
INTRODUCTION
Prostate cancer is the most frequently diagnosed malignancy and the second leading cause of cancer death among men in the United States [1]. It is estimated that in the year 2016, there will be 180,890 new cases and 26,120 deaths due to prostate cancer [1]. This cancer is a disease of aging as 1 in 325 persons will develop invasive prostate cancer prior to the age of 50, but drastically rises to 1 in 48 for those aged 50 to 59, 1 in 17 aged 60 to 69, and 1 in 10 aged 70 years and older [1]. Despite these high rates, the genetic basis of this disease is not well understood.
Recent meta-analysis showed a close association between several cytochrome P450 (CYP) 1A1 polymorphisms and prostate cancer risk [2, 3], suggesting that CYP1A1 may contribute to prostate cancer tumorigenesis. The carcinogenic potential of CYP1A1 is thought to be associated with metabolic activation of procarcinogens such as polycyclic aromatic hydrocarbons (PAHs), which can form PAH-DNA adducts in several types of malignancies [4, 5]. Although PAHs produce significant levels of PAH-DNA adducts in prostate cancer cells, its correlation with prostate cancer risk is controversial [6, 7]. Interestingly, recent studies have shown that CYP1A1 promotes breast cancer progression even in the absence of xenobiotics [8] and suggests the possibility that this gene may be involved in other carcinogenic mechanisms.
CYP1A1 is located in chromosome 15q24.1 region, consists of 7 exons and is roughly 6 kilobases in length. The protein localizes mainly to the endoplasmic reticulum and is composed of 512 amino acids with a size of 58 kDa. Under normal physiologic conditions, CYP1A1 expression is induced by PAHs via activation of the aryl hydrocarbon receptor (AhR). The AhR complex then translocates to the nucleus and binds to its partner protein, aryl hydrocarbon receptor nuclear translocator (ARNT). The AhR/ARNT heterodimer binds to specific DNA recognition sites termed xenobiotic responsive elements (XREs) located upstream of the transcription start site and initiates CYP1A1 transcription [3, 9]. Thus CYP1A1 expression is regulated by direct interaction between the AhR/ARNT heterodimer and XREs.
Studies have shown that epigenetic changes can regulate the expression of several tumor-specific genes [10, 11]. We have demonstrated that DNA hypermethylation of CpG islands involving the promoter of tumor suppressor genes can lead to functional loss of these genes in several types of malignancies, including prostate cancer [12–14]. Also, DNA hypomethylation of oncogenic genes is thought to be associated with prostate cancer development and progression [15]. Previous studies have shown that the expression level of CYP1A1 is frequently up-regulated in several human tissues due to hypomethylation of XRE sites which may promote binding of the AhR/ARNT heterodimer [16–19].
One putative mechanism affecting XRE methylation status of CYP1A1 is tobacco smoking as demonstrated in human lung [17, 18]. It is widely accepted that tobacco smoking can cause lung cancer, and smoking-induced CYP1A1 gene alterations may contribute to the initiation of lung carcinogenesis [17, 18]. Importantly, recent studies have shown a close association of smoking with the risk of prostate cancer [20–23]. Therefore we hypothesized that smoking may affect CYP1A1 expression in human prostate tissue through the alteration of XRE CpG methylation in the enhancer region of this gene.
In this study, we assessed whether CYP1A1 levels were elevated in prostate cancer compared to normal prostate or benign prostatic hyperplasia (BPH) using tissue microarray (TMA) of human specimens as well as prostatic cell lines (cancer versus BPH-1). Also, we evaluated the methylation level of XRE sites of the CYP1A1 enhancer in cell lines and clinical samples and determined the effects of smoker status. Finally, we knocked the CYP1A1 gene down in prostate cancer cell lines by RNA interference and performed functional analysis to evaluate its biological role in tumorigenesis.
RESULTS
CYP1A1 expression in prostate cell lines and clinical samples
Initially we measured mRNA and protein expression levels of CYP1A1 in 3 prostate cancer cell lines (PC-3, LNCaP and DU145) and as a comparison, measured expression in BPH-1 cells. Both mRNA (Figure 1A) and protein (Figure 1B) were up-regulated with variable increases in cancerous cells with DU145 showing the largest elevation of expression compared with nonmalignant BPH-1 cells. Next we investigated the expression of CYP1A1 by immunohistochemical staining in 102 primary prostate cancers, 14 normal prostate and 70 BPH samples obtained from TMAs. While CYP1A1 expression was weak or not detected in most of the normal prostate (0.79 ± 0.11) and BPH (0.57 ± 0.07) tissues, the majority of prostate cancer samples showed much higher CYP1A1 immunoreactivity with an average staining score of 1.82 ± 0.08 (P < 0.001, Figure 1C). Thus CYP1A1 is up-regulated in prostate cancer cell lines and tissues.
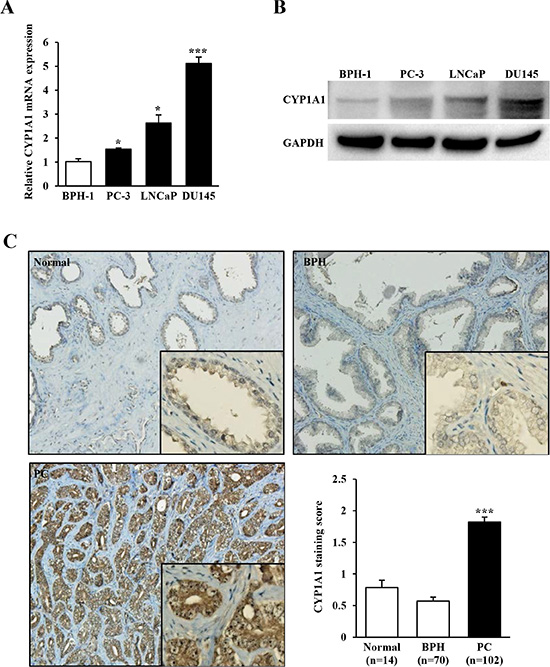
Figure 1: CYP1A1 expression in prostate cancer cell lines and tissues. (A) Relative mRNA expression levels of CYP1A1 in prostate cancer cell lines (PC-3, LNCaP, and DU145) and benign cells (BPH-1) as measured by real-time PCR. The expression level of CYP1A1 mRNA was significantly up-regulated in prostate cancer cell lines. Experiments done in triplicate; *P < 0.05, ***P < 0.001. (B) Representative immunoblot displaying CYP1A1 (top gel) and GAPDH (bottom gel) in BPH-1, PC-3, LNCaP and DU145. (C) Representative immunostaining of CYP1A1 in clinical samples (normal prostate, benign prostatic hyperplasia (BPH), and prostate cancer (PC) obtained from tissue microarray. Bar graph: CYP1A1 protein expression in prostate cancer (n = 102) samples was significantly higher than that of normal (n = 14) or BPH (n = 70) tissues. ***P < 0.001.
