
Number of years of smoking, mean ± SD |
35.4± 13.0 |
33.3± 13.2 |
0.05 |
Number of pack-years of smoking, mean ± SD |
29.7± 22.3 |
27.6± 20.1 |
0.24 |
a 2-sided Ps were derived from Student t-tests for continuous variables and Chi-square test for categorical variables.
After excluding a monomorphic SNP (rs1801279) in the study population, we analyzed 6 candidate SNPs and a tag SNP of the NAT2 gene. Among control subjects, minor allele frequencies (MAF) ranged from 0.03 to 0.45 (Table 2). SNPs 803A>G (rs1208), 481C>T (rs1799929) and 341T>C (rs1801280) were highly correlated with each other (all pairwise r2 ≥0.94), whereas a moderate correlation was observed between 282 C>T (rs1041983) and 590 G>A (rs1799930) (r2=0.44). No correlation was observed between other pairs of the candidate SNPs among controls. The tag SNP rs1495741 was only correlated with 282 C>T (rs1041983) (r2=0.77) (Supplementary Table S1 and Supplementary Figure S1). Allele frequencies and linkage disequilibrium (LD) observed in the present study population were similar to those of other Chinese populations but very different from those of Caucasians published in the HapMap database (Supplementary Figure S2A, S2B, and S2C).
Table 2: The geometric means of urinary caffeine metabolite ratio (CMR) by NAT2 genotypes in bladder cancer patients (cases) and control subjects (controls), The Shanghai Bladder Cancer Study
NAT2 Genotypes |
MAF a |
Cases |
Controls |
% Diff c |
Pd |
|||
|---|---|---|---|---|---|---|---|---|
N |
Geometric Means of CMR (95% CI) b |
N |
Geometric Means of CMR (95% CI) b |
|||||
All Subjects |
N/A |
478 |
0.42 (0.40-0.44) |
473 |
0.46 (0.44-0.48) |
-8.7 |
0.03 |
|
Individual SNP of 6-SNP Panel |
||||||||
282 C>T |
CC |
0.41 |
188 |
0.52 (0.48-0.56) |
151 |
0.50 (0.46-0.54) |
4.0 |
0.48 |
(rs1041983) |
CT |
221 |
0.40 (0.40-0.46) |
251 |
0.48 (0.44-0.50) |
-12.5 |
0.006 |
|
TT |
69 |
0.22 (0.20-0.26) |
71 |
0.32 (0.28-0.36) |
-31.3 |
0.005 |
||
P-trend |
1.2×10−32 |
2.1×10−8 |
||||||
590 G>A |
GG |
0.24 |
293 |
0.46 (0.44-0.50) |
271 |
0.48 (0.44-0.50) |
-4.1 |
0.44 |
(rs1799930) |
GA |
170 |
0.38 (0.34-0.40) |
176 |
0.44 (0.42-0.48) |
-13.6 |
0.0009 |
|
AA |
15 |
0.24 (0.18-0.30) |
26 |
0.28 (0.22-0.34) |
-14.3 |
0.54 |
||
P-trend |
2.6×10−10 |
4.0×10−4 |
||||||
857 G>A |
GG |
0.16 |
336 |
0.46 (0.44-0.50) |
327 |
0.46 (0.44-0.50) |
0 |
0.83 |
(rs1799931) |
GA |
126 |
0.36 (0.32-0.38) |
137 |
0.42 (0.38-0.46) |
-14.3 |
0.002 |
|
AA |
16 |
0.22 (0.16-0.26) |
9 |
0.32 (0.22-0.44) |
-31.3 |
0.14 |
||
P-trend |
4.0×10−16 |
0.01 |
||||||
803 A>G |
AA |
0.03 |
450 |
0.42 (0.40-0.44) |
442 |
0.46 (0.42-0.48) |
-8.7 |
0.03 |
(rs1208) |
AG |
26 |
0.38 (0.30-0.46) |
30 |
0.42 (0.34-0.50) |
-9.5 |
0.44 |
|
GG |
2 |
0.42 (0.22-0.84) |
1 |
0.08 (0.04-0.24) |
N/A e |
N/A |
||
P-trend |
0.27 |
0.04 |
||||||
481 C>T |
CC |
0.03 |
453 |
0.42 (0.40-0.44) |
443 |
0.46 (0.44-0.48) |
-8.7 |
0.02 |
(rs1799929) |
CT |
24 |
0.38 (0.32-0.48) |
29 |
0.42 (0.34-0.50) |
-4.8 |
0.68 |
|
TT |
1 |
0.50 (0.20-1.32) |
1 |
0.08 (0.04-0.24) |
N/A e |
N/A |
||
P-trend |
0.52 |
0.05 |
||||||
341 T>C |
TT |
0.03 |
452 |
0.42 (0.40-0.44) |
443 |
0.46 (0.44-0.48) |
-8.7 |
0.02 |
(rs1801280) |
TC |
25 |
0.38 (0.32-0.46) |
29 |
0.42 (0.34-0.50) |
-9.5 |
0.6 |
|
CC |
1 |
0.50 (0.20-1.34) |
1 |
0.08 (0.04-0.24) |
N/A e |
N/A |
||
P-trend |
0.44 |
0.05 |
||||||
6-SNP panel inferred acetylation status |
||||||||
Rapid f |
N/A |
170 |
0.52 (0.50-0.56) |
135 |
0.50 (0.46-0.54) |
4.0 |
0.28 |
|
Int |
231 |
0.44 (0.42-0.46) |
260 |
0.50 (0.46-0.52) |
-12.0 |
0.0006 |
||
Slow |
77 |
0.24 (0.22-0.26) |
78 |
0.30 (0.26-0.32) |
-20.0 |
0.03 |
||
P-trend |
4.4×10−34 |
5.6×10−11 |
||||||
Tag SNP |
GG |
0.45 |
163 |
0.52 (0.50-0.56) |
134 |
0.50 (0.46-0.54) |
4.0 |
0.23 |
(rs1495741) |
GA |
240 |
0.44 (0.42-0.46) |
250 |
0.50 (0.46-0.52) |
-12.0 |
0.0007 |
|
AA |
75 |
0.24 (0.22-0.26) |
89 |
0.32 (0.28-0.34) |
-25.0 |
0.005 |
||
P-trend |
2.1×10−33 |
4.4×10−10 |
||||||
a MAF: Minor allele frequency among controls.
