
INTRODUCTION
Identified as a class I carcinogen [1], Helicobacter pylori (H. pylori) may play important roles in the multi-stage process of gastric carcinogenesis [2]. Our previous studies in a high-risk area of gastric cancer (GC) revealed that H. pylori infection increased the risks of GC and precancerous gastric lesions [3, 4], and eradication treatment inhibited or reversed gastric lesion progression [5, 6].
The aberrant epigenetic alterations induced by reactive oxygen species (ROS) and nitric oxide (NO) after H. pylori infection in gastric mucosa may serve as one of the main mechanisms in carcinogenesis [7, 8]. For example, hypermethylation of tumor suppressor genes Cyclin-dependent kinase inhibitor 2A (CDKN2A) [9], Cadherin 1 [10] and Runt-related transcription factor 3 (RUNX3) [11] was found not only in GC but also in H. pylori infected precancerous gastric lesions [12]. Accumulating genome-wide analyses were performed to describe the large-scale characteristics in GC [13], while little is known about the methylation profiles specific for H. pylori infection.
Previously, identification of epigenetic biomarkers was mainly focused on tissue samples, however, an increasing number of studies are extracting DNA from non-invasive body fluids such as peripheral blood [14]. Aberrant methylation of blood leukocyte DNA was first reported in lung cancer [15]. Our population-based study also found that the methylation levels of Insulin-like growth factor 2 (IGF2) and Tumor suppressor candidate 3 (TUSC3) in blood leukocytes were significantly increased ahead of clinical diagnosis, which may serve as early biomarkers for GC [16]. Further study showed that the presence of H. pylori antibodies was associated with a higher methylation level of TUSC3 in blood leukocytes, suggesting that the methylation status of peripheral blood may also be affected by H. pylori infection [17].
Interestingly, the evidences for origin of blood leukocyte DNA methylation and the relationship with target tissue are still controversial [18]. At present study, we compared the genome-wide methylation profiles prior to and after H. pylori eradication in blood leukocytes as well as in gastric mucosa, and further systematically selected and validated novel H. pylori associated differentially methylated CpGs/genes in gastric mucosa and/or in blood leukocytes.
RESULTS
Comparison of genome-wide methylation profiles between gastric mucosa and blood leukocyte DNA
General characteristics of the 8 subjects for methylation array detection were described in Supplementary Table S1. For correlation analysis between methylation levels in gastric mucosa and blood leukocytes, the baseline H. pylori positive samples of the 6 successfully and 2 unsuccessfully eradicated subjects were combined and calculated the average methylation levels (β values). The methylation levels of more than 480,000 CpGs in the Infinium methylation array were highly correlated between gastric mucosa and blood leukocyte DNA with the R2 as 0.89 (Figure 1).
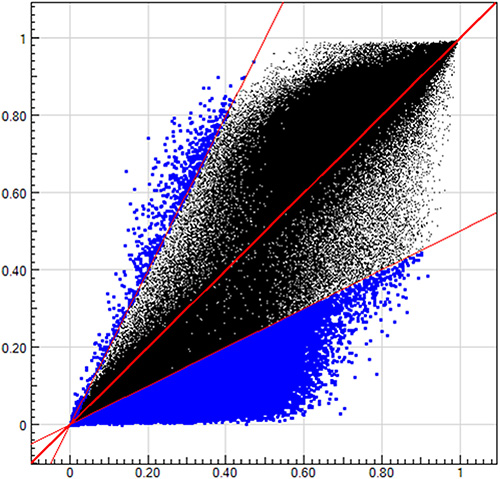
Figure 1: The methylation levels in gastric mucosa and blood leukocyte DNA. The blue dots in upper left region represent the CpGs with (methylation level in gastric mucosa)/(methylation level in blood leukocytes) ≤ 0.5. The blue dots in lower right region represent the CpGs with (methylation level in gastric mucosa)/(methylation level in blood leukocytes) ≥ 2. The black dots in the middle region represent the CpGs with (methylation level in gastric mucosa)/(methylation level in blood leukocytes) > 0.5 and < 2.
Selection of differentially methylated CpGs before and after H. pylori eradication
To identify differentially methylated CpGs in gastric mucosa, we selected 3026 significant CpGs (|Δβ| ≥ 10%, P < 0.05) prior to and after successful H. pylori eradication. Among them, 26 were excluded for overlapping with the significant CpGs in 2 unsuccessfully eradicated subjects. In the 3000 selected H. pylori associated differentially methylated CpGs (covering 1699 genes), 1570 (52.3%) were hypomethylated (Δβ ≥ 10%) and 1430 (47.7%) were hypermethylated (Δβ ≤ −10%) after eradication. Figure 2 showed a hierarchical cluster analysis of the 568 top significant CpGs (P < 0.01) in 6 pairs of gastric mucosa samples prior to and after successful H. pylori eradication. The methylation levels of these significant CpGs were distinguished between H. pylori positive gastric mucosa prior to treatment and negative samples after eradication.
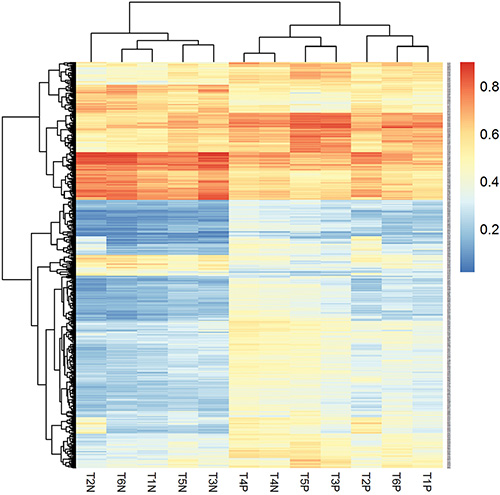
Figure 2: Hierarchical cluster analysis of the differentially methylated CpGs in gastric mucosa before and after H. pylori successful eradication. “T*P” represents H. pylori positive gastric mucosa before eradication and “T*N” represents H. pylori negative gastric mucosa after successful eradication. The methylation levels of the significant CpGs were distinguished between H. pylori positive gastric mucosa prior to treatment and negative samples after eradication. Methylation level is shown as a continuous variable (0 to 1) from a blue to red color.