Increased expression of CYP1A1 in prostate cancer cell lines after 5-aza-dC treatment
We utilized 5-aza-deoxycytidine (5-aza-dC) to screen the methylation status of CYP1A1 in prostate cancer cell lines. As shown in Figure 2A, CYP1A1 mRNA expression was markedly increased in all cancer cells after treatment indicating that promoter CpG methylation affects CYP1A1 expression. To validate the relationship between CpG methylation and expression of the CYP1A1 transcript, we performed methylation-specific PCR (MSP) analysis of region A (MSP-A) of the enhancer region which contains the XRE-1383 site (see Figure 6A, 6B) using bisulfite-modified DNA of treatment groups. In control cells, densitometry measurements of MSP-A and USP-A bands show higher levels of methylation in all cancer lines with PC-3 having 100% (Figure 2B). Bisulfite DNA sequencing confirmed that most CpG sites in region A area were completely methylated in untreated PC-3 cells (Figure 2B). After 5-aza-dC treatment however, a dramatic reduction in % methylation of this site were observed in all cancer lines. These results show that expression of CYP1A1 is inversely regulated by methylation of region A of the promoter in human prostate cancer cell lines.
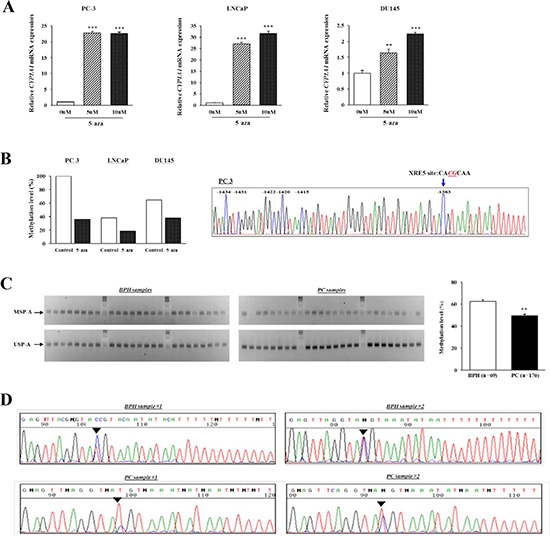
Figure 2: Analysis of CYP1A1 methylation in region A (XRE-1383). (A) CYP1A1 expression before and after demethylation (5-aza-dC) treatment in human prostate cancer cell lines as measured by real-time PCR. CYP1A1 mRNA expression was significantly increased after 5-aza-dC treatment as compared with that before demethylation in PC-3, LNCaP, and DU145. Experiments done in triplicate; **P < 0.01, ***P < 0.001. (B) After 5-aza-dC treatment and performing methylation-specific PCR (MSP) on bisulfite-modified DNA, densitometry of MSP-A and USP-A bands were measured using Image J software and methylation calculated as MSP/[MSP+USP]. The amount of methylation dramatically decreased in all prostate cancer cell lines compared with untreated controls. Chromatogram: DNA sequencing shows CpG sites around region A were completely methylated in PC-3 cells. (C) Representative results of MSP-A and USP-A of CYP1A1 enhancer in benign prostate hyperplasia (BPH) and prostate cancer (PC) samples. Bisulfite-modified DNA of clinical specimens underwent MSP. Top and bottom gels show MSP-A and USP-A bands, respectively. Each column is samples from the same patient. Bar graph: Methylation of CYP1A1 was significantly lower in prostate cancer (PC, n = 176) than BPH (n = 69) samples as measured by densitometry of gel bands. **P < 0.01. (D) Typical bisulfite DNA sequencing chromatograms in BPH and prostate cancer (PC) samples. Though the region A site was partially methylated in samples, methylation was more prominent in BPH than in prostate cancer.
Methylation level of the CYP1A1 enhancers in prostate tissues
Since Figure 1C indicated increased expression of CYP1A1 in prostate cancer regions, we then determined whether cancer tissues also have lower methylation levels. MSP of region A in 176 prostate cancer and 69 BPH samples were analyzed. Representative MSP-A and USP-A bands of 24 BPH and 24 prostate cancer tissues are shown in Figure 2C. The relative methylation amount of the CYP1A1 promoter in region A was significantly lower in prostate cancer (49.2 ± 1.6%) than in BPH samples (62.5 ± 1.3%; P < 0.01). Also, bisulfite DNA sequencing was performed to confirm whether the MSP bands reflect the true methylation status of the CYP1A1 enhancer. Representative sequence chromatograms of two BPH and two prostate cancer samples in region A are shown in Figure 2D and methylation of the XRE-1383 site was generally higher in BPH than in prostate cancer tissues. These results indicate that methylation analysis by MSP was consistent with bisulfite DNA sequencing.
MSP analysis of other CYP1A1 enhancers containing XRE-983 (region B) and XRE-895 (region C) (see Figure 6A, 6B) was also performed. As shown in Figure 3A, most BPH tissues showed both MSP-B and USP-B bands whereas many prostate cancer tissues had only a USP-B band. The relative methylation level of CYP1A1 in region B was significantly lower in prostate cancer (18.5 ± 2.1%) than BPH samples (32.1 ± 2.0%; P < 0.001). Likewise, prostate cancer tissues have lower CYP1A1 methylation in region C in comparison with BPH samples (18.0 ± 2.3% and 40.7 ± 4.2%, respectively; P < 0.001, Figure 3B). Therefore DNA hypomethylation of three CYP1A1 XRE sites was more common in prostate cancer than BPH tissues. We further evaluated the relationship between the methylation level of the CYP1A1 enhancer and pathological status; however, no significant association was found for either grade or stage of cancer (data not shown).
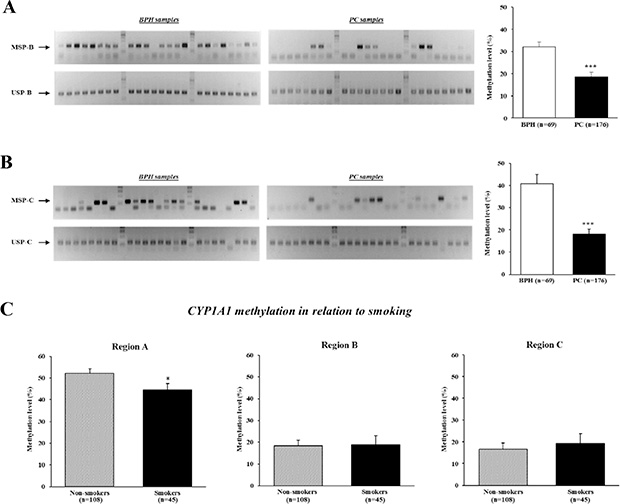
Figure 3: Analysis of CYP1A1 methylation in enhancer regions B (XRE-983) and C (XRE-895), and effect of smoking on methylation levels. DNA of clinical specimens underwent bisulfite modification followed by methylation-specific PCR (MSP) analyses. (A) Representative gel results of MSP-B (top gel) and unmethylation-specific PCR (USP)-B (bottom gel) of CYP1A1 enhancer in benign prostate hyperplasia (BPH) and prostate cancer (PC) samples. Bar graph: The prevalence of CYP1A1 methylation was significantly lower in prostate cancer (n = 176) than BPH (n = 69) samples as measured by densitometry of gel bands. ***P < 0.001. (B) Representative gel results of MSP-C (top gel) and USP-C (bottom gel) of CYP1A1 enhancer in BPH and prostate cancer (PC) samples. Bar graph: A significant reduction in CYP1A1 methylation levels in prostate cancer (PC) samples were also observed for this site. ***P < 0.001. (C) CYP1A1 methylation levels in relation to tobacco smoker status at all 3 regions. Smoking habit affected the methylation level of CYP1A1 region A. Non-smokers n = 108, Smokers n = 45; *P < 0.05.
