
INTRODUCTION
Cannabinoids are classified into 3 types: phytocannabinoids, which are extracted from cannabis sativa, endocannabinoids that are produced in the animal body, and synthetic cannabinoids that are synthesized in the laboratories. Endocannabinoid system consists of cannabinoids, cannabinoid receptors and the enzymes that metabolize the endogenous cannabinoids, such as fatty acid amide hydrolase (FAAH) and monoacyl glycerol lipase (MAGL). Cannabinoids act primarily on G-protein coupled receptors (GPCRs): cannabinoid receptors 1 and 2 (CNR1 and CNR2) [1]. CNR2 (CB2) is a component of the endocanabinoid system. Different endocannabinoids such as anandamide (AEA) have been shown to possess affinity towards it [2]. Endocannabinoid compounds have been shown to inhibit breast cancer cell proliferation and migration through different mechnisms [3]. Synthetic and phytocannabinoids have also been shown to possess anti-proliferative and anti-invasive potentials in vitro and in vivo [4–7]. In the present study, we analyzed the anti-tumorigenic properties of CNR2 specific agonist synthetic cannabinoid (JWH-015) against different breast cancer subtypes.
CNR2 is highly expressed in most immune cells [8, 9]. Different studies suggest a potential role of CNR2 in immune system. CNR2 knock out mice showed significantly less memory B-cells, CD4+ and CD8+ T-cells compared to heterozygous mice [10]. In addition to immune cells, CNR2 has also been shown to be expressed in breast cancer tissues and cell lines [6]. CNR2 specific agonists (JWH-015 and JWH-133) inhibit growth through different mechanism including inhibition of CXCL12/CXCR4 axis, cyclo-oxygenase-2 (COX-2) expression and induction of apoptosis in different cancer types [1, 5, 6]. Although there is a strong evidence of the anti-tumorogenic properties of CNR2 agonists, not much is known about the role of CNR2 activation on different growth factor receptor, such as epidermal growth factor receptor (EGFR) and insulin-like growth factor 1 receptor (IGF-IR)-mediated tumorigenic events in breast cancer.
Overexpression of EGFR and IGF-IR, lack of hormonal targeted cancer therapy, low survival rate and poor patient prognosis are hallmark features of the estrogen receptor alpha-negative (ERα-) breast cancer subtype [11–15]. The standard therapy for ERα- breast cancer using anthracyclins, taxanes and platinum compounds has shown unresponsiveness and rapid development of resistance [16–20]. Insulin growth factor receptor (IGF-IR) is predominantly activated in ERα+ as well as ERα- breast cancer subtypes [21, 22]. IGF-I has shown a great importance in breast cancer progression through its anti-apoptotic, mitogenic, and invasive potential in both ERα+ and ERα- breast cancer cells [23–25]. ERα+ breast cancer cells are associated with high expression of IGF-IR [26]. IGF-IR activation is, in turn, associated with increased invasion, secretion of metalloproteinases (MMPs) and activation of EMT process in ERα+ cells [24]. ERα- breast cancer cells are also associated with hyperactivation of IGF-IR [22]. IGF-IR activation and phosphorylation is associated with poor prognosis in many breast cancer subtypes including ERα+ and ERα- subtypes [27].
In this study, we analyzed the effect of the CNR2 activation on different tumorigenic related events. First, we analyzed the effect of CNR2 specific agonist on EGF-induced tumorigenic events in ERα- breast cancer cells. Second we analyzed the effect of CNR2 activation on IGF-I induced tumorigenesis in ERα+ and ERα- breast cancer cells. We also studied the effect of CNR2 activation on breast cancer growth using ERα+ and ERα- breast cancer mouse model systems.