b Adjusted for age at reference date and sex.
c Percentage of differences: cases’ geometric mean minus controls’ geometric mean divided by controls’ geometric mean, then multiplied by 100.
d 2-sided Ps were derived from analysis of covariance comprising the difference in geometric means of caffeine metabolite ratio between cases and controls with adjustment for age and sex.
e N/A, not applicable due to small number of subjects in this category.
f Acetylation status inferred by genotypes of the 6-candidate SNPs, as described in details in the Methods section; Int: intermediate.
NAT2 genotypes partially reflected hepatic N-acetylation function measured by CMR. Among control subjects, minor alleles of all individual SNPs were associated with lower level of CMR compared to major alleles (Table 2, P-trends ≤ 0.05). Similar genotype-phenotype associations were found in bladder cancer patients except for the three SNPs with extremely low MAF. NAT2 acetylation status inferred genotypically by the tag SNP and the 6-SNP panel were highly correlated in both controls (r2=0.87) and cases (r2=0.96) (P < 1.4×10−30) (Supplementary Table S2), indicating that the single tag SNP rs1495741 captured almost all genetic variation of the NAT2 exon 2 region in Chinese people. The SNP-inferred NAT2 slow acetylation status, either based on the tag SNP or 6-SNP panel, resulted in a similar ~40% reduction in CMR in both controls and cases (P-trends ≤ 4.4×10−10) (Table 2). Smoking, gender and age did not modify the influencing effects of genetic variants on the CMR (Supplementary Table S3).
Patients with bladder cancer overall had statistically significant 8.7% lower CMR than controls (P = 0.03) (Table 2). Compared to their counterparts in control group, the CMR was 12% and ≥20% lower in case patients with intermediate and slow acetylation status inferred by NAT2 genotype (both P values < 0.03). Among subjects with the SNP-inferred NAT2 rapid acetylation status, however, cases had a 4% higher in CMR than controls, but the difference was not statistically significant (P = 0.28).
Overall SNP-inferred slow acetylation status was significantly associated with reduced risk of bladder cancer compared with rapid acetylation status (Table 3). This risk association was slightly stronger for acetylation status inferred by the tag SNP (P-trend < 0.03) than by the 6-SNP panel approach (P-trend = 0.06). On the other hand, low level of CMR (i.e., slow N-acetylation status) was associated with an increased risk of bladder cancer compared to high level of CMR (Table 3). Controlling for variation in hepatic N-acetylation capacity (i.e., CMR) significantly strengthened the association between SNP-inferred acetylation status and bladder cancer risk; the slow acetylation status was associated with approximately 50% reduced risk of bladder cancer compared with the rapid acetylation status (P-trends ≤ 0.003). Conversely, the adjustment for SNP-determined acetylation status also strengthened the CMR-bladder cancer risk association (Table 3). No significant collinearity was observed between NAT2 genotype and CMR in the same model (tolerance > 0.86). The association between the SNP-inferred acetylation status and bladder cancer risk was much stronger in never smokers than that in ever smokers. Furthermore, the association between SNP-inferred acetylation status and bladder cancer became even stronger in lifelong never smokers who were not exposed to environmental tobacco smoke (ETS), determined by non-detectable total cotinine in urine (data not shown).
Table 3: Associations between NAT2 acetylation status inferred by genotype and bladder cancer risk, The Shanghai Bladder Cancer Study
NAT2 Status |
All Samples |
Never Smokers |
Ever Smokers |
||||||
|---|---|---|---|---|---|---|---|---|---|
Ca/Coa |
OR (95%CI) b |
OR (95%CI)c |
Ca/Co |
OR (95%CI) b |
OR (95%CI)c |
Ca/Co |
OR (95%CI) b |
OR (95%CI)c |
|
6-SNP Panel |
|||||||||
Rapid d |
170/135 |
1.00 |
1.00 |
65/64 |
1.00 |
1.00 |
105/71 |
1.00 |
1.00 |
Intermediate |
231/260 |
0.70(0.52-0.94) |
0.69(0.52-0.93) |
81/108 |
0.74(0.47-1.16) |
0.72(0.46-1.14) |
150/152 |
0.66(0.45-0.97) |
0.66(0.45-0.97) |
Slow |
77/78 |
0.75(0.51-1.11) |
0.55(0.35-0.85) |
19/36 |
0.51(0.26-0.98) |
0.31(0.14-0.67) |
58/42 |
0.93(0.56-1.55) |
0.74(0.42-1.31) |
P for trend |
0.06 |
0.003 |
0.04 |
0.004 |
0.48 |
0.12 |
|||
Tag SNP rs1495741 |
|||||||||
GG (rapid) |
163/134 |
1.00 |
1.00 |
64/63 |
1.00 |
1.00 |
99/71 |
1.00 |
1.00 |
GA (Intermediate) |
240/250 |
0.79(0.59-1.06) |
0.78(0.58-1.04) |
82/107 |
0.76(0.48-1.19) |
0.75(0.47-1.18) |
158/143 |
0.79(0.54-1.17) |
0.78(0.53-1.15) |
AA (Slow) |
75/89 |
0.66(0.45-0.98) |
0.46(0.30-0.72) |
19/38 |
0.48(0.25-0.93) |
0.29(0.13-0.62) |
56/51 |
0.80(0.49-1.31) |
0.60(0.34-1.04) |
P for trend |
0.03 |
0.0009 |
0.03 |
0.006 |
0.31 |
0.06 |
|||
Caffeine metabolite ratio |
|||||||||
Rapid(CMR≥ 0.34) |
355/384 |
1.00 |
1.00 |
129/175 |
1.00 |
1.00 |
226/209 |
1.00 |
1.00 |
Slow(CMR< 0.34) |
123/89 |
1.50(1.10-2.06) |
1.98(1.37-2.85) |
36/33 |
1.55(0.91-2.62) |
2.54(1.34-4.82) |
87/56 |
1.49(1.00-2.20) |
1.74(1.11-2.71) |
P |
0.01 |
0.0003 |
0.11 |
0.005 |
0.05 |
0.05 |
|||
Abbreviations: Int, intermediate; OR, odds ratio; CI, confidence interval; CMR, caffeine metabolite ratio.