Methylation of CYP1A1 enhancers in smoker and non-smoker prostate cancer tissues
We then evaluated the association between smoker status and the methylation level of CYP1A1 enhancers in prostate cancer patients. Of 176 patients who underwent radical prostatectomy, 78 never smoked, 30 were ex-smokers who had quit smoking 7 or more days before surgery, 45 were current smokers, and the remaining 23 were unknown. A past history of smoking was not significantly associated with the methylation level in each region (data not shown). Interestingly however, the methylation level of CYP1A1 in the region A site was significantly higher in 108 nonsmokers (never smokers and ex-smokers for > 7 days combined) than 45 smokers (52.2 ± 2.1% and 44.6 ± 2.9%, respectively; P < 0.05, Figure 3C). Cohen’s d value for this difference is calculated to be 0.36 which is moderate. No differences were found in regions B and C. Thus tobacco smoking may affect the methylation of the CYP1A1 enhancer region A.
Effect of CYP1A1 knockdown on prostate cancer cell growth
The higher basal expression levels of CYP1A1 in prostate cancer tissues led us to examine the functional significance of CYP1A1 in prostate cancer. We determined this by testing the effects of CYP1A1 reduction on prostate cancer cell viability. LNCaP and DU145 were selected as these showed higher constitutive levels of CYP1A1 (Figure 1A and 1B). After transfection with two different CYP1A1 siRNAs (#1 and #2), significant reduction of CYP1A1 mRNA was found in both LNCaP and DU145 cells (Figure 4A). CYP1A1 protein was also lowered as well (see Figure 5C). As shown in Figure 4B, knockdown of CYP1A1 caused the growth of these cells to be significantly inhibited in a time-dependent manner as compared with controls.
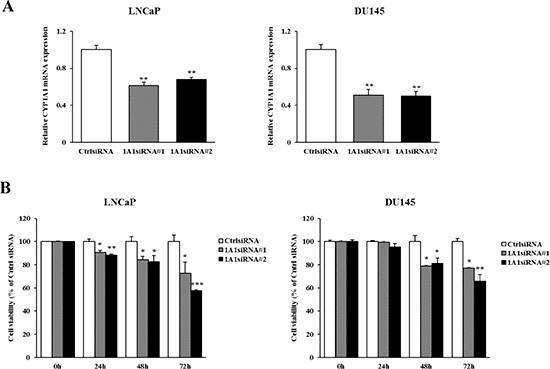
Figure 4: Effect of CYP1A1 knockdown on cell proliferation in prostate cancer cell lines. (A) Knockdown of CYP1A1 levels in LNCaP and DU145 were determined by real-time PCR at 48 hours after transfection with two different CYP1A1 siRNAs. Both siRNA’s significantly reduced CYP1A1 expression. Experiments done in triplicate; **P < 0.01. (B) Cell viability was analyzed by the MTS cell proliferation assay at 0, 24, 48 and 72 hours after siRNA treatment. In both cell lines, the attenuation of CYP1A1 significantly inhibited cell viability in a time dependent manner. Experiments done in triplicate; *P < 0.05, **P < 0.01, ***P < 0.001.
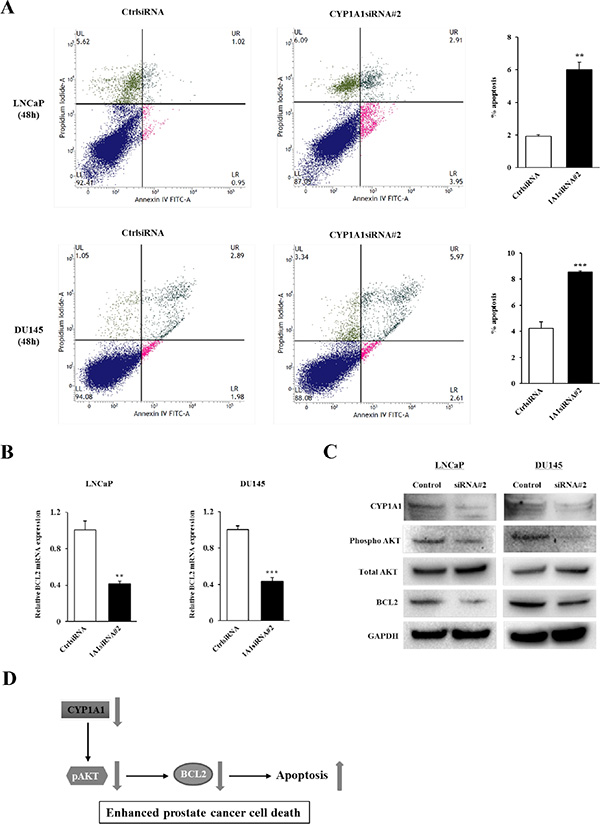
Figure 5: Effect of CYP1A1 knockdown on apoptosis. (A) Apoptosis assays with LNCaP (upper graphs) and DU145 (lower graphs) cells were measured 48 hours post-siRNA#2 transfection by flow cytometry. Representative biparametric histograms showing cell populations in early (bottom right quadrant) and late (top right quadrant) apoptotic, and viable (bottom left quadrant) states for each treatment. Bar graph: The percentage of apoptotic cell fractions (early plus late apoptotic cells) in CYP1A1 siRNA#2 transfectants were significantly elevated compared to controls. Experiments done in triplicate; **P < 0.01, ***P < 0.001. (B) Verification of cDNA microarray data. Among 5 genes significantly altered due to siRNA#2 (see Supplementary Table S1), only BCL2 was confirmed to be down-regulated by real-time PCR using Taqman probe in both LNCaP and DU145. No differences in remaining genes were observed. Experiments done in triplicate; **P < 0.01, ***P < 0.001. (C) Immunoblotting analysis of CYP1A1, phospho AKT, total AKT and BCL2 in control and CYP1A1 siRNA#2-transfected LNCaP and DU145 cells. GAPDH was used as a loading control. (D) Schematic representation of the proposed effect of CYP1A1 down-regulation in prostate cancer. CYP1A1 knockdown inhibits BCL2 expression through the inactivation of AKT, leading to prostate cancer cell death.
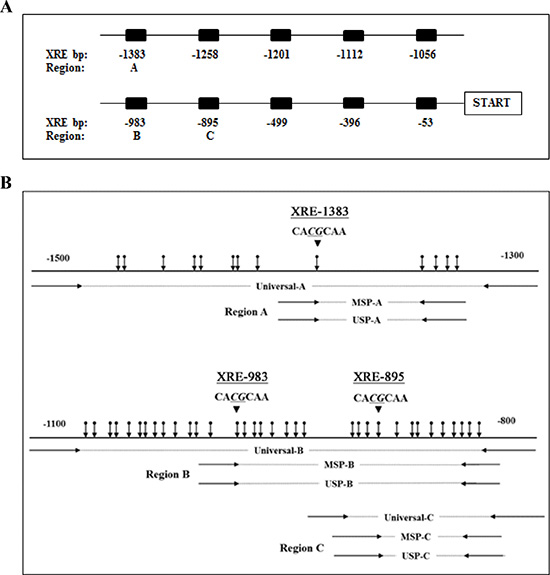
Figure 6: Diagram of the CYP1A1 enhancers and locations of primers. (A) Schematic representation of 10 xenobiotic responsive elements (XRE) located upstream of the transcription start site. Numbers represent bp prior to start site for each XRE. DNA methylation levels of CYP1A1 enhancer region A (-1383 bp), B (-983 bp) and C (-895 bp) were evaluated in this study. (B) Location of designed primers utilized for amplification of regions A, B and C of the CYP1A1 enhancer are depicted. Methylation-specific PCR (MSP) and unmethylation-specific PCR (USP) primers contain several CpG sites within the primer sequence whereas universal primers do not contain any CpG sites. Vertical arrows indicate CpG sites and large arrowheads indicate XRE core sequence.