RESULTS
CNR2 is expressed in ERα- and ERα+ breast cancer subtypes and associated with better prognosis
In this study, we analyzed CNR2, IGF-IR and EGFR expression in ERα- and ERα+ breast cancer cells. As shown, EGFR is highly expressed in different ERα- cell lines compared to ERα+ breast cancer cell lines however; CNR2 is highly expressed in both breast cancer subtypes (Figure 1-A). IGF-IR is highly expressed in different ERα+ cell lines especially MCF-7 cells; it is also expressed in ERα- breast cancer cells but it might be at lower levels (Figure 1-A). Thereafter, we used SUM159 and MDA-MB231 cells as representatives of ERα- breast cancer cells [28] and MCF-7 as a representative of ERα+ breast cancer cells [29]. In order to analyze the correlation of CNR2 to the prognosis of the breast cancer patients, we used Kaplan Meier publically available datasets. We observed that higher CNR2 expression correlates with significantly better recurrence free survival (RFS) in ERα-, basal, ERα+ and luminal A breast cancer patients (Figure 1B-1C and Supp. Figure 1A-1B).
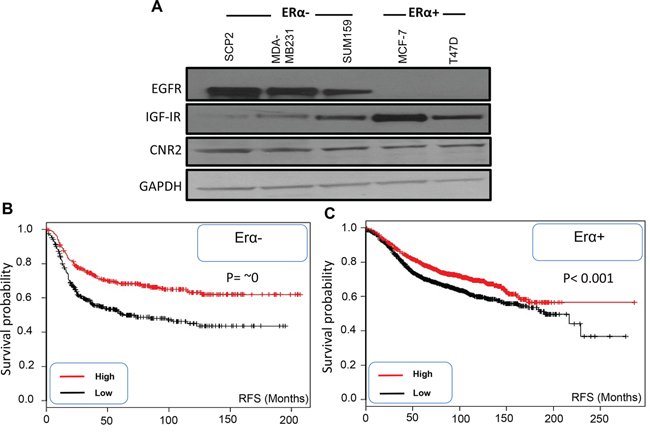
Figure 1: Expression of EGFR, IGF-IR and CNR2 proteins in ERα- and ERα+ breast cancer cells and correlation of CNR2 to breast cancer patient prognosis. A. Western blot image shows expression of EGFR, IGF-IR and CNR2 proteins in different ERα- (SCP2, MDA-MB231 and SUM159) and ERα+ (MCF-7 and T47D) breast cancer cell lines. GAPDH was used as a loading control. B. Kaplan Meier blot showing recurrence free survival (RFS) of high/low expressing CNR2 breast cancer patients of ERα- subtype. P value = ~ zero. C. Kaplan Meier blot showing recurrence free survival (RFS) of high/low expressing CNR2 breast cancer patients of ERα+ subtype. P value = 1e-05.
These results show that EGFR is highly expressed in ERα- breast cancer cells whereas; IGF-IR is highly expressed in ERα+ breast cancer cells. CNR2 is expressed in both ERα+ and ERα- breast cancer cells and associated with better prognosis in both subtypes.
CNR2 activation inhibits EGF-induced tumorigenic events in ERα- breast cancer cells
To analyze the possible role of CNR2 activation on EGF/EGFR pathway, we studied the effect CNR2 specific agonist (JWH-015) [1] on EGF-induced tumorigenic events in ERα- cell lines. We found that JWH-015 significantly inhibits EGF-induced migration as well as invasion of SUM159 and MDA-MB231 cells (Figure 2A:2D). NF-kB activation is crucial for cancer cells to promote their migration and invasive potentials especially for ERα- cells [30, 31]. Moreover, EGFR overexpression strongly correlates with NF-kB activation [32]. Therefore, we analyzed the effect of CNR2 agonist on NF-kB activation by using luciferase reporter assay. As shown, JWH-015 significantly inhibits NF-kB active units after EGF stimulation (Figure 2E). To test the specificity of JWH-015 towards CNR2, we used CNR2-specific antagonist (SR144528), and we found that CNR2 blocking significantly abrogated JWH-015-mediated inhibition of migration and invasion of SUM159 and MDA-MB231 cells after EGF stimulation (Figure 2A:2D). To further analyze the molecular mechanism, we investigated CNR2 agonist’s effect on EGF/EGFR axis. As shown, JWH-015 inhibits EGFR activation and its downstream targets STAT3, AKT and ERK (Figure 2F).