a Number of bladder cancer patients (Ca) and control subjects (Co).
b Adjusted for age at reference date (continuous) and sex. Additionally adjusted for smoking status (never, former, current), number of cigarettes per day (continuous), and number of years of smoking (continuous) for all subjects and ever smokers.
c In addition to variables in b, hepatic N-acetylation status determined by CMR (<0.34 versus ≥0.34) was adjusted for the genotype-disease association; and genotype of Tag SNP rs1495741 was adjusted for the CMR-disease association.
d The acetylation status inferred by the 6-candidate SNP genotypes of NAT2, as described in details in the Methods section.
We further examined the associations between the SNP-inferred acetylation status and bladder cancer risk stratified by CMR levels (Table 4). Among subjects with CMR ≥ 0.34, the SNP-inferred slow acetylation status was associated with a statistically significantly reduced risk of bladder cancer; ORs (95% CIs) for the slow acetylation status inferred by the 6-SNP panel and the tag SNP were 0.31 (0.16-0.61) and 0.25 (0.13-0.48), respectively (both P-trends ≤ 6.0×10−5). The associations were not strengthened by further stratification by smoking status, possibly due to limited number of subjects in each category (data not shown). In contrast, among subjects with CMR < 0.34, the SNP-inferred slow acetylation status was associated with statistically non-significant increase in risk of bladder cancer. The difference in the genotype-disease relationship between the high and low CMR groups was statistically significant (P-interactions ≤ 0.01).
Table 4: The association between NAT2 genotype-inferred acetylation status and risk of bladder cancer stratified by hepatic N-acetylation status determined by caffeine metabolic ratio (CMR), The Shanghai Bladder Cancer Study
NAT2 genotype |
Rapid hepatic N-acetylators (CMR ≥ 0.34) |
Slow hepatic N-acetylators (CMR < 0.34) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Cases |
Controls |
OR (95%CI)b |
Cases |
Controls |
OR (95%CI)b |
|||||
N |
CMRa |
N |
CMRa |
N |
CMRa |
N |
CMRa |
|||
6-SNP Panel |
||||||||||
Rapid c |
151 |
0.58 |
116 |
0.56 |
1.00 |
19 |
0.22 |
19 |
0.18 |
1.00 |
Intermediate |
189 |
0.50 |
232 |
0.52 |
0.62 (0.45-0.85) |
42 |
0.24 |
28 |
0.22 |
1.48 (0.65-3.35) |
Slow |
15 |
0.44 |
36 |
0.54 |
0.31 (0.16-0.61) |
62 |
0.20 |
42 |
0.16 |
1.44 (0.66-3.14) |
P for trend |
1.2 × 10−14 |
0.03 |
6.0 × 10−5 |
0.14 |
0.21 |
0.43 |
||||
P interaction=0.01 |
||||||||||
Tag SNP rs1495741 |
||||||||||
GG (rapid) |
145 |
0.58 |
116 |
0.56 |
1.00 |
18 |
0.22 |
18 |
0.18 |
1.00 |
GA (Intermediate) |
196 |
0.50 |
224 |
0.52 |
0.70 (0.51-0.95) |
44 |
0.24 |
26 |
0.22 |
1.73 (0.75-4.01) |
AA (Slow) |
14 |
0.44 |
44 |
0.54 |
0.25 (0.13-0.48) |
61 |
0.20 |
45 |
0.16 |
1.33 (0.60-2.95) |
P for trend |
1.2 × 10−13 |
0.09 |
4.0 × 10−5 |
0.19 |
0.36 |
0.54 |
||||
P interaction=0.008 |
||||||||||
Abbreviations: Int, intermediate; OR, odds ratio; CI, confidence interval.
a Adjusted for age at reference date (continuous) and sex.
bAdjusted for age at reference date (continuous), sex, smoking status (never, former, current), number of cigarettes per day (continuous), and years of smoking (continuous).
cNAT2 acetylation status inferred by the 6-candidate SNP genotypes of NAT2, as described in details in the Methods section.
DISCUSSION
The present study with a relatively large sample size in an Asian population demonstrates several novel findings. 1) The acetylation status inferred by a single tag SNP captures almost all genetic variations in the exon 2 region of the NAT2 gene in Chinese people; 2) Both the single tag SNP and multiple candidate SNPs reflect variation in CMR, a measure of hepatic N-acetylation capacity; 3) The SNP-inferred slow acetylation status is significantly associated with a reduced risk of bladder cancer whereas the CMR-determined slow acetylation status is significantly associated with increased risk of bladder cancer as reported previously in this study population [20] as well as in Western populations [19]. The association between the SNP-inferred slow acetylation status and reduced risk of bladder cancer becomes stronger when the inter-individual variation in CMR is controlled for, suggesting that the O-acetylation rather than N-acetylation plays an important role in the observed SNP-bladder cancer risk association. Conversely, the effect of CMR-determined slow acetylation status on bladder cancer risk was greatly increased after controlling for genetic variation in the NAT2, further suggesting hepatic N-acetylation measured by CMR is a detoxification pathway of bladder carcinogen arylamines. 4) The SNP-inferred slow acetylation and reduced risk of bladder cancer was mainly confined to never smokers, further suggesting the potential role of NAT2 in the activation of non-tobacco bladder carcinogens. These novel and intriguing findings demonstrate a more complex function of the NAT2 gene in bladder carcinogenesis than previously appreciated.