Effect of CYP1A1 knockdown on apoptosis
Since attenuation of CYP1A1 significantly inhibited proliferation of prostate cancer cell lines, we hypothesized that this may be due to apoptosis induction. Apoptosis was examined in control siRNA and CYP1A1 siRNA#2-treated cells and results for LNCaP and DU145 cells at 48 hours post-transfection are shown in Figure 5A. In both cell lines, the apoptotic and early apoptotic fractions (upper right and lower right quadrants, respectively) were significantly greater in CYP1A1-depleted cells compared to control cells. Similar results were also observed in both LNCaP and DU145 cells at 72 hours after siRNA transfection (data not shown). This supports an anti-apoptotic role for CYP1A1 suggesting that it regulates tumorigenicity by affecting the apoptotic pathway.
To further understand the precise mechanism of the pro-apoptotic effect on prostate cancer cells induced by CYP1A1 knockdown, we performed array analyses to determine which apoptotic genes are altered due to CYP1A1 in LNCaP cells. There were 3 genes up-regulated and 2 genes down-regulated two-fold or greater after CYP1A1 depletion (Supplementary Table S1). Among these 5 genes, only BCL2 was found to be significantly altered in both LNCaP and DU145 cells by real-time PCR using Taqman probes (Figure 5B). Also, BCL2 protein levels were significantly decreased in CYP1A1 siRNA#2-treated cells compared with control in both cell lines (Figure 5C). These data indicate that attenuation of CYP1A1 expression caused reduction of BCL2 expression with increased apoptotic effects in prostate cancer. Furthermore, we sought to determine whether CYP1A1 knockdown inhibits AKT phosphorylation since a recent study showed that CYP1A1 is associated with the PI3K-AKT signaling pathway in breast cancer cell lines [8]. As shown in Figure 5C, CYP1A1 knockdown reduced AKT phosphorylation in both cell lines indicating CYP1A1 may be an upstream activator of the PI3K-AKT signaling pathway. Figure 5D describes a proposed simplified schematic representation of the effect of CYP1A1 down-regulation in prostate cancer cells.
DISCUSSION
In this study, we demonstrate that prostate cancer has high expression of CYP1A1 through DNA hypomethylation of XRE sites. It is well known that methylation of CpG sites within the gene promoter is significantly related to down-regulation or silencing of several types of genes. Previous studies have shown that the expression of human CYP1A1 gene may, at least in part, be regulated by methylation of the XRE core sequence 5′-GCGTG-3′ (or 5′-CACGC-3′) located in the 5′-upstream region [16–19]. Our study on prostate cancer also supports regulation by methylation as treatment of PC-3, LNCaP, and DU145 with demethylating agent caused an enhanced increase of CYP1A1 expression with dramatic loss of methylation as shown by MSP/USP analyses of the enhancer XRE-1383 site. These results are in agreement with Sterling and Cutroneo [24] who find CYP1A1 protein to be expressed constitutively in all of these cancer lines; and Okino et al [16]. observes LNCaP to be methylated at enhancer sites of the CYP1A1 promoter. It is worthy to note that despite the presence of methylation at the XRE-1383 site, all untreated cancer cells displayed increased though variable constitutive levels of CYP1A1 (compared to BPH-1). However paradoxical, we can point out that PC-3 cells that showed complete methylation by MSP/USP densitometry and bisulfite sequencing had only a small increase in CYP1A1 expression, whereas LNCaP and DU145 displayed partial methylation that can lead to relatively higher gene expression. Compared to LNCaP, it is not known why DU145 had increased CYP1A1 expression despite elevated levels of methylation. Other epigenetic processes such as histone acetylation and deacetylation has been shown to affect gene transcription [25] and further studies are needed to determine if this could also play a role. Nonetheless, 5-aza-dC treatment showed a clear and pronounced inverse effect between expression and methylation of CYP1A1.
In tissue, a similar pattern was also observed as TMA specimens displayed a significant increase of CYP1A1 expression in prostate cancer compared to normal and BPH; and methylation analyses of clinical specimens exhibited cancer to be hypomethylated at all three enhancer sites (XRE-1383, XRE-983, XRE-895) compared to BPH. In contrast to our results, Okino et al. [16] did not detect methylation in the enhancer region of all 30 non-cancerous prostates whereas 11 of 30 tumor specimens showed methylation. Reasons for discrepancy with our study are not known but in their study, expression of CYP1A1 was not measured and would be of interest if a correlation existed with methylation in their clinical specimens. In support of our results however, in other studies that utilized normal prostate tissues only, Martin et al. [26] determined that all 10 specimens were devoid of CYP1A1 by Western analysis and John et al. [7] observed minimal gene expression by real-time PCR. Thus taken together with our results of prostatic tissues and cell lines, these findings strongly suggest that DNA hypomethylation of the CYP1A1 enhancer is a frequent event in prostate cancer and involved in the induction of CYP1A1 expression.
Recently several epidemiologic reports using relatively large cohorts suggest that tobacco smoking, an important risk factor for several cancers [27, 28], may also be associated with prostate cancer [20–23]. It is postulated that the induction of several carcinogen relevant genes, especially CYP1A1, could play important roles in smoking-induced tumorigenesis [29, 30]. In the prostate, studies by Sterling and Cutroneo [24] and Hruba et al. [6] showed that CYP1A1 expression was enhanced by benzo[a]pyrene, a genotoxin in tobacco smoke. These reports led us to determine the possible association between smoking and methylation levels of the CYP1A1 enhancer in prostate cancer patients. Here, we clearly show for the first time that methylation of the CYP1A1 enhancer encompassing the XRE-1383 site was especially affected by smoking with a medium effect size difference as measured by Cohen’s d value; and this observation is in agreement with previous experiments on human lung tissues [17, 18]. Though we haven’t measured the correlation of CYP1A1 expression in tissue with smoker status, the evidence is strong to predict that CYP1A1 levels would be higher in smokers due to lower methylation. Thus smoking could influence the pathogenesis of prostate cancer via the epigenetic dysregulation of CYP1A1. The putative mechanisms underlying tobacco smoking and regulation of CYP1A1 methylation is that smoking promotes AhR/ARNT heterodimer binding to XREs which leads to loss of methylation by eliminating DNA methyltransferases from the CYP1A1 enhancer [19]. Therefore tobacco smoke may indirectly enhance demethylation of the CYP1A1 enhancer.