Using the same strategy, only 2 significant CpGs with |Δβ| ≥ 10% (P < 0.05) were found in blood leukocyte DNA. To select more potential differentially methylated CpGs in blood leukocytes, we used a smaller cut-off |Δβ| as 5% and found a total of 386 differentially methylated CpGs (covering 282 genes) in blood leukocytes (|Δβ| ≥ 5%, P < 0.05), with 71 (18.4%) hypomethylated and 315 (81.6%) hypermethylated after H. pylori eradication. Furthermore, to select more potential common candidate significant CpGs overlapped between gastric mucosa and blood leukocytes, the smaller cut-off |Δβ| of 5% in blood leukocytes was used. A total of 33459 (from gastric mucosa) and 386 (from blood leukocytes) significant CpGs (|Δβ| ≥ 5%, P < 0.05) were compared, and 17 CpGs (involving 11 genes) were overlapped with the same trends of methylation changes after eradication between gastric mucosa and blood leukocyte DNA (Table 1).
Table 1: Differentially methylated CpGs overlapped between blood leukocytes and gastric mucosa
ID |
Average Δβ in blood leukocyte |
Average Δβ in gastric mucosa |
Chr. |
CpG site |
Gene name |
Location with gene |
Location with CGI |
|---|---|---|---|---|---|---|---|
cg12643083 |
−7.38% |
−9.24% |
1 |
159743172 |
FCGR2A |
Body |
|
cg22813165 |
−6.69% |
−5.46% |
19 |
40794451 |
HAUS5 |
TSS1500 |
N_Shore |
cg10180440 |
−6.58% |
−5.49% |
10 |
102315476 |
|
|
S_Shelf |
cg23772352 |
−6.15% |
−6.65% |
12 |
124909736 |
|
|
N_Shelf |
cg05018460 |
−6.14% |
−5.93% |
15 |
78475133 |
|
|
|
cg05025071 |
−5.83% |
−6.71% |
19 |
6838530 |
EMR1 |
TSS200 |
|
cg13686044 |
−5.63% |
−6.25% |
17 |
14677092 |
|
|
|
cg25025181 |
−5.58% |
−10.32% |
1 |
244445133 |
SMYD3 |
Body |
|
cg22534374 |
−5.45% |
−5.54% |
1 |
199778233 |
|
|
S_Shelf |
cg08904369 |
−5.39% |
−5.94% |
6 |
167624178 |
UNC93A |
TSS1500 |
|
cg06518781 |
−5.25% |
−5.55% |
19 |
22495123 |
|
|
S_Shelf |
cg14183176 |
−5.13% |
−7.34% |
3 |
123982893 |
HSPBAP1 |
Body |
|
cg24202468 |
−5.12% |
−7.61% |
13 |
97993340 |
STK24 |
Body |
|
cg01711344 |
−5.10% |
−11.86% |
11 |
67526980 |
UNC93B1 |
Body |
N_Shore |
cg05590451 |
5.57% |
8.15% |
16 |
20798035 |
DCUN1D3 |
5′UTR |
|
cg01785046 |
6.43% |
5.16% |
7 |
91348383 |
MTERF |
TSS1500 |
S_Shore |
cg06522206 |
6.73% |
7.10% |
12 |
116135682 |
NOS1 |
3′UTR |
|
According to the manufacturer information, the CpGs in Infinium methylation array were located 7.8% in the 1st exon, 15.0% in 5′ un-translated regions (UTR), 12.9% in 200 bp upstream of transcription start site (TSS200), 17.9% in TSS1500, 4.4% in 3′UTR and 42.0% in gene body, respectively. Comparing to the array distribution, the significant CpGs in gastric mucosa were found more frequently in 5′UTR (21.2%) and TSS200 (20.4%), and less frequently in gene body (35.2%), P = 6.7 × 10−56. Furthermore, the hypomethylated CpGs were found more frequently in 5′UTR (23.6% vs. 18.2%) and TSS200 (27.2% vs. 12.0%) than hypermethylated ones, P = 1.8 × 10−35 (Figure 3A). In contrast with the tendency in gastric mucosa, the significant CpGs in blood leukocytes were located less frequently in 1st exon (2.8%) and TSS200 (8.8%), and more frequently in gene body (49.5%), P = 6.7 × 10−4. In addition, there was no statistical difference between the distributions of hypo- and hypermethylated CpGs after eradication in blood leukocyte DNA, P = 0.665 (Figure 3C).
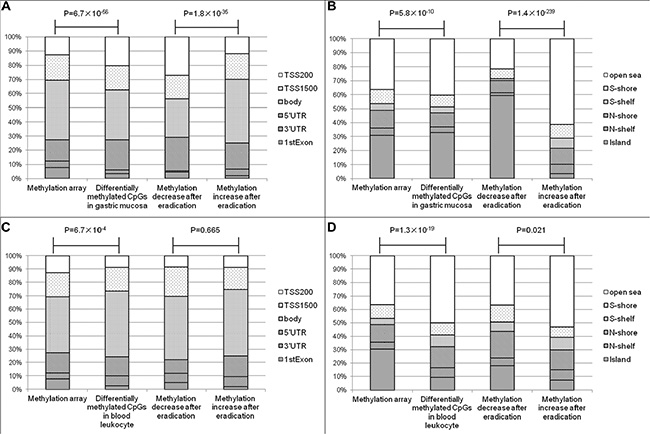
Figure 3: The relationships between differentially methylated CpGs and genes or CGIs. (A) The relationships between differentially methylated CpGs of gastric mucosa and genes. (B) The relationships between differentially methylated CpGs of gastric mucosa and CGIs. (C) The relationships between differentially methylated CpGs of blood leukocytes and genes. (D) The relationships between differentially methylated CpGs of blood leukocytes and CGIs.