Urinary CMR primarily measures hepatic N-acetylation activity of NAT2 whereas NAT2 genotypes potentially reflect both N- and O-acetylation activities [6, 28, 29]. Dual functions of NAT enzymes that involve carcinogenic detoxification (N-acetylation) and activation (O-acetylation) have been long described in the liver [30, 31]. It has been shown in various animal models and in human tissue that functional levels of NAT2 expression are also detected in the urinary bladder [32–34]. The literature is mixed on NAT2 catalytic activity in the urinary bladder, which is partially due to the strong NAT1 catalytic activity in the tissue [34]. However, NAT2-dependent 4-ABP and 2-naphthylamine N-acetyltransferase activities have been reported in human bladder [28, 35, 36]. Moreover, p-Aminobenzoic acid N-acetyltransferase activity (selective for NAT1) and N-hydroxy-ABP O-acetyltransferase activities (not selective for NAT1 or NAT2) in human bladder cytosols do not correlate [35]. These findings provide compelling evidence for the existence of NAT2 O-acetylation function in the local urothelium.
The increased risk of bladder cancer associated with low CMR observed in the present study as well as by others supported an important role of hepatic N-acetylation in the detoxification of bladder carcinogens [9, 19, 20, 37]. On the other hand, most NAT2 variant alleles share one or more common missense SNPs that decrease gene expression, enzyme activity or enzyme stability that affect both N- or O-acetylation functions [5, 38]. Therefore, NAT2 SNP-inferred slow acetylation status potentially measured variations in both N- and O-acetylation catalytic capacities [28], and was associated with decreased risk of bladder cancer in our study. The adjustment for CMR resulted in significant strengthening of the association, which indirectly demonstrated an important role of O-acetylation in the activation of bladder carcinogens, leading to elevated risk of bladder cancer for individuals who carried high functional alleles of the NAT2 gene. This was further supported by a stronger association between the SNP-inferred slow acetylation status and decreased bladder cancer risk among individuals with higher CMR.
Different carcinogens are targeted by the N- and O-acetylation pathways [39]. Therefore, the net effect of SNP-inferred NAT2 acetylation status on risk of bladder cancer would depend on the individual’s exposure to specific type of bladder carcinogens. Smokers are exposed primarily to arylamines, substrates of the N-acetylation pathway. Epidemiological studies and genome-wide association (GWA) studies in Europe consistently reported higher risk of bladder cancer associated with the SNP-inferred NAT2 slow acetylation status among ever smokers [9-12, 23, 40, 41]. Compared to ever smokers, never smokers experience two- to three-fold lower risk of bladder cancer [2], and thus may be more sensitive to diffused environmental carcinogens as substrates of the O-acetylation pathway.
Metabolism of heterocyclic amines (HCAs) involves primarily activation through local O-acetylation of NAT2 but not detoxification through hepatic N-acetylation due to the steric hindrance of the exocyclic amine [5]. Although the findings were not all consistent, NAT2-catalyzed O-acetylation of N-hydroxy-heterocyclic amines accounted for the association between NAT2 rapid acetylation status and increased risk of colorectal cancer among people who frequently consumed well-done meat [42–45]. A positive association between meat intake and risk of bladder cancer has also been demonstrated among never smokers with genotype-inferred NAT2 rapid acetylation status, suggesting a possible activation effect of NAT2 on HCA through O-acetylation pathway in the urinary bladder [46]. Indeed, the fry-cooking of meat, a common food preparation by Chinese, produces high levels of HCAs (~49.95 ng/day) [47]. Future studies are warranted to investigate the interaction between NAT2 and exposure to HCA carcinogens on risk of bladder cancer.
Notable strengths of the study included population based design, relatively large sample size and homogenous study population (only Han Chinese). We collected detailed information on various aspects of participants’ characteristics and controlled potential confounding effects such as smoking. Furthermore, urinary total cotinine was quantified and active and passive smoking status was confirmed. More importantly, the function of the SNPs in the NAT2 gene was confirmed by CMR, a functional measure of hepatic N-acetylation. The simultaneous measurements of both CMR and the NAT2 SNPs (reflecting both N- and O-acetylation) allowed for the assessment of the putative association between the O-acetylation status and bladder cancer, which provides insight on the potential biological mechanism of NAT2 genetic variants on bladder carcinogenesis.
Our study has several potential limitations. One of limitations was the retrospective study design. Urine samples were collected from patients after their diagnosis of bladder cancer. The disease status or progress might have potential impact on hepatic function of caffeine metabolism. However, our data did not show any significant difference in CMR by disease stage. Among bladder cancer patients, the CMR levels were comparable across different patients by tumor stages and grades at diagnosis (Supplementary Table S3). Another concern about the retrospective study design was the potential impact of lifestyle changes due to bladder cancer diagnosis on the hepatic N-acetylation function. There was a high proportion of patients with bladder cancer quit smoking after their cancer diagnosis (Supplementary Table S3). However, smoking status for cases was determined at two years prior to cancer diagnosis. Furthermore, we did not detect any difference in CMR between smoking statuses at urine collection among either controls or cases (Supplementary Table S3). Genotype of the NAT2 gene would not be altered by the disease status, thus the observed association between the SNP-inferred acetylation status and bladder cancer risk are unlikely to be confounded by the retrospective study design. Nonetheless, the observed association may be due to genetic linkage to other as-yet-unidentified risk genes for bladder cancer.