It should be noted that the expression level of many genes induced by smoking can be reduced after smoking cessation [31]. In the case of CYP1A1, a study by Anttila et al. [17] showed the methylation level of its promoter to increase within 1–7 days after quitting smoking in lung cancer which may inhibit the binding of AhR/ARNT heterodimer and subsequently lead to a reduction of CYP1A1 expression. In our study, ex-smokers who had quit smoking 7 or more days before surgery combined with never-smokers showed higher CYP1A1 methylation levels compared with current smokers, indicating smoking to be a modifiable risk factor for prostate cancer that is dependent on CYP1A1 expression. This observation can help to add to the recommendation for tobacco smoking cessation.
Significant up-regulation of CYP1A1 has been observed in several types of malignancies, i.e. esophagus, breast, urinary bladder, and brain tumor, and is associated with poor prognosis [32–36]. These findings are conceivable since CYP1A1 is known to play an important role in the formation of PAH-DNA adducts which may induce mutations in several cancer-related genes that contribute to carcinogenesis [37, 38]. Though previous research has failed to demonstrate a role for PAH-DNA adducts as a cancer factor in the prostate [6, 7], our results showing that prostate cancer has high CYP1A1 expression as compared to normal/benign prostate tissues suggests that CYP1A1 may contribute to prostate cancer pathogenesis. We observe that reduction of basal CYP1A1 expression leads to significant inhibition of cell proliferation through induction of apoptosis in prostate cancer cells, indicating CYP1A1 expression is critical for cell growth, and this is in concordance with Hruba et al. [6] who showed that enhanced CYP1A1 levels induced by benzo[a]pyrene displayed no effect on either apoptosis or cell cycle arrest in LNCaP cells. Interestingly, a recent study has also revealed that the attenuation of CYP1A1 could prevent breast cancer progression even in the absence of xenobiotics [8]. Thus CYP1A1 appears to have both canonical carcinogen metabolic function as well as its own oncogenic function.
We further identified that the effects of CYP1A1 knockdown on increased apoptosis in prostate cancer cell lines were correlated with reduction of anti-apoptotic BCL2, the primary gatekeeper of the intrinsic (mitochondrial) apoptotic pathway. This is expected since metabolites of CYP1A1 such as heterocyclic aromatic amines have been shown to cause increase in BCL2 mRNA and protein [39]. BCL2 is commonly overexpressed in prostate cancer compared to normal prostate [40] and elevation of BCL2 expression is associated with poor prognosis and suspected in the development of castration-resistant prostate cancer [41–43]. Currently several clinical trials targeting BCL2 expression in prostate cancer are ongoing [44]; therefore CYP1A1 may be a promising target for prostate cancer treatment since it is closely associated with BCL2 expression. What remain unclear are the mechanisms of CYP1A1 regulation of BCL2. Expression of BCL2 is involved in the PI3K-AKT signaling pathway implicated in the development and progression of prostate cancer [45–47] and indeed, CYP1A1 metabolites have been shown to lead to AKT phosphorylation [48]. AKT activated by PI3K leads to the activation of NFkB, which then translocates to the nucleus and transcribes BCL2. In this study, we show that CYP1A1 knockdown can cause reduction of AKT phosphorylation in prostate cancer cells and this effect was also shown in breast cancer cells [8]. This inhibition of AKT could then promote FOXO3a and Par-4 activation leading to apoptosis as was demonstrated in prostate cancer cells by Das et al. [49]. Also in a report by Zhu et al. [50], silencing of prostatic PC3 cells for AKT resulted in reductions of growth promoting RANKL, PTHrP, and BMP-2. Taken together, our results suggest that CYP1A1 may act as an upstream activator of the PI3K-AKT signaling pathway and enhance the transcriptional activation of anti-apoptotic genes that include BCL2; and further studies are needed to verify this hypothesis.
In summary, we show that CYP1A1 is an important oncogene in prostate cancer that is up-regulated by DNA promoter hypomethylation. Tobacco smoking can cause a further reduction of methylation at the CYP1A1 distal enhancer that may lead to carcinogen activation through increased CYP1A1 expression. Furthermore, CYP1A1 is shown to have its own oncogenic function and promote prostate cancer proliferation and survival via dysregulation of BCL2. Altogether our findings demonstrate that tobacco smoking may contribute to the pathogenesis of prostate cancer and that inhibition of CYP1A1 may have therapeutic potential.
MATERIALS AND METHODS
Tissue microarray
In total, 102 primary prostate cancer tissues composed of 10 specimens from TMA PR804, 36 from TMA PR956, and 56 from TMA PR208 (all acquired from US Biomax, Rockville, MD) were evaluated. Also, 14 normal prostate and 70 BPH specimens were included in these 3 TMAs.
Clinical samples
A total of 176 newly diagnosed prostate cancer tissues from radical prostatectomy and 69 pathologically proven BPH samples from transurethral resection were obtained from a urology tissue bank at the Veterans Affairs (VA) Medical Center. The pathological background of the prostate cancer patients includes Gleason < 4 – 33 cases, Gleason 5 – 33 cases, Gleason 6 – 31 cases, Gleason 7 – 47 cases and Gleason > 8 – 32 cases; pT2 – 117 cases, pT3 – 56 cases and pT4 – 3 cases. The median age (range) of prostate cancer and BPH patients were 69 (49–80 years) and 75 (54–87 years), respectively. Our routine strategy to diagnose prostate cancer included serum PSA level, transrectal ultrasonography, color Doppler ultrasonography [51] and MRI that enabled us to accurately localize prostate cancer before radical prostatectomy. A portion of each tissue sample was fixed in 10% buffered formalin (pH 7.0) and embedded in paraffin wax. Sections (5 μm) were used for hematoxylin and eosin staining for histological evaluation. Another portion of tissue was frozen fresh and stored at –80°C until analyzed. Written informed consent was obtained from each patient for molecular analyses of the resected specimens and this study was approved by the Clinical Research Office of the San Francisco VA Medical Center and the Institutional Review Board of the University of California at San Francisco.
Cell lines and reagents
Human prostate cancer cell lines (PC-3, LNCaP, and DU145) were purchased from the American Type Culture Collection (Manassas, VA). Keratinocyte serum-free medium, bovine pituitary extract and human recombinant epidermal growth factor were purchased from Invitrogen (Carlsbad, CA). RPMI 1640, Opti-minimum essential medium and penicillin/streptomycin were obtained from the UCSF Cell Culture Facility (San Francisco, CA). Fetal bovine serum was a product of Atlanta Biologicals (Lawrenceville, GA). All cell lines were cultured in RPMI 1640 medium supplemented with 10% FBS. Cells were maintained in a humidified atmosphere of 5% CO2/95% air at 37°C.
Nucleic acid extraction
Genomic DNA from prostate cancer and BPH tissue samples were extracted using a DNA extraction kit (Qiagen, Valencia, CA). Genomic DNA from cell line samples were extracted using DNAzol reagent (Life Technologies, San Diego, CA) and total RNA was extracted with TRI reagent (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s instructions. The RNA pellet obtained after isopropanol and ethanol precipitation was dried, resuspended in RNase-free water, and stored in aliquots of 25 μL at −80°C until reverse-transcribed. The concentrations of DNA and RNA were determined with a spectrophotometer and their integrity was checked by gel electrophoresis.
cDNA preparation and gene quantification
Using 1 μg of RNA, 0.5 μg of oligo-dT primer and 0.5 units of RNase inhibitor, cDNA was constructed using reverse transcriptase (Promega, Madison, WI). The mRNA transcript levels of CYP1A1, CD70, MCL1, TNFRSF11B, FAS and BCL2 were measured by the 7500 Fast Real-Time PCR System (Applied BioSystems, Foster City, CA) with GAPDH used as the reference gene. The data were analyzed by the delta-delta Ct method to calculate the fold-change.