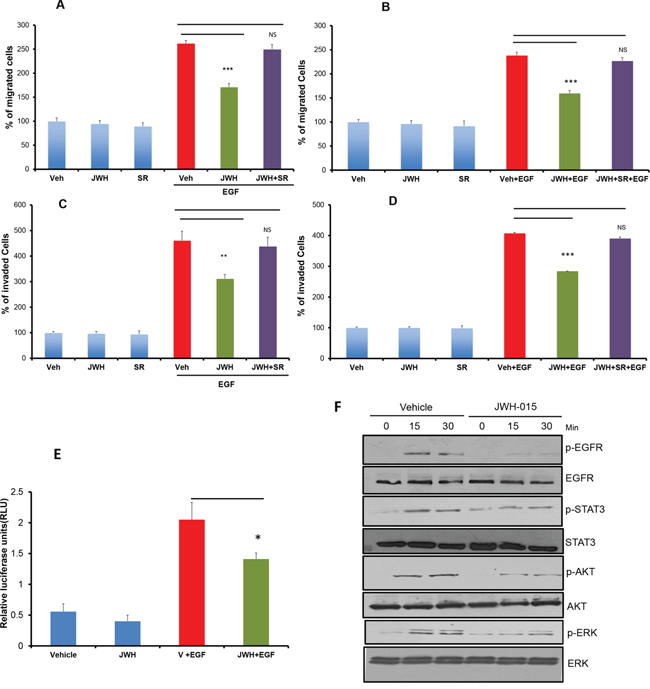
Figure 2: CNR2 activation inhibits EGF/EGFR signaling in ERα- breast cancer cells. SUM159 A. or MDA-MB231 B. cells were treated with JWH-015 for 24 hours and subjected to transwell migration assay in presence or absence of EGF (100 ng/ml) and specific CNR2 blocker SR-144528. SUM159 C. or MDA-MB231 D. cells were treated with JWH-015 for 24 hours and subjected to transwell invasion assay in presence or absence of EGF (100 ng/ml) and specific CNR2 blocker SR-144528. Number of migrated or invaded cells were counted and plotted as % of control. NS = non significant. E. SUM159 cells were treated with vehicle or JWH-015 in presence or absence of EGF and subjected to NF-kB luciferase reporter assay. F. SUM159 cells were treated with vehicle or JWH-015, stimulated with EGF (100 ng/ml) for 15 or 30 minutes then cell lysates were used for western blot analysis for the indicated proteins.
These results indicate that CNR2 triggering has the ability to inhibit EGFR activation and its downstream targets and therefore suppresses EGF-induced tumorigenic events in ERα- breast cancer cells.
CNR2 activation inhibits IGF-I-induced tumorigenic events in ERα- breast cancer cells
ERα- breast cancer cells are also associated with hyperactivation of IGF-IR [22]. IGF-IR activation and phosphorylation is associated with poor prognosis in many breast cancer subtypes including ERα+ and ERα- subtypes [27]. We have shown that ERα- breast cancer cells also express IGF-IR (Figure 1A). Therefore, we investigated the role of JWH-015 on IGF-I-induced tumorigenic events in SUM159 and MDA-MB231 cells (ERα-). We found that JWH-015 significantly inhibits IGF-I-induced migration as well as invasion of SUM159 and MDA-MB231 cells (Figure 3A:3D). At the molecular level, we found that JWH-015 inhibits IGF-I-induced activation of IGF-IR and its downstream target AKT in SUM159 cells (Figure 3E and Supp. Figure 2A-2B). We also found that JWH-015 inhibits secretion of metalloproteinase-2 (MMP2) in SUM-159 cells (Supp. Figure 4). To confirm the specificity of JWH-015 towards CNR2 in these studies, we used a CNR2-specific antagonist (SR144528), and we found that CNR2 blocking has significantly abrogated JWH-015 potential in inhibition of migration and invasion induced by IGF-I in SUM159 and MDA-MB231 cells (Figure 3A:3D). These results indicate that CNR2 activation has the ability to inhibit IGF-IR activation and its downstream key molecule (AKT), which might explain the inhibitory effect of JWH-015 on IGF-I-induced migration and invasion in ERα- cells.