In summary, this study demonstrates a statistically significant reduced risk of bladder cancer associated with the SNP-inferred NAT2 slow acetylation status, but an increased risk of bladder cancer with the CMR-determined slow acetylation status. The apparent opposite direction for phenotypic and genetic measures of acetylation statuses with bladder cancer risk may reflect dual functions of NAT2 in bladder carcinogenesis because the CMR only measures the systemic detoxification capacity through the hepatic N-acetylation pathway while the SNP-inferred NAT2 acetylation represents both hepatic detoxification and local carcinogen activation through the O-acetylation pathway at local urothelium. The observed net protective effect of the SNP-inferred NAT2 slow acetylation status on bladder cancer risk may reflect both the high prevalence of NAT2 rapid acetylation status and exposure to bladder carcinogens that are uniquely catalyzed through the O-acetylation activation pathway in the study population. Future studies are warranted to ascertain the specific role of N- and O-acetylation in bladder carcinogenesis, particularly in populations exposed to different types of bladder carcinogens.
MATERIALS AND METHODS
Study participants
The Shanghai Bladder Cancer Study has been described in detail elsewhere [20]. Briefly, bladder cancer patients were identified through the Shanghai Cancer Registry, a population-based cancer registry monitoring approximately 8 million residents in the urban area of Shanghai, China. All cases diagnosed with bladder cancer between July 1st of 1995 and June 30th of 1998 were eligible to participate in the study. The registry identified 708 cases aged 25 to 74 years at diagnosis of bladder cancer, among whom 56 died before contacting, 29 refused to be interviewed and 42 were untraceable. We interviewed the remaining 581 (82%) eligible patients between July 1996 and June 1999. The diagnosis of bladder cancer of 531 (91%) patients was based on histopathological evidence and diagnosis of the remaining 50 (9%) patients was based on positive computerized tomography scan and/or ultrasonography with consistent clinical history.
Control subjects were randomly selected from urban residents of Shanghai through the City Residents Registry. Controls were chosen to match the frequency distribution by sex and 5-year age groups of patients with bladder cancer. Among the 750 randomly selected control subjects, 74 were untraceable and 72 refused to participate in the study. The remaining 604 (80%) subjects were interviewed between July 1996 and June 1999. The study has been approved by institutional review boards at the Shanghai Cancer Institute, the University of Minnesota and the University of Pittsburgh. Consent forms have been signed by all study participants.
Data collection
In-depth in-person interviews using a structured questionnaire were conducted to collect information from each eligible subject on a variety of factors known or suspected to be related to bladder cancer. The questionnaire included background information, demographics, history of tobacco smoke, history of passive smoking (for non-smokers only), history of alcohol, coffee, tea, soft drinks and plain water drinking, use of hormone replacement therapy (for women only), medical history, usual adult diet and occupational history, from birth to up to 2 years before the diagnosis of bladder cancer for cases and 2 years before the date of interview for controls [20].
All study participants were asked to donate blood and urine samples at the end of the interview. One 10-ml blood sample was collected from each study participant in a heparin coated tube and fractioned into plasma, buffy coat and erythrocytes on the day of the sample collection. All components of the blood samples were stored at −80°C. For the collection of overnight urine sample, each subject was given two packets of Nestle instant coffee or two cans of Coca-Cola classic drink (about 70 mg of caffeine) to be drunk between 3 pm and 6 pm. An overnight urine sample was collected by the subject, picked up by the interviewer in the following morning, processed, acidified (400 mg of ascorbic acid per 20 ml of urine) and stored at −80°C until analysis. A total of 535 (92%) of the 581 interviewed cases and 543 (90%) of the 604 interviewed control subjects donated blood and urine samples. After excluding subjects with missing information on any of the SNPs genotyped (3 cases and 2 controls) or urinary CMR (13 cases and 32 controls), 478 bladder cancer cases and 473 control subjects were included in the present study.
Laboratory measurements
NAT2 phenotype by CMR
The measurement of urinary caffeine metabolites, i.e. 5-acetylamino-6-amino-3-methyluracil (AAMU), 1-methylxanthin (MX), 1-methyluric acid (MU) and 1,7-dimethylxanthin (17X) has been described in detail elsewhere [20]. Briefly, levels of AAMU in urine were determined using high-performance size exclusion chromatography [48]. Quantification of MX, MU and 17X in urine was conducted using a modified method of Grant et al [29]. All analyses were performed with appropriate internal standards. Concentrations of all metabolites were calculated using calibration curves. NAT2 hepatic N-acetylation function was determined phenotypically by ratio of urinary caffeine metabolites (CMR) using the following formula: AAMU/(AAMU+ MX+MU). The ratios enabled differentiation at a cut-off of 0.34 and study participants were classified as slow (ratio < 0.34) or rapid (ratio ≥ 0.34) acetylators [48].
Urinary total cotinine was measured by the standard gas chromatographic-mass spectrometric method as described previously [20]. For the subgroup analysis by smoking status at the time of urine collection, we classified study subjects into current smokers and lifelong never smokers. Current smokers were those who self-reported as daily smokers at reference date with urinary total cotinine greater than 75 ng/ml. Lifelong never smokers were self-reported never smokers at reference date with urinary total cotinine <75 ng/ml. Fourteen self-reported never smokers but whose urinary total cotinine were greater than 75 ng/mL were excluded from this subgroup analysis (7 cases and 7 controls). We further classified lifelong never smokers into those were ETS-exposed (urinary total cotinine 1-75 ng/ml) or ETS non-exposed.
NAT2 Genotype
Genomic DNA was extracted in batches from peripheral blood buffy coats using QIAmp DNA mini kit according to manufacturer’s protocol (Qiagen Inc, Valencia, CA, US). Quality and quantity of purified DNA were evaluated using Nanodrop UV-spectrometer (Thermo Fisher Scientific Inc., Wilmington, DE, US). DNA samples were stored at −80°C until analysis and plated and genotyped at the Genomics Core Facility at University of Minnesota using MassARRAY technology and iPLEX Gold Assay (Sequenom Inc., San Diego, CA, US). Five percent duplicates and two sets of in-house trio samples were included for quality control purposes. The concordance among blind duplicate pairs was >99.95%. The average successful genotyping rate for each sample and SNP was 100%.