Western blot analysis
Whole cell extracts from cell lines were prepared using radio-immunoprecipitation assay buffer (RIPA; Thermo Scientific, Rockford, IL) containing protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Protein quantification was done using a BCA protein assay kit (Thermo Scientific) according to the manufacturer’s instructions. Total cell protein (15–20 μg) was used for Western blotting. Samples were transferred to PVDF membranes that were immersed in 3% skim milk containing antibody against CYP1A1 (polyclonal #ab3568, Abcam, Cambridge, MA), AKT (monoclonal #4691, Cell Signaling Technology, Danvers, MA), phospho-AKT (monoclonal #4060, Cell Signaling Technology), BCL2 (polyclonal sc-492, Santa Cruz Biotechnology, Dallas, Texas), and GAPDH (monoclonal sc-47724, Santa Cruz Biotechnology) overnight at 4°C. Blots were washed in TBS containing 0.1% Tween20 and labeled with horseradish peroxidase conjugated, secondary anti-rabbit antibody (Cell Signaling Technology). Specific complexes were visualized with an enhanced chemiluminescence detection system (GE Healthcare, Little Chalfont, UK) using the Chemidoc imaging system (Bio-Rad, Hercules, CA). GAPDH was used as control for equal protein loading.
5′-Aza-2-deoxycytidine (5-aza-dC) treatment
Prostate cancer cell lines were treated with 5-aza-dC (Sigma-Aldrich, St Louis, MO) to screen for epigenetic alterations. 5-aza-dC was added to fresh cell culture medium at a concentration of 5 μM in duplicate wells. The cultured cells were harvested after 4 days of treatment. The mRNA transcripts before and after 5-aza-dC treatment were analyzed by real-time PCR after cDNA conversion.
Bisulfite DNA sequencing and methylation analysis
Genomic DNA (100 ng) was modified with sodium bisulfite (Sigma-Aldrich) using a commercial kit (Life Technologies). In the CYP1A1 promoter, up to 10 high affinity binding sites for the AHR complex has been identified [52] and are shown in Figure 6A. Based on previous findings the methylation of the -1383 bp CpG site located within the distal XRE was significantly affected by smoking [17, 18] and shown to regulate CYP1A1 expression [18], and our methylation analysis was focused on this CpG site (XRE-1383, region A, Figure 6A). In addition, we evaluated region B which contains XRE at -983 bp (XRE-983) that also is affected by smoking [18] as well as region C that contains XRE at the -895 bp site (XRE-895, Figure 6A). Universal, methylation-, and unmethylation-specific PCR primers were designed using a MethPrimer program (http://itsa.ucsf.edu/~urolab/methprimer) according to our previous studies [12–14]. The 3 regions (A, B, and C) amplified by these primers have 5, 22 and 13 CpG sites, respectively, and a schematic diagram of the location of these primers and CpG sites are shown in Figure 6B. For MSP (methylation-specific PCR), a second round of nested PCR (MSP and USP (unmethylation-specific PCR)) was done using the universal PCR product amplified by universal-sense and universal-antisense primers as a template. The universal primer sets contain no CpG sites in either the forward or reverse primer. In each amplification, the absence of a DNA template served as a negative control. The universal primer sequences for regions A through C were 5′-GGGATTATTTTTTGGTTTGGATTA-3′ (sense) and 5′-CATAACCTAACTACCTACCTCC-3′ (antisense), 5′-AGGTTGGTTTTTTAAGAGTTT-3′ (sense) and 5′-ATTAACAAAACACAAAAATCC-3′ (antisense), and 5′-TTTGTTTTTTAGAGGGATGT-3′ (sense) and 5′-CTTTAATTAACAAAACACAAAAAT-3′ (antisense), respectively. The MSP primer sequences for regions A-C were 5′- GGGTTAGGTGAGTTAGGTCG-3′ (sense) and 5′- CCGACGCTATCCCGCCCTCCG-3′ (antisense), 5′- CGGGTTTTCGGTTTTTTTTAC-3′ (sense) and 5′- CCCGACTCTAACTTACGTACG-3′ (antisense), and 5′- TAGAGGGATGTCGTCGGCGTAC-3′ (sense) and 5′- TAACAAAACACAAAAATCCGACGACG-3′ (antisense), respectively. The USP primer sequences for regions A-C were 5′- TGGGGTTAGGTGAGTTAGGTTG-3′ (sense) and 5′- TCCAACACTATCCCACCCTCCA-3′ (antisense), 5′- GTGGGTTTTTGGTTTTTTTTATGT-3′ (sense) and 5′- CCCCAACTCTAACTTACATACACC-3′ (antisense), and 5′- TAGAGGGATGTTGTTGGTGTAT-3′ (sense) and 5′- TAACAAAACACAAAAATCCAACAACA-3′ (antisense), respectively. The MSP and USP products were analyzed by 2% agarose gel electrophoresis. Density of each band was calculated by Image J software (http://rsb.info.nih.gov/ij) and the relative methylation level in each sample was determined using the following formula; MSP ratio (%) = MSP /[MSP+USP]. DNA sequencing was also performed on bisulfite-modified DNA. One μl of modified DNA was amplified using a pair of universal primers in a total volume of 20 μL. Sequencing of the PCR products using either a forward or reverse universal primer was done according to the manufacturer’s instructions (Applied Biosystems).
Knockdown of CYP1A1 in LNCaP and DU145 cells
siRNA oligonucleotides against human CYP1A1 and mismatch control oligonucleotides were purchased from Life Technologies. For inhibition of CYP1A1, 5 μl of siRNA oligonucleotides (siRNA-CYP1A1 (s3800 (designated #1), s3801 (designated #2), or siRNA-control) and 5 μl of lipofectamine RNAiMAX reagent (Life Technologies) were diluted with 250 μl of Opti-MEM (Gibco, Carlsbad, CA). Cells were then transfected with lipofectamine+siRNA-CYP1A1 (#1 or #2) or siRNA-control. Transfection was terminated after 5 hours by aspirating the transfection medium including non-adherent cells and adding fresh RPMI 1640 containing 10% FBS. The remaining cells were incubated at 37°C.
MTS assay
Cells were plated in triplicate in 96-well microplates at a density of 3 × 103 cells per well. After treatment with CYP1A1 siRNA, the number of viable cells was determined over time by adding 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium-based CellTiter 96 Aqueous One Solution Reagent (Promega) to each well and measuring the absorbance at 490 nm on SPECTRA MAX 190 plate reader (Molecular Devices, Sunnyvale, CA).
Apoptosis assay
Fluorescence-activated cell-sorting (FACS) analysis for apoptosis was done 48 hours post-transfection using an annexin V-fluorescein isothiocyanate (FITC)/7-amino-actinomycin D (7-AAD) staining system obtained from BD Biosciences (San Jose, CA) and a BD FACSVerseTM flow cytometer (BD Biosciences). Cells were stained with annexin V-FITC only (early apoptotic) or both annexin V-FITC and 7-AAD (late apoptotic), and the combined sum was considered to be the total apoptotic cell fraction.