Because aberrant methylation has been frequently found in CpG islands (CGIs), shores (regions up to 2 kb from the islands) and shelves (2–4 kb from the islands), we further investigated the relationship between the differentially methylated CpGs and CGIs by comparing with the distribution of methylation array [30.9% in CGI, 5.1% in N (north)-shelf, 4.6% in S (south)-shelf, 13.0% in N-shore, 10.1% in S-shore and 36.3% in open sea]. For gastric mucosa, the significant CpGs were located more frequently in CGIs (32.8%), P = 5.8 × 10−10, and hypomethylated CpGs were found more frequently in CGIs (59.4%) than hypermethylated CpGs (3.6%), P = 1.4 × 10−239 (Figure 3B). For the significant CpGs in blood leukocytes, only 9.6% were found in CGIs, which was much less than the composition of methylation array, P = 1.3 × 10−19. The hypomethylated CpGs were located more frequently in CGI (18.3%) than hypermethylated CpGs (7.6%) after eradication, P = 0.021 (Figure 3D).
Gene ontology and pathway categories of differentially methylated CpGs after H. pylori eradication
For Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, we selected the differentially methylated CpGs located from TSS to TSS1500 and in CGIs. The correlation between methylation levels at the selected CpGs and their target gene expressions was analyzed in 230 GC tissues collected from The Cancer Genome Atlas (TCGA) dataset (http://cancergenome.nih.gov/) by Illumina HumanMethylation 450 K BeadChip array and RNA-sequencing platform. A total of 294 hypo- and 51 hypermethylated CpGs after successful eradication were negatively associated with their target gene expressions (R ≤ −0.2, P < 0.05), which were enrolled in the GO and KEGG pathway analyses.
After successful eradication in gastric mucosa, the hypermethylated CpGs were associated with the inflammatory process and pathways, such as positive regulation of immune system process (Supplementary Table S2), B and T cell receptor signaling pathway and leukocyte transendothelial migration (Supplementary Table S3). While, the hypomethylated CpGs were enriched in tube morphogenesis and development, embryonic morphogenesis and development, homophilic cell adhesion, response to hormone stimulus, epithelium development and so on (Supplementary Table S2). For the differentially methylated CpGs in blood leukocyte DNA, GO and KEGG pathway analyses showed the P values to be more than 0.05 and not significantly enriched.
Stage I validation of candidate differentially methylated genes
From the list of top differentially methylated CpGs located in 5′UTR, TSS200 or 1st exon (Supplementary Table S4), 15 and 3 CpGs (covering 12 and 3 genes) were selected from gastric mucosa and blood leukocyte DNA for Stage I validation. From the 11 candidate overlapped significant genes between gastric mucosa and blood leukocytes, only MTERF and HAUS5 genes were detected in Stage I, because the involving significant CpGs were near to the promoter (TSS1500) and CGIs (shore). The CpGs in the promoters of the candidate genes were amplified using CpG-free primer sets, which were analyzed quantitatively by denaturing high performance liquid chromatography (DHPLC) for methylation levels in the two-stage validation.
A total of 16 subjects were selected for Stage I validation, including 8 successfully and 8 unsuccessfully H. pylori eradicated cases. The baseline gastric lesions included 7 superficial gastritis, 4 chronic atrophic gastritis, 3 intestinal metaplasia and 2 mild dysplasia. Among 12 candidate genes from gastric mucosa, the methylation levels of SPI1 and PRIC285 were increased from 34.9% to 53.8% (P = 0.022) and 35.5% to 50.1% (P = 0.024) after successful eradication, respectively (Table 2). While in unsuccessful eradication group, no statistical methylation change was found for these genes. In addition, although the P values showed no statistical differences (0.132 and 0.069), S1PR4 and CELSR3 were also selected for Stage II validation because of the methylation increasing of 15.5% and decreasing of 9.6% after eradication, respectively.
Table 2: Stage I self-comparison validation for candidate differentially methylated genes
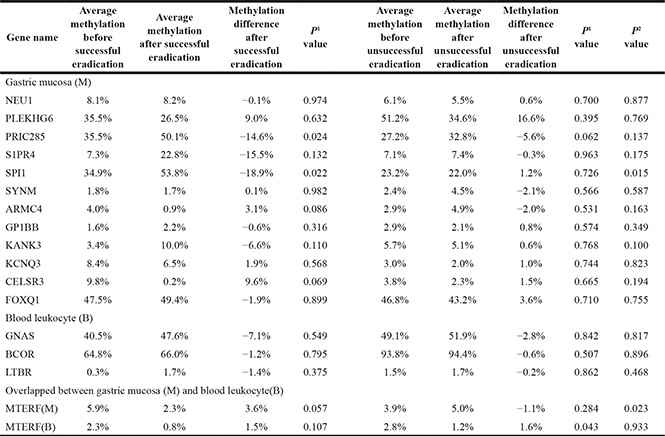
1Paired t test, comparing the methylation level before and after H. pylori eradication in successful and unsuccessful groups.
2t test, comparing the average methylation differences between successful and unsuccessful eradication groups.
For the 3 candidate genes from blood leukocytes, no significant methylation change was found either in successful or unsuccessful eradication group (Table 2). As a candidate gene overlapped between gastric mucosa and blood leukocytes, MTERF was hypomethylated both in gastric mucosa (5.9% to 2.3%) and in blood leukocytes (2.3% to 0.8%) after eradication, although the P values (0.057 and 0.107) showed no statistical significance (Table 2). While for HAUS5, no positive result was found in gastric mucosa and blood leukocyte DNA.