A conventional panel of seven SNPs in exon 2 of the NAT2 gene was used to determine NAT2 haplotypes and infer acetylation status [49]. The panel includes four SNPs that are known to reduce NAT2 activity: 191G>A (rs1801279), 341T>C (rs1801280), 590 G>A (rs1799930) and 857 G>A (rs1799931); and three SNPs that do not change NAT2 activity but are required to infer acetylation status accurately: 803 A>G (rs1208), 481 C>T (rs1799929) and 282 C>T (rs1041983). One of the seven SNPs, NAT2 191G>A (rs1801279), was not polymorphic in our study population and hence was excluded from further analysis. The genotype frequencies for the other six SNPs were all in Hardy-Weinberg equilibrium among the control population. Individuals carrying two of the NAT2 alleles associated with rapid acetylation status, i.e. NAT2*4 (wild type), NAT2*12A (803 A>G) or NAT2*13A (282 C>T), were classified as rapid acetylators. Individuals carrying two of the NAT2 alleles associated with slow acetylation status, i.e. NAT2*5A (341T>C and 481 C>T), NAT2*5B (341T>C, 481 C>T and 803 A>G), NAT2*5C (341T>C and 803 A>G), NAT2*6A (282 C>T and 590 G>A), NAT2*7B (282 C>T and 857 G>A) were classified as slow acetylators. Individuals carrying one rapid acetylation allele and one slow acetylation allele were classified as intermediate acetylators [8] (Supplementary Table S4).
In addition to the 6-SNP panel, a tag SNP (rs1495741) recently identified among European populations at the 3′ end of the NAT2 gene was also genotyped [10]. This SNP has been demonstrated to correlate with NAT2 acetylation status in European populations [7]. The distribution of this SNP was found to be in Hardy-Weinberg equilibrium among control subjects.
Statistical analysis
Chi-square test was used to examine the difference in distributions of categorical or nominal variables and Student t-test for continuous variables between cases and controls. Pearson’s correlation coefficient, r2 and corresponding P values were calculated for measuring pairwise correlation between two SNPs. Linkage disequilibrium map for the tag SNP rs1495741 in relation to 6 candidate SNPs were generated using Haploview [50]. Analysis of covariance (ANCOVA) method was used to compare the difference in geometric means of CMR between cases and controls, or across different genotypes with adjustment for age and sex.
Unconditional logistic regression was used to calculate odds ratios (ORs), their corresponding 95% confidence intervals (CIs) and associated P values to measure the strength of associations between the SNP-inferred or CMR-determined acetylation status and bladder cancer risk with adjustment for age, sex, smoking status, number of cigarettes per day, and number of years of smoking [51]. Similar multivariable logistic regression models with further adjustment for CMR or stratified by CMR were used to assess indirectly the effect of O-acetylation on bladder cancer risk. We assumed additive genetic effects and homozygous wild-type, heterozygous, and homozygous variant genotypes were coded as 0, 1, and 2, respectively, in the trend test. To test collinearility between NAT2 genotype and CMR, the tolerance of each variable in the model was requested, with a value less than 0.20 indicating the presence of collinearity.
All analyses were conducted using SAS 9.4 (SAS Institute, Cary CA). All P values were 2-sided. Values of P less than 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
The authors would like to thank the patients, clinical investigators, and laboratory staff who participated in the study.
COMPETING INTERESTS
The authors declare no competing interests.
GRANT SUPPORT
The research study was supported by R01 CA144034 and UM1 CA182876 (to J.M. Yuan) and P30 CA077598 (to H.H. Nelson) from the National Cancer Institute, USA; partially supported by the Major Science and Technology Project of Guangdong Province 2014B020210001 and the Fundamental Research Funds for the Central Universities D215185w (to L. Quan).
REFERENCES
1. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
2. Yu MC, Skipper PL, Tannenbaum SR, Chan KK and Ross RK. Arylamine exposures and bladder cancer risk. Mutation research. 2002; 506-507:21-28.
3. Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nature reviews Cancer. 2003; 3:733-744.
4. Hein DW, Trinidad A, Yerokun T, Ferguson RJ, Kirlin WG and Weber WW. Genetic control of acetyl coenzyme A-dependent arylamine N-acetyltransferase, hydrazine N-acetyltransferase, and N-hydroxy-arylamine O-acetyltransferase enzymes in C57BL/6J, A/J, AC57F1, and the rapid and slow acetylator A.B6 and B6.A congenic inbred mouse. Drug metabolism and disposition. 1988; 16:341-347.
5. Hein DW. Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutation research. 2002; 506-507:65-77.
6. Gross M, Kruisselbrink T, Anderson K, Lang N, McGovern P, Delongchamp R and Kadlubar F. Distribution and concordance of N-acetyltransferase genotype and phenotype in an American population. Cancer epidemiology, biomarkers & prevention. 1999; 8:683-692.
7. Garcia-Closas M, Hein DW, Silverman D, Malats N, Yeager M, Jacobs K, Doll MA, Figueroa JD, Baris D, Schwenn M, Kogevinas M, Johnson A, Chatterjee N, Moore LE, Moeller T, Real FX, et al. A single nucleotide polymorphism tags variation in the arylamine N-acetyltransferase 2 phenotype in populations of European background. Pharmacogenetics and genomics. 2011; 21:231-236.
8. Moore LE, Baris DR, Figueroa JD, Garcia-Closas M, Karagas MR, Schwenn MR, Johnson AT, Lubin JH, Hein DW, Dagnall CL, Colt JS, Kida M, Jones MA, Schned AR, Cherala SS, Chanock SJ, et al. GSTM1 null and NAT2 slow acetylation genotypes, smoking intensity and bladder cancer risk: results from the New England bladder cancer study and NAT2 meta-analysis. Carcinogenesis. 2011; 32:182-189.