Apoptosis-related gene array analyses
cDNAs from control and CYP1A1 siRNA#2-treated cells were evaluated for gene expression using the RT2 ProfilerTM PCR Array PAHS-012ZC (Human Apoptosis PCR Array, Qiagen) on the ABI Fast 7500 Real-Time PCR System with RT2 Real-Time SYBR Green PCR master mix according to the manufacturer’s protocol.
Immunohistochemical analyses
Immunostaining of CYP1A1 was performed on TMA (tissue microarray) slides using the UltraVision Detection System (Thermo Scientific) according to the manufacturer’s instructions. After 12 hours incubation with rabbit polyclonal antibody for CYP1A1 (1:200, #ab3568, Abcam), 3,3′-diaminobenzidine was added as chromogen followed by counterstaining with hematoxylin. The degree of immunostaining was evaluated by two independent observers. Cytoplasmic expression was analyzed by the intensity of positive cells using Image J software (http://rsb.info.nih.gov/ij) and was ranked on an overall scale from 0 to 3; with 0 indicating the absence of staining; 1, weak staining; 2, moderate staining; and 3, strong staining according to our previous study [53].
Statistical analyses
Values are presented as the mean ± standard error based on results obtained from at least three independent experiments. All data were analyzed using StatView 5 statistical software (SAS Institute, Inc., Cary, NC). The relationship between two variables was analyzed using the two-tailed unpaired Student’s t-test. Cohen’s d value was calculated to determine effect size difference between groups. A p-value of less than 0.05 was considered to be statistically significant.
ACKNOWLEDGMENTS
We thank Dr. Roger Erickson for his support and assistance with the preparation of the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
GRANT SUPPORT
This research study was supported by the National Institutes of Health (R21CA185003; UO1CA184966-01A1; RO1CA138642; RO1CA160079) and the Department of Veterans Affairs Merit Review and Program Project awards.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Han G, Ma Y, Liu P, Wei X, Zhang X, Zhu F. Quantitative synthesis of the association between the cytochrome P450 1A1 Ile462Val polymorphism and prostate cancer risk. Tumour Biol. 2013; 34:1511–1516.
3. Wu B, Liu K, Huang H, Yuan J, Yuan W, Wang S, Chen T, Zhao H, Yin C. MspI and Ile462Val polymorphisms in CYP1A1 and overall cancer risk: a meta-analysis. PLoS One. 2013; 8:e85166.
4. Nebert DW, Dalton TP. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006; 6:947–960.
5. Androutsopoulos VP, Tsatsakis AM, Spandidos DA. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer. 2009; 9:187.
6. Hruba E, Trilecova L, Marvanova S, Krcmar P, Vykopalova L, Milcova A, Libalova H, Topinka J, Starsichova A, Soucek K, Vondracek J, Machala M. Genotoxic polycyclic aromatic hydrocarbons fail to induce the p53-dependent DNA damage response, apoptosis or cell-cycle arrest in human prostate carcinoma LNCaP cells. Toxicol Lett. 2010; 197:227–235.
7. John K, Ragavan N, Pratt MM, Singh PB, Al-Buheissi S, Matanhelia SS, Phillips DH, Poirier MC, Martin FL. Quantification of phase I/II metabolizing enzyme gene expression and polycyclic aromatic hydrocarbon-DNA adduct levels in human prostate. Prostate. 2009; 69:505–519.
8. Rodriguez M, Potter DA. CYP1A1 regulates breast cancer proliferation and survival. Mol Cancer Res. 2013; 11:780–792.
9. Puga A, Ma C, Marlowe JL. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem Pharmacol. 2009; 77:713–722.
10. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11:726–734.
11. Gao Y, Tollefsbol TO. Impact of Epigenetic Dietary Components on Cancer through Histone Modifications. Curr Med Chem. 2015; 22:2051–2064.
12. Mitsui Y, Shiina H, Hiraki M, Arichi N, Hiraoka T, Sumura M, Honda S, Yasumoto H, Igawa M. Tumor suppressor function of PGP9.5 is associated with epigenetic regulation in prostate cancer—novel predictor of biochemical recurrence after radical surgery. Cancer Epidemiol Biomarkers Prev. 2012; 21:487–496.
13. Mitsui Y, Yasumoto H, Nagami T, Hiraki M, Arichi N, Ishikawa N, Araki A, Maruyama R, Tanaka Y, Dahiya R, Shiina H. Extracellular activation of Wnt signaling through epigenetic dysregulation of Wnt inhibitory factor-1 (Wif-1) is associated with pathogenesis of adrenocortical tumor. Oncotarget. 2014; 5:2198–2207. doi: 10.18632/oncotarget.1889.
14. Mitsui Y, Hirata H, Arichi N, Hiraki M, Yasumoto H, Chang I, Fukuhara S, Yamamura S, Shahryari V, Deng G, Saini S, Majid S, Dahiya R, et al. Inactivation of bone morphogenetic protein 2 may predict clinical outcome and poor overall survival for renal cell carcinoma through epigenetic pathways. Oncotarget. 2015; 6:9577–9591. doi: 10.18632/oncotarget.3445.
15. Zelic R, Fiano V, Grasso C, Zugna D, Pettersson A, Gillio-Tos A, Merletti F, Richiardi L. Global DNA hypomethylation in prostate cancer development and progression: a systematic review. Prostate Cancer Prostatic Dis. 2015; 18:1–12.
16. Okino ST, Pookot D, Li LC, Zhao H, Urakami S, Shiina H, Igawa M, Dahiya R. Epigenetic inactivation of the dioxin-responsive cytochrome P4501A1 gene in human prostate cancer. Cancer Res. 2006; 66:7420–7428.
17. Anttila S, Hakkola J, Tuominen P, Elovaara E, Husgafvel-Pursiainen K, Karjalainen A, Hirvonen A, Nurminen T. Methylation of cytochrome P4501A1 promoter in the lung is associated with tobacco smoking. Cancer Res. 2003; 63:8623–8628.
18. Tekpli X, Zienolddiny S, Skaug V, Stangeland L, Haugen A, Mollerup S. DNA methylation of the CYP1A1 enhancer is associated with smoking-induced genetic alterations in human lung. Int J Cancer. 2012; 131:1509–1516.
19. Shen ES, Whitlock JP, Jr. The potential role of DNA methylation in the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 1989; 264:17754–17758.
20. Carter BD, Abnet CC, Feskanich D, Freedman ND, Hartge P, Lewis CE, Ockene JK, Prentice RL, Speizer FE, Thun MJ, Jacobs EJ. Smoking and mortality—beyond established causes. N Engl J Med. 2015; 372:631–640.
21. Rohrmann S, Linseisen J, Allen N, Bueno-de-Mesquita HB, Johnsen NF, Tjonneland A, Overvad K, Kaaks R, Teucher B, Boeing H, Pischon T, Lagiou P, Trichopoulou A, et al. Smoking and the risk of prostate cancer in the European Prospective Investigation into Cancer and Nutrition. Br J Cancer. 2013; 108:708–714.