Stage II validation of candidate differentially methylated genes
In Stage II validation, 25 H. pylori positive and 25 negative subjects were selected for case-control comparison and 50 pairs of gastric mucosa samples before and after eradication treatment were enrolled for self-comparison, respectively. For the case-control comparison, 4 candidate genes (SPI1, PRIC285, S1PR4 and CELSR3) were selected from Stage I validation. As shown in Supplementary Table S5, the frequency of chronic atrophic gastritis was higher in H. pylori positive (52.0%) than in negative groups (16.0%, P = 0.021). No significant difference in age, sex, cigarette smoking, and alcohol consumption was found between two groups.
The univariate analysis showed significant lower methylation levels of SPI1, PRIC285 and S1PR4 in H. pylori positive than in negative groups (SPI1: 39.7% vs. 81.1%, P < 0.001; PRIC285: 44.5% vs. 74.6%, P = 0.002; S1PR4: 34.8% vs. 66.7%, P = 0.002). Using the methylation medians in negative group (SPI1: 89.9%, PRIC285: 79.5%, S1PR4: 80.3%) as cut-off values, we divided the methylation status into hyper- and hypo-methylation groups and found that SPI1 and S1PR4 methylation were decreased significantly in H. pylori positive subjects [SPI1: Odd ratio (OR), 0.12; 95% confidence interval (CI), 0.02–0.70, S1PR4: OR, 0.15; 95%CI, 0.03–0.75] (Table 3).
Table 3: Associations between methylation status and H. pylori infection in gastric mucosa in Stage II case-control validation
|
H. pylori negative n (%) |
H. pylori positive n (%) |
P value |
ORa |
95% CIa |
|
|---|---|---|---|---|---|---|
SPI1 (cut-off value 89.9%) |
||||||
Hypermethylated |
12 (48.0) |
2 (8.0) |
0.018 |
0.12 |
0.02–0.70 |
|
Hypomethylated |
13 (52.0) |
23 (92.0) |
|
|
|
|
PRIC285 (cut-off value 79.5%) |
||||||
Hypermethylated |
12 (48.0) |
5 (20.0) |
0.238 |
0.41 |
0.09–1.81 |
|
Hypomethylated |
13 (52.0) |
17 (68.0) |
|
|
|
|
Missing |
|
3 (12.0) |
|
|
|
|
S1PR4 (cut-off value 80.3%) |
||||||
Hypermethylated |
13 (52.0) |
3 (12.0) |
0.021 |
0.15 |
0.03–0.75 |
|
Hypomethylated |
10 (40.0) |
20 (80.0) |
|
|
|
|
Missing |
2 (8.0) |
2 (8.0) |
|
|
|
|
CELSR3 (cut-off value 0%) |
||||||
Methylated |
8 (32.0) |
9 (36.0) |
0.969 |
0.97 |
0.24–3.97 |
|
Unmethylated |
15 (60.0) |
14 (56.0) |
|
|
|
|
Missing |
2 (8.0) |
2 (8.0) |
|
|
|
|
aUnconditional logistic regression analysis, adjusted for age, sex, smoking, drinking and pathologic diagnosis.
Among 50 self-comparison subjects, 37 were successfully H. pylori eradicated. The distribution of age, gender, smoking, drinking and baseline pathology showed no significant difference between the two groups (Supplementary Table S6). After successful eradication, the methylation levels of SPI1, PRIC285 and S1PR4 were increased significantly (SPI1: 34.6% to 75.2%, P < 0.001; PRIC285: 46.8% to 70.4%, P < 0.001; S1PR4: 12.9% to 26.7%, P = 0.003). In unsuccessful group, S1PR4 and PRIC285 genes showed no remarkable change (17.0% to 14.0%, P = 0.664; 44.8% to 45.0%, P = 0.979, respectively), while SPI methylation level decreased after treatment (59.0% to 33.1%, P = 0.027). The methylation level changes of the three genes showed significant differences between successful and unsuccessful eradication groups (all P < 0.05).
The clinical relevance of SPI1, PRIC285, S1PR4 methylation and correlation with expression in GC by TCGA database
To describe a clinical relevance of SPI1, PRIC285 and S1PR4 genes, we investigated their promoter methylation status and expression levels in TCGA public genome database (http://cancergenome.nih.gov/) of 29 normal and 230 gastric tumors, which were divided into four subtypes by molecular classification [19]. The methylation levels of CpGs on promoters (cg07698783, cg15982099, cg07675031 on SPI1; cg11779113, cg06064964, cg09844573 on PRIC285 and cg02716776, cg02996471, cg27121758 on S1PR4) were dramatically lower in the majority of GCs than in normal tissues (Figure 4A–4C).