9. Rothman N, Garcia-Closas M and Hein DW. Commentary: Reflections on G. M. Lower and colleagues’ 1979 study associating slow acetylator phenotype with urinary bladder cancer: meta-analysis, historical refinements of the hypothesis, and lessons learned. International journal of epidemiology. 2007; 36:23-28.
10. Rothman N, Garcia-Closas M, Chatterjee N, Malats N, Wu X, Figueroa JD, Real FX, Van Den Berg D, Matullo G, Baris D, Thun M, Kiemeney LA, Vineis P, De Vivo I, Albanes D, Purdue MP, et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nature genetics. 2010; 42:978-984.
11. Garcia-Closas M, Rothman N, Figueroa JD, Prokunina-Olsson L, Han SS, Baris D, Jacobs EJ, Malats N, De Vivo I, Albanes D, Purdue MP, Sharma S, Fu YP, Kogevinas M, Wang Z, Tang W, et al. Common genetic polymorphisms modify the effect of smoking on absolute risk of bladder cancer. Cancer research. 2013; 73:2211-2220.
12. Figueroa JD, Han SS, Garcia-Closas M, Baris D, Jacobs EJ, Kogevinas M, Schwenn M, Malats N, Johnson A, Purdue MP, Caporaso N, Landi MT, Prokunina-Olsson L, Wang Z, Hutchinson A, Burdette L, et al. Genome-wide interaction study of smoking and bladder cancer risk. Carcinogenesis. 2014; 35:1737-1744.
13. Humans IMWGotEoCRt. Some aromatic amines, organic dyes, and related exposures. IARC monographs on the evaluation of carcinogenic risks to humans. 2010; 99:1-658.
14. Hayes RB, Bi W, Rothman N, Broly F, Caporaso N, Feng P, You X, Yin S, Woosley RL and Meyer UA. N-acetylation phenotype and genotype and risk of bladder cancer in benzidine-exposed workers. Carcinogenesis. 1993; 14:675-678.
15. Carreon T, Ruder AM, Schulte PA, Hayes RB, Rothman N, Waters M, Grant DJ, Boissy R, Bell DA, Kadlubar FF, Hemstreet GP, 3rd, Yin S and LeMasters GK. NAT2 slow acetylation and bladder cancer in workers exposed to benzidine. International journal of cancer. 2006; 118:161-168.
16. Skipper PL, Kim MY, Sun HL, Wogan GN and Tannenbaum SR. Monocyclic aromatic amines as potential human carcinogens: old is new again. Carcinogenesis. 2010; 31:50-58.
17. Ploeg M, Aben KK and Kiemeney LA. The present and future burden of urinary bladder cancer in the world. World journal of urology. 2009; 27:289-293.
18. Giovino GA, Mirza SA, Samet JM, Gupta PC, Jarvis MJ, Bhala N, Peto R, Zatonski W, Hsia J, Morton J, Palipudi KM, Asma S and Group GC. Tobacco use in 3 billion individuals from 16 countries: an analysis of nationally representative cross-sectional household surveys. Lancet. 2012; 380:668-679.
19. Yuan JM, Chan KK, Coetzee GA, Castelao JE, Watson MA, Bell DA, Wang R and Yu MC. Genetic determinants in the metabolism of bladder carcinogens in relation to risk of bladder cancer. Carcinogenesis. 2008; 29:1386-1393.
20. Tao L, Xiang YB, Chan KK, Wang R, Gao YT, Yu MC and Yuan JM. Cytochrome P4501A2 phenotype and bladder cancer risk: The Shanghai bladder cancer study. International journal of cancer. 2012; 130:1174-1183.
21. Golka K, Prior V, Blaszkewicz M and Bolt HM. The enhanced bladder cancer susceptibility of NAT2 slow acetylators towards aromatic amines: a review considering ethnic differences. Toxicology letters. 2002; 128:229-241.
22. Song DK, Xing DL, Zhang LR, Li ZX, Liu J and Qiao BP. Association of NAT2, GSTM1, GSTT1, CYP2A6, and CYP2A13 gene polymorphisms with susceptibility and clinicopathologic characteristics of bladder cancer in Central China. Cancer detection and prevention. 2009; 32:416-423.
23. Tsukino H, Nakao H, Kuroda Y, Imai H, Inatomi H, Osada Y and Katoh T. Glutathione S-transferase (GST) M1, T1 and N-acetyltransferase 2 (NAT2) polymorphisms and urothelial cancer risk with tobacco smoking. European journal of cancer prevention. 2004; 13:509-514.
24. Kim WJ, Lee HL, Lee SC, Kim YT and Kim H. Polymorphisms of N-acetyltransferase 2, glutathione S-transferase mu and theta genes as risk factors of bladder cancer in relation to asthma and tuberculosis. The Journal of urology. 2000; 164:209-213.
25. Hsieh FI, Pu YS, Chern HD, Hsu LI, Chiou HY and Chen CJ. Genetic polymorphisms of N-acetyltransferase 1 and 2 and risk of cigarette smoking-related bladder cancer. British journal of cancer. 1999; 81:537-541.
26. Ishizu S, Hashida C, Hanaoka T, Maeda K and Ohishi Y. N-acetyltransferase activity in the urine in Japanese subjects: comparison in healthy persons and bladder cancer patients. Japanese journal of cancer research. 1995; 86:1179-1181.
27. Horai Y, Fujita K and Ishizaki T. Genetically determined N-acetylation and oxidation capacities in Japanese patients with non-occupational urinary bladder cancer. European journal of clinical pharmacology. 1989; 37:581-587.
28. Hein DW. N-acetyltransferase 2 genetic polymorphism: effects of carcinogen and haplotype on urinary bladder cancer risk. Oncogene. 2006; 25:1649-1658.
29. Grant DM, Tang BK and Kalow W. Polymorphic N-acetylation of a caffeine metabolite. Clinical pharmacology and therapeutics. 1983; 33:355-359.