22. Rieken M, Shariat SF, Kluth LA, Fajkovic H, Rink M, Karakiewicz PI, Seitz C, Briganti A, Roupret M, Loidl W, Trinh QD, Bachmann A, Pourmand G. Association of Cigarette Smoking and Smoking Cessation with Biochemical Recurrence of Prostate Cancer in Patients Treated with Radical Prostatectomy. Eur Urol. 2015; 68:949–956.
23. Zapata DF, Howard LE, Aronson WJ, Kane CJ, Terris MK, Amling CL, Cooperberg MR, Freedland SJ. Smoking is a predictor of adverse pathological features at radical prostatectomy: Results from the Shared Equal Access Regional Cancer Hospital database. Int J Urol. 2015; 22:658–662.
24. Sterling KM, Jr, Cutroneo KR. Constitutive and inducible expression of cytochromes P4501A (CYP1A1 and CYP1A2) in normal prostate and prostate cancer cells. J Cell Biochem. 2004; 91:423–429.
25. Lehrmann H, Pritchard LL, Harel-Bellan A. Histone acetyltransferases and deacetylases in the control of cell proliferation and differentiation. Adv Cancer Res. 2002; 86:41–65.
26. Martin FL, Patel, II, Sozeri O, Singh PB, Ragavan N, Nicholson CM, Frei E, Meinl W, Glatt H, Phillips DH, Arlt VM. Constitutive expression of bioactivating enzymes in normal human prostate suggests a capability to activate pro-carcinogens to DNA-damaging metabolites. Prostate. 2010; 70:1586–1599.
27. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108.
28. Carbone D. Smoking and cancer. Am J Med. 1992; 93:13S-17S.
29. Gumus ZH, Du B, Kacker A, Boyle JO, Bocker JM, Mukherjee P, Subbaramaiah K, Dannenberg AJ, Weinstein H. Effects of tobacco smoke on gene expression and cellular pathways in a cellular model of oral leukoplakia. Cancer Prev Res. 2008; 1:100–111.
30. Zhang L, Lee JJ, Tang H, Fan YH, Xiao L, Ren H, Kurie J, Morice RC, Hong WK, Mao L. Impact of smoking cessation on global gene expression in the bronchial epithelium of chronic smokers. Cancer Prev Res. 2008; 1:112–118.
31. Penning TM, Lerman C. Genomics of smoking exposure and cessation: lessons for cancer prevention and treatment. Cancer Prev Res. 2008; 1:80–83.
32. Murray GI, Shaw D, Weaver RJ, McKay JA, Ewen SW, Melvin WT, Burke MD. Cytochrome P450 expression in oesophageal cancer. Gut. 1994; 35:599–603.
33. Murray GI, Taylor VE, McKay JA, Weaver RJ, Ewen SW, Melvin WT, Burke MD. Expression of xenobiotic metabolizing enzymes in tumours of the urinary bladder. Int J Exp Pathol. 1995; 76:271–276.
34. Murray GI, Patimalla S, Stewart KN, Miller ID, Heys SD. Profiling the expression of cytochrome P450 in breast cancer. Histopathology. 2010; 57:202–211.
35. Vinothini G, Nagini S. Correlation of xenobiotic-metabolizing enzymes, oxidative stress and NFkappaB signaling with histological grade and menopausal status in patients with adenocarcinoma of the breast. Clin Chim Acta. 2010; 411:368–374.
36. Wahid M, Mahjabeen I, Baig RM, Kayani MA. Expression of CYP1A1 and GSTP1 in human brain tumor tissues in Pakistan. Asian Pac J Cancer Prev. 2013; 14:7187–7191.
37. Luch A, Kudla K, Seidel A, Doehmer J, Greim H, Baird WM. The level of DNA modification by (+)-syn-(11S,12R,13S,14R)- and (-)-anti-(11R,12S,13S,14R)-dihydrodiol epoxides of dibenzo[a,l]pyrene determined the effect on the proteins p53 and p21WAF1 in the human mammary carcinoma cell line MCF-7. Carcinogenesis. 1999; 20:859–865.
38. Hussain SP, Amstad P, Raja K, Sawyer M, Hofseth L, Shields PG, Hewer A, Phillips DH, Ryberg D, Haugen A, Harris CC. Mutability of p53 hotspot codons to benzo(a)pyrene diol epoxide (BPDE) and the frequency of p53 mutations in nontumorous human lung. Cancer Res. 2001; 61:6350–6355.
39. Pezdirc M, Zegura B, Filipic M. Genotoxicity and induction of DNA damage responsive genes by food-borne heterocyclic aromatic amines in human hepatoma HepG2 cells. Food Chem Toxicol. 2013; 59:386–394.
40. Mazaris E, Tsiotras A. Molecular pathways in prostate cancer. Nephrourol Mon. 2013; 5:792–800.
41. Bubendorf L, Sauter G, Moch H, Jordan P, Blochlinger A, Gasser TC, Mihatsch MJ. Prognostic significance of Bcl-2 in clinically localized prostate cancer. Am J Pathol. 1996; 148:1557–1565.
42. McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, Tu SM, Campbell ML. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992; 52:6940–6944.
43. Apakama I, Robinson MC, Walter NM, Charlton RG, Royds JA, Fuller CE, Neal DE, Hamdy FC. bcl-2 overexpression combined with p53 protein accumulation correlates with hormone-refractory prostate cancer. Br J Cancer. 1996; 74:1258–1262.
44. Zielinski RR, Eigl BJ, Chi KN. Targeting the apoptosis pathway in prostate cancer. Cancer J. 2013; 19:79–89.
45. Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem. 2006; 281:13694–13707.
46. Shukla S, Maclennan GT, Marengo SR, Resnick MI, Gupta S. Constitutive activation of P I3 K-Akt and NF-kappaB during prostate cancer progression in autochthonous transgenic mouse model. Prostate. 2005; 64:224–239.
47. Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002; 14:381–395.
48. Solhaug A, Refsnes M, Holme JA. Role of cell signalling involved in induction of apoptosis by benzo[a]pyrene and cyclopenta[c,d]pyrene in Hepa1c1c7 cells. J Cell Biochem. 2004; 93:1143–1154.
49. Das TP, Suman S, Alatassi H, Ankem MK, Damodaran C. Inhibition of AKT promotes FOXO3a-dependent apoptosis in prostate cancer. Cell Death Dis. 2016; 7:e2111.
50. Zhu W, Hu X, Xu J, Cheng Y, Shao Y, Peng Y. Effect of PI3K/Akt Signaling Pathway on the Process of Prostate Cancer Metastasis to Bone. Cell Biochem Biophys. 2015; 72:171–177.
51. Shigeno K, Igawa M, Shiina H, Wada H, Yoneda T. The role of colour Doppler ultrasonography in detecting prostate cancer. BJU Int. 2000; 86:229–233.
52. Kress S, Reichert J, Schwarz M. Functional analysis of the human cytochrome P4501A1 (CYP1A1) gene enhancer. European journal of biochemistry. 1998; 258:803–812.
53. Chang I, Mitsui Y, Fukuhara S, Gill A, Wong DK, Yamamura S, Shahryari V, Tabatabai ZL, Dahiya R, Shin DM, Tanaka Y. Loss of miR-200c up-regulates CYP1B1 and confers docetaxel resistance in renal cell carcinoma. Oncotarget. 2015; 6:7774–7787. doi: 10.18632/oncotarget.3484.