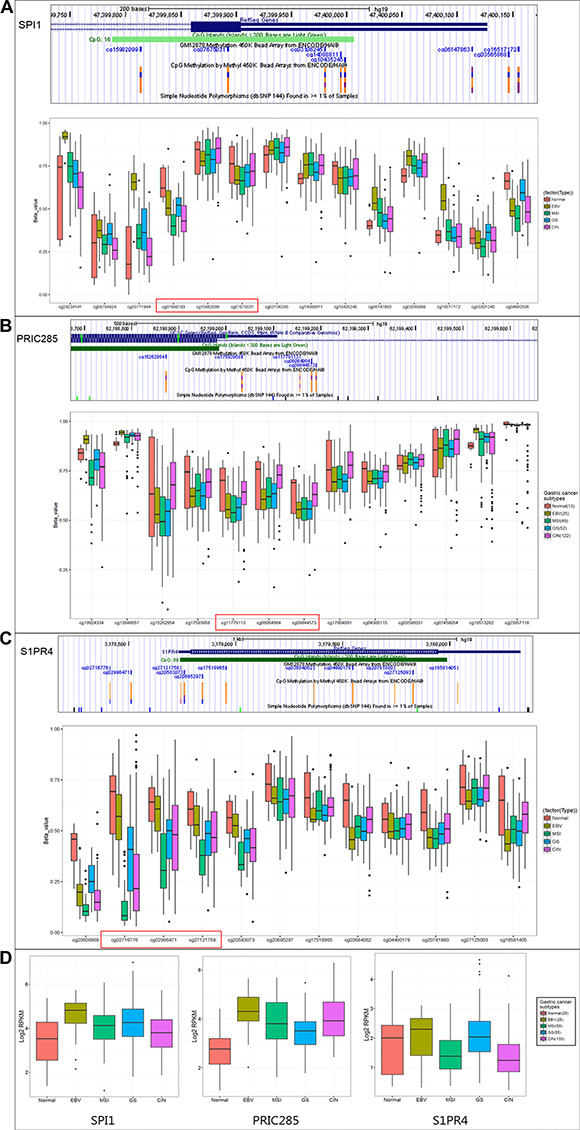
Figure 4: The clinical relevance of SPI1, PRIC285, S1PR4 methylation and correlation with expression in GC by TCGA database. The promoter methylation status (A, B, C) and RNA expression levels (D) of SPI1, PRIC285 and S1PR4 genes in TCGA public genome database of 29 normal and 230 GC subjects, which were divided into four subtypes (EBV: Epstein–Barr virus-positive; MSI: microsatellite instability high status; GS: genomically stable; CIN: chromosomal instability).
From RNA-sequencing data of TCGA, we also found that the expressions of SPI1 or PRIC285 were increased in all types of GC compared to normal tissues and S1PR4 was increased in Epstein-Barr virus-positive and genomically stable subtypes (Figure 4D), suggesting that up-regulation of these genes in GCs might be associated with the decrease of promoter CpGs methylation.
DISCUSSION
Using Infinium HumanMethylation 450 K BeadChip, a comprehensive assessment of genome-wide epigenetic alterations was conducted by comparing blood leukocyte DNA as well as gastric mucosa DNA prior to and after anti-H. pylori treatment, respectively. Correlations were evaluated firstly between the methylation levels of gastric mucosa and blood leukocyte DNA, and novel H. pylori associated differentially methylated CpGs/genes were selected and validated.
Recently, some large-scale comparisons of DNA methylation profiles between blood leukocytes and matched target tissues (such as adipose and brain) suggested that the concordant genes might be tissue specific [20–22]. The present comprehensive array analysis enabled us to compare the methylation levels between blood leukocytes and gastric mucosa systematically for the first time. We found the methylation levels were highly correlated between the two kinds of tissues at baseline (R2 = 0.89), which implied that blood leukocyte DNA methylation might serve as a potential surrogate biomarker for the methylation status of gastric mucosa.
Recent high-throughput technologies enabling global DNA methylome analysis have been used in various cancers to elucidate epigenetic mechanisms [23, 24]. In 2012, a study found that 11,740 CpGs were differentially methylated in GC tissues with 83% hypermethylated using Infinium 27 K methylation array [13], which covered a total of 27,578 CpGs and 14,495 gene promoters in the human genome. In our present study, the CpGs were not limited to the promoter and CGIs and also involved in intragenic regions and gene deserts by Infinium 450 K methylation array. The comparison results found 3000 differentially methylated CpGs with 52.3% hypomethylated and 47.7% hypermethylated after eradication in gastric mucosa, suggesting that H. pylori infection may be associated with hypermethylation as well as hypomethylation in the early stage of gastric lesion evolution.
The distributions of differentially methylated CpGs may provide important information about epigenetic mechanism in human carcinogenesis. For example, “tandem control” mechanism was suggested for epigenetic regulation, in which promoter hypermethylation and gene body hypomethylation may lead to repression, or promoter hypomethylation and gene body hypermethylation may lead to activation [13]. In our study, H. pylori infection might more likely induce genes repression by hypermethylation of promoter and hypomethylation of gene body in gastric mucosa. In addition, several tumor studies showed that significantly hypermethylated CpGs were found more frequently at CGIs, while most hypomethylated CpGs were located at open sea regions [25–27]. Our current findings for H. pylori associated differentially methylated CpGs were consistent with the above results, which suggested the epigenetic alterations induced by H. pylori might be related to gastric carcinogenesis.
To date, few studies have examined the association between methylation levels in blood leukocyte DNA and risk of GC [17, 28]. We found 386 differentially methylated CpGs (|Δβ| ≥ 5%) that may be associated with H. pylori infection in blood leukocyte DNA. Compared to those in gastric mucosa, the number of the differentially methylated CpGs in blood leukocytes was much less, with more hypermethylation (81.6%) and less hypomethylation (18.4%). On the other hand, the significant CpGs from blood leukocyte DNA were located more frequently at gene body and open sea region, which was different with those from gastric mucosa. In addition, the 17 overlapped significant CpGs between gastric mucosa and blood leukocyte DNA were also found outside the promoter and CGIs regions. These distribution discrepancies and higher frequency of hypermethylation in blood leukocyte DNA after eradication were consistent with the previous findings of genomic hypomethylation in H. pylori positive subjects [29, 30], suggesting that H. pylori associated epigenetic alterations in blood leukocyte DNA might be more related with global hypomethylation rather than the aberrant methylation of specific promoter CGI loci.
GO and KEGG pathway analyses of the differentially methylated CpGs in gastric mucosa showed that hypermethylated CpGs in promoter occurred at immune response process and pathways, consisting with the active process of the inflammation after H. pylori infection. Our study also showed that hypomethylated CpGs in promoter were enriched in various biological processes and molecular functions. Among them, cell-cell adhesion was noticed for the vital roles to inhibit individual epithelial cell motility and to provide homeostatic gastric tissue architecture. Being a crucial factor for cell-cell adhesion function, E-cadherin (encoded by CDH1 gene) was found to be silenced by H. pylori infection and the average methylation level was decreased 10.1% after eradication in our present study.