30. Hanna PE. N-acetyltransferases, O-acetyltransferases, and N,O-acetyltransferases: enzymology and bioactivation. Advances in pharmacology. 1994; 27:401-430.
31. Hein DW. Acetylator genotype and arylamine-induced carcinogenesis. Biochimica et biophysica acta. 1988; 948:37-66.
32. Husain A, Zhang X, Doll MA, States JC, Barker DF and Hein DW. Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug metabolism and disposition. 2007; 35:721-727.
33. Barker DF, Walraven JM, Ristagno EH, Doll MA, States JC and Hein DW. Quantitative tissue and gene-specific differences and developmental changes in Nat1, Nat2, and Nat3 mRNA expression in the rat. Drug metabolism and disposition. 2008; 36:2445-2451.
34. Hein DW. N-acetyltransferase SNPs: emerging concepts serve as a paradigm for understanding complexities of personalized medicine. Expert opinion on drug metabolism & toxicology. 2009; 5:353-366.
35. Badawi AF, Hirvonen A, Bell DA, Lang NP and Kadlubar FF. Role of aromatic amine acetyltransferases, NAT1 and NAT2, in carcinogen-DNA adduct formation in the human urinary bladder. Cancer research. 1995; 55:5230-5237.
36. Frederickson SM, Hatcher JF, Reznikoff CA and Swaminathan S. Acetyl transferase-mediated metabolic activation of N-hydroxy-4-aminobiphenyl by human uroepithelial cells. Carcinogenesis. 1992; 13:955-961.
37. Cartwright RA, Glashan RW, Rogers HJ, Ahmad RA, Barham-Hall D, Higgins E and Kahn MA. Role of N-acetyltransferase phenotypes in bladder carcinogenesis: a pharmacogenetic epidemiological approach to bladder cancer. Lancet. 1982; 2:842-845.
38. Zang Y, Doll MA, Zhao S, States JC and Hein DW. Functional characterization of single-nucleotide polymorphisms and haplotypes of human N-acetyltransferase 2. Carcinogenesis. 2007; 28:1665-1671.
39. Hein DW, Millner LM, Leggett CS and Doll MA. Relationship between N-acetyltransferase 2 single-nucleotide polymorphisms and phenotype. Carcinogenesis. 2010; 31:326-327; author reply 328-329.
40. Gu J, Liang D, Wang Y, Lu C and Wu X. Effects of N-acetyl transferase 1 and 2 polymorphisms on bladder cancer risk in Caucasians. Mutation research. 2005; 581:97-104.
41. Garcia-Closas M, Malats N, Silverman D, Dosemeci M, Kogevinas M, Hein DW, Tardon A, Serra C, Carrato A, Garcia-Closas R, Lloreta J, Castano-Vinyals G, Yeager M, Welch R, Chanock S, Chatterjee N, et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. 2005; 366:649-659.
42. Lilla C, Verla-Tebit E, Risch A, Jager B, Hoffmeister M, Brenner H and Chang-Claude J. Effect of NAT1 and NAT2 genetic polymorphisms on colorectal cancer risk associated with exposure to tobacco smoke and meat consumption. Cancer epidemiology, biomarkers & prevention. 2006; 15:99-107.
43. Ognjanovic S, Yamamoto J, Maskarinec G and Le Marchand L. NAT2, meat consumption and colorectal cancer incidence: an ecological study among 27 countries. Cancer causes & control. 2006; 17:1175-1182.
44. Cotterchio M, Boucher BA, Manno M, Gallinger S, Okey AB and Harper PA. Red meat intake, doneness, polymorphisms in genes that encode carcinogen-metabolizing enzymes, and colorectal cancer risk. Cancer epidemiology, biomarkers & prevention. 2008; 17:3098-3107.
45. Shin A, Shrubsole MJ, Rice JM, Cai Q, Doll MA, Long J, Smalley WE, Shyr Y, Sinha R, Ness RM, Hein DW and Zheng W. Meat intake, heterocyclic amine exposure, and metabolizing enzyme polymorphisms in relation to colorectal polyp risk. Cancer epidemiology, biomarkers & prevention. 2008; 17:320-329.
46. Lumbreras B, Garte S, Overvad K, Tjonneland A, Clavel-Chapelon F, Linseisen JP, Boeing H, Trichopoulou A, Palli D, Peluso M, Krogh V, Tumino R, Panico S, Bueno-De-Mesquita HB, Peeters PH, Lund E, et al. Meat intake and bladder cancer in a prospective study: a role for heterocyclic aromatic amines? Cancer causes & control. 2008; 19:649-656.
47. Wong KY, Su J, Knize MG, Koh WP and Seow A. Dietary exposure to heterocyclic amines in a Chinese population. Nutrition and cancer. 2005; 52:147-155.
48. Tang BK, Kadar D and Kalow W. An alternative test for acetylator phenotyping with caffeine. Clinical pharmacology and therapeutics. 1987; 42:509-513.
49. Selinski S, Blaszkewicz M, Lehmann ML, Ovsiannikov D, Moormann O, Guballa C, Kress A, Truss MC, Gerullis H, Otto T, Barski D, Niegisch G, Albers P, Frees S, Brenner W, Thuroff JW, et al. Genotyping NAT2 with only two SNPs (rs1041983 and rs1801280) outperforms the tagging SNP rs1495741 and is equivalent to the conventional 7-SNP NAT2 genotype. Pharmacogenetics and genomics. 2011; 21:673-678.
50. Barrett JC, Fry B, Maller J and Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005; 21:263-265.
51. Tao L, Xiang YB, Wang R, Nelson HH, Gao YT, Chan KK, Yu MC and Yuan JM. Environmental tobacco smoke in relation to bladder cancer risk--the Shanghai bladder cancer study [corrected]. Cancer epidemiology, biomarkers & prevention. 2010; 19:3087-3095.