The current array-based methylation profiling identified a large number of H. pylori associated differentially methylated CpGs in gastric mucosa and blood leukocyte DNA. In the final larger sample size validation, SPI1, PRIC285 and S1PR4 methylation levels in gastric mucosa were decreased when H. pylori positive subjects were compared with negative cases, and increased after successful eradication by self-comparison. The consistent results from the case-control and self-comparison studies suggested that methylation alterations of SPI1, PRIC285 and S1PR4 in gastric mucosa might be associated with H. pylori infection. We also identified several differentially methylated CpGs overlapped between gastric mucosa and blood leukocyte DNA, although a negative validation result was found after the detection of overall methylation status in promoter region by DHPLC rather than the methylation levels of specific CpGs by probes in methylation array.
According to the reports, SPI1 is important for the transcriptional regulation of interleukin-1β, involving in inflammatory and immunoregulatory process [31]. PRIC285 acts as a nuclear receptor coactivator by stimulating peroxisome proliferators-activated receptor α (PPARα)-mediated transcription [32], and was down-regulated in the leukocyte fraction of epithelial ovarian cancer subjects [33]. S1PR4 is one of the five G protein-coupled receptors of sphingosine-1-phosphate (S1P) [34], which has received much attention for its potential roles in tumorigenesis and therapies [35, 36]. No epigenetic regulation mechanism was reported for SPI1, PRIC285 or S1PR4 genes in gastric lesions or GC, until our methylation array screening and validations, together with TCGA database analysis, suggested that the up-regulation of the three genes by the decrease of promoter methylation might be associated with H. pylori infection and gastric carcinogenesis. However, further functional studies are still needed to verify the relationships and mechanisms between the aberrant methylation and expression in gastric mucosa or H. pylori infection.
The major strengths of our study lie in a clinical trial design by comparing the methylation levels prior to and after H. pylori eradication, which can control other confounders, such as age, gender, environmental and exposure factors. To exclude the effects on methylation alterations by intervention drugs, we also compared the methylation changes between successful and unsuccessful eradication groups. The correlations between the methylation levels in gastric mucosa and blood leukocyte DNA were evaluated in large-scale for the first time, and provided important clues for surrogate biomarker identification in blood leukocyte DNA. A limitation in our study may be the fact that the case number for validation study was small. For the subsequent validation of aberrant methylation genes in blood leukocyte DNA, a new candidate gene selection strategy will be needed because of the different methylation alteration trend and different distribution of significant CpGs between gastric mucosa and blood leukocyte DNA.
In summary, we have comprehensively characterized genome-wide DNA methylation patterns in gastric mucosa and blood leukocyte DNA and identified a large subset of candidate CpGs/genes associated with H. pylori infection. Several differentially methylated CpGs were overlapped between gastric mucosa and blood leukocyte DNA, although a negative validation result was currently shown. The validations in gastric mucosa identified three novel potential aberrant methylation genes (SPI1, PRIC285 and S1PR4) associated with H. pylori, which still need to confirm in a larger sample size investigation and further functional study.
MATERIALS AND METHODS
Study population
A large intervention trial to prevent GC by H. pylori eradication was launched in March 2011 and completed in September 2013 in Linqu County [37]. During this period, an endoscopic screening project was conducted in Linqu, in which 33 and 108 H. pylori positive subjects participated in the both projects in 2012 and 2013, respectively. For our current study, blood and fresh gastric tissue samples were collected in the endoscopic screening project prior to anti-H. pylori treatment at baseline and a repeat endoscopy examination at six months after the treatment. For genome-wide methylation profiling and Stage I validation, we totally selected 16 subjects (8 successfully and 8 unsuccessfully eradicated) from 33 participants in 2012. Among them, 6 successfully and 2 unsuccessfully eradicated subjects with mild gastric lesions were used for methylation profiling, and all of the 16 subjects were enrolled in Stage I validation. For Stage II validation, 25 H. pylori positive and 25 negative subjects were selected randomly from the baseline endoscopic screening in 2013 for case-control design. Furthermore, 50 subjects with blood and fresh gastric tissue samples prior to and after treatment were selected at random from 108 participants in 2013 for self-comparison design.
The detailed procedure of endoscopy has been described elsewhere [6]. Briefly, upper endoscopy examinations were conducted by four experienced gastroenterologists. The gastric mucosa was examined and five biopsies were obtained from standard sites of the stomach according to the Updated Sydney System [38]. For the present study, one or two additional biopsies were obtained from the lesser curvature of antrum or angulus adjacent and frozen in liquid nitrogen immediately. All specimens were reviewed by a panel of three pathologists according to the Updated Sydney System [38] and Padova International Classification [39]. The detailed information of H. pylori screening and treatment has been reported on our baseline results of large intervention trial [37].
A written informed consent was obtained from each subject and the study was approved by the Institutional Review Board of Peking University Cancer Hospital.
Genome-wide methylation profiling in gastric mucosa and blood leukocyte
The DNA from gastric mucosa and blood leukocytes was extracted using Dneasy Tissue and Blood kits (Qiagen) and bisulfite-converted using EZ DNA methylation kit (Zymo Research) according to the manufacturer’s protocol.
Genome-wide methylation profiling was performed using Infinium HumanMethylation450 BeadChip array (Illumina, San Diego, CA, USA). After whole-genome amplification with 200ng of input bisulfite-converted DNA, the product was fragmented, purified and applied to the BeadChips using Illumina-supplied reagents and conditions. After extension, the array was stained fluorescently, and scanned with an iSan System (Illumina). The data were analyzed by GenomeStudio Methylation Module Software (Illumina). A CpG site was considered to be informative if the sum of the signals for methylated and unmethylated sequence at the CpG site was significantly higher (detection P value < 0.05) than signals of the negative control probes on the same array. For each CpG site, the β value reflects the methylation level, which was computed by β = (max (M, 0))/(|U| + |M|+100). A β value of 0–1.0 indicates the percent methylation from 0% to 100%, respectively.
Identification of differentially methylated CpGs and genes
To select candidate differentially methylated CpGs before and after H. pylori eradication, the methylation levels of 484,543 and 484,435 target CpGs in methylation array with detection P values ≤ 0.05 were analyzed in 6 successfully eradicated subjects. When the methylation differences before and after eradication were ≥ 10% in gastric mucosa (≥ 5% in blood leukocyte) with P values less than 0.05, the CpGs were selected as H. pylori associated differentially methylated ones by excluding those overlapped by unsuccessful eradication group. To identify candidate H. pylori associated differentially methylated genes for further validation, the genes with top differentially methylated CpGs in 5′UTR, TSS200, exon-1 regions and CGIs were focused on due to their well-known correlations with gene transcription regulation.
Hot-start PCR and DHPLC analysis
CpG-free universal primer sets and bisulfite-modified DNA were used to amplify the selected candidate genes. The PCR reaction mixture (20 μL) included 20 ng DNA template, 0.5 mmol/L dNTP, 0.5 μmol/L of each primer, and 1.0 U of HotStart Taq DNA polymerase (Qiagen GmbH). The PCR products were then analyzed quantitatively by DHPLC. Hypermethylated and hypomethylated PCR products were separated using a DNASep analytical column (Transgenomic) at the corresponding partial denaturing temperature. The primer sequence and partial denaturing temperature were listed in the Supplementary Table S7. M. SssI-methylated genomic DNA, obtained from blood samples, was used as a positive control. A sample containing a methylated PCR product peak was defined as methylation-positive. The peak areas corresponding to the methylated and unmethylated PCR products were used to calculate the percentage of methylated copies (proportion of hypermethylated copies = methylated peak area/total peak area) for each candidate gene (Supplementary Figure S1A). The methylation of the CpGs was further confirmed using traditional bisulfite sequencing. The bisulfite sequencing results were consistent with the DHPLC analysis (Supplementary Figure S1B).
Statistical analysis
The student paired t test was used to identify the differentially methylated CpGs and genes before and after H. pylori eradication by methylation array or DHPLC detections. The Mann-Whitney U test was used to compare the methylation level differences between successful and unsuccessful eradication groups. In Stage II case-control validation, χ2 test was used to analyze the differences in age (separated into two groups according to median age), sex, smoking, drinking and baseline pathological diagnosis between H. pylori positive and negative groups. The differences of methylation levels between H. pylori positive and negative groups were calculated by Mann-Whitney U test. The frequency of hyper- or hypo-methylated candidate genes (according to the median methylation levels in negative group) between H. pylori positive and negative groups was compared by unconditional logistic regression, adjusting age, sex, smoking, drinking and baseline pathological diagnosis. All statistical analyses were carried out using Statistical Product and Service Solutions (version 16.0).
Abbreviations
CGI, CpG island; CI, confidence interval; GC, gastric cancer; GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; H. pylori, Helicobacter pylori; OR, odds ratio; UTR, un-translated region; TCGA, The Cancer Genome Atlas; TSS, transcription start site.
CONFLICTS OF INTEREST
All authors disclosed no potential conflicts.
GRANT SUPPORT
A3 Foresight Program from Natural Science Foundation of China (30921140311 to W. You and Y. Kim); National Natural Science Foundation of China (81171989 to K. Pan, and 30801346 to Y. Zhang); and National Basic Research Program of China (973 Program: 2010CB529303 to W. You).
REFERENCES
1. Schistosomes, liver flukes, Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994; 61:1–241.
2. Correa P. A human model of gastric carcinogenesis. Cancer Res. 1988; 48:3554–3560.
3. You WC, Zhang L, Gail MH, Chang YS, Liu WD, Ma JL, Li JY, Jin ML, Hu YR, Yang CS, Blaser MJ, Correa P, Blot WJ, et al. Gastric dysplasia and gastric cancer: Helicobacter pylori, serum vitamin C, and other risk factors. J Natl Cancer Inst. 2000; 92:1607–1612.
4. Zhang L, Blot WJ, You WC, Chang YS, Kneller RW, Jin ML, Li JY, Zhao L, Liu WD, Zhang JS, Ma JL, Samloff IM, Correa P, et al. Helicobacter pylori antibodies in relation to precancerous gastric lesions in a high-risk Chinese population. Cancer Epidemiol Biomarkers Prev. 1996; 5:627–630.
5. You WC, Brown LM, Zhang L, Li JY, Jin ML, Chang YS, Ma JL, Pan KF, Liu WD, Hu Y, Crystal-Mansour S, Pee D, Blot WJ, et al. Randomized double-blind factorial trial of three treatments to reduce the prevalence of precancerous gastric lesions. J Natl Cancer Inst. 2006; 98:974–983.
6. Wong BC, Zhang L, Ma JL, Pan KF, Li JY, Shen L, Liu WD, Feng GS, Zhang XD, Li J, Lu AP, Xia HH, Lam S, et al. Effects of selective COX-2 inhibitor and Helicobacter pylori eradication on precancerous gastric lesions. Gut. 2012; 61:812–818.
7. Obst B, Wagner S, Sewing KF, Beil W. Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis. 2000; 21:1111–1115.
8. Hmadcha A, Bedoya FJ, Sobrino F, Pintado E. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med. 1999; 190:1595–1604.
9. Shim YH, Kang GH, Ro JY. Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Lab Invest. 2000; 80:689–695.
10. Chan AO, Lam SK, Wong BC, Wong WM, Yuen MF, Yeung YH, Hui WM, Rashid A, Kwong YL. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003; 52:502–506.
11. Hu SL, Kong XY, Cheng ZD, Sun YB, Shen G, Xu WP, Wu L, Xu XC, Jiang XD, Huang DB. Promoter methylation of p16, Runx3, DAPK and CHFR genes is frequent in gastric carcinoma. Tumori. 2010; 96:726–733.
12. Lu XX, Yu JL, Ying LS, Han J, Wang S, Yu QM, Wang XB, Fang XH, Ling ZQ. Stepwise cumulation of RUNX3 methylation mediated by Helicobacter pylori infection contributes to gastric carcinoma progression. Cancer. 2012; 118:5507–5517.
13. Zouridis H, Deng N, Ivanova T, Zhu Y, Wong B, Huang D, Wu YH, Wu Y, Tan IB, Liem N, Gopalakrishnan V, Luo Q, Wu J, et al. Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci Transl Med. 2012; 4:156ra140.
14. Shivapurkar N, Gazdar AF. DNA methylation based biomarkers in non-invasive cancer screening. Curr Mol Med. 2010; 10:123–132.
15. Woodson K, Mason J, Choi SW, Hartman T, Tangrea J, Virtamo J, Taylor PR, Albanes D. Hypomethylation of p53 in peripheral blood DNA is associated with the development of lung cancer. Cancer Epidemiol Biomarkers Prev. 2001; 10:69–74.
16. Zhang Y, Su HJ, Pan KF, Zhang L, Ma JL, Shen L, Li JY, Liu WD, Oze I, Matsuo K, Yuasa Y, You WC. Methylation status of blood leukocyte DNA and risk of gastric cancer in a high-risk Chinese population. Cancer Epidemiol Biomarkers Prev. 2014; 23:2019–2026.
17. Yuasa Y, Nagasaki H, Oze I, Akiyama Y, Yoshida S, Shitara K, Ito S, Hosono S, Watanabe M, Ito H, Tanaka H, Kang D, Pan KF, et al. Insulin-like growth factor 2 hypomethylation of blood leukocyte DNA is associated with gastric cancer risk. Int J Cancer. 2012; 131:2596–2603.
18. Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, Apostolidou S, Jones A, Lechner M, Beck S, Jacobs IJ, Widschwendter M. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE. 2009; 4:e8274.
19. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–209.
20. Huang YT, Chu S, Loucks EB, Lin CL, Eaton CB, Buka SL, Kelsey KT. Epigenome-wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics. 2016:1–10.
21. Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, Al-Sarraj S, Dobson R, Schalkwyk LC, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012; 13:R43.
22. Farre P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin. 2015; 8:19.
23. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, Campagne F, Mazumdar M, Greally JM, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010; 17:13–27.
24. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010; 17:510–522.
25. Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan JB, Shen R. High density DNA methylation array with single CpG site resolution. Genomics. 2011; 98:288–295.
26. Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, Yu MW, Chen CJ, Santella RM. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012; 55:1799–1808.
27. Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H, Santella RM. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics. 2013; 8:34–43.
28. Hou L, Wang H, Sartori S, Gawron A, Lissowska J, Bollati V, Tarantini L, Zhang FF, Zatonski W, Chow WH, Baccarelli A. Blood leukocyte DNA hypomethylation and gastric cancer risk in a high-risk Polish population. Int J Cancer. 2010; 127:1866–1874.
29. Kosumi K, Baba Y, Ishimoto T, Harada K, Miyake K, Izumi D, Tokunaga R, Murata A, Eto K, Sugihara H, Shigaki H, Iwagami S, Sakamoto Y, et al. Relationship between LINE-1 hypomethylation and Helicobacter pylori infection in gastric mucosae. Med Oncol. 2015; 32:117.
30. Saito M, Suzuki K, Maeda T, Kato T, Kamiyama H, Koizumi K, Miyaki Y, Okada S, Kiyozaki H, Konishi F. The accumulation of DNA demethylation in Sat alpha in normal gastric tissues with Helicobacter pylori infection renders susceptibility to gastric cancer in some individuals. Oncol Rep. 2012; 27:1717–1725.
31. Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol. 1996; 16:4717–4725.
32. Surapureddi S, Yu S, Bu H, Hashimoto T, Yeldandi AV, Kashireddy P, Cherkaoui-Malki M, Qi C, Zhu YJ, Rao MS, Reddy JK. Identification of a transcriptionally active peroxisome proliferator-activated receptor alpha -interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc Natl Acad Sci U S A. 2002; 99:11836–11841.
33. Pils D, Tong D, Hager G, Obermayr E, Aust S, Heinze G, Kohl M, Schuster E, Wolf A, Sehouli J, Braicu I, Vergote I, Van Gorp T, et al. A combined blood based gene expression and plasma protein abundance signature for diagnosis of epithelial ovarian cancer—a study of the OVCAD consortium. Bmc Cancer. 2013; 13:178.
34. Saba JD. Lysophospholipids in development: Miles apart and edging in. J Cell Biochem. 2004; 92:967–992.
35. Huwiler A, Pfeilschifter J. New players on the center stage: sphingosine 1-phosphate and its receptors as drug targets. Biochem Pharmacol. 2008; 75:1893–1900.
36. Murph M, Mills GB. Targeting the lipids LPA and S1P and their signalling pathways to inhibit tumour progression. Expert Rev Mol Med. 2007; 9:1–18.
37. Pan KF, Zhang L, Gerhard M, Ma JL, Liu WD, Ulm K, Wang JX, Zhang Y, Bajbouj M, Zhang LF, Li M, Vieth M, Liu RY, et al. A large randomised controlled intervention trial to prevent gastric cancer by eradication of Helicobacter pylori in Linqu County, China: baseline results and factors affecting the eradication. Gut. 2015.
38. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996; 20:1161–1181.
39. Rugge M, Correa P, Dixon MF, Hattori T, Leandro G, Lewin K, Riddell RH, Sipponen P, Watanabe H. Gastric dysplasia: the Padova international classification. Am J Surg Pathol. 2000; 24:167–176.