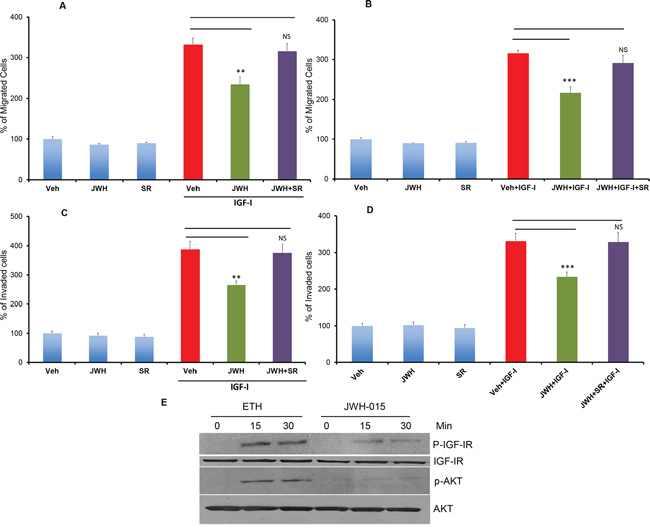
Figure 3: CNR2 activation inhibits IGF/IGF-IR signaling in ERα- breast cancer cells. SUM159 A. or MDA-MB231 B. cells were treated with JWH-015 for 24 hours and subjected to transwell migration assay in presence or absence of IGF-I (50 ng/ml) and specific CNR2 blocker SR-144528. SUM159 C. or MDA-MB231 D. cells were treated with JWH-015 for 24 hours and subjected to transwell invasion assay in presence or absence of IGF-I (50 ng/ml) and specific CNR2 blocker SR-144528. Number of migrated or invaded cells were counted and plotted as % of control. NS = non significant. E. SUM159 cells were treated with vehicle or JWH-015, stimulated with IGF-I (50 ng/ml) for 15 or 30 minutes then cell lysates were used for western blot analysis for the indicated proteins.
CNR2 activation inhibits IGF-I-induced tumorigenic events in ERα+ breast cancer cells
ERα+ breast cancer cells are associated with hyperactivation of IGF-IR [26]. IGF-IR activation is, in turn, associated with increased invasion, secretion of metalloproteinases (MMPs) and activation of EMT process in ERα+ cells [24].
Therefore, in this study, we analyzed the role of CNR2 activation on IGF-I-induced tumorigenic events in ERα+ breast cancer cells. In MCF-7 cells (ERα+), we found that JWH-015 significantly inhibits IGF-I-induced migration and invasion (Figure 4A-4B). We subsequently analyzed the molecular mechanism through investigation CNR2 agonist’s effect on IGF-I/IGF-IR pathway. As shown, JWH-015 inhibits the activation of IGF-IR and its downstream target AKT (Figure 4D and Supp. Figure 2C-2D). To investigate the role of CNR2 activation on IGF-I-induced secretion of metalloproteinases, we used zymogram technique. Interestingly, we found that JWH-015 inhibits IGF-I-induced secretion of metalloproteinase-9 (MMP9) in MCF-7 cells (Figure 4C). To confirm the specificity of JWH-015 towards CNR2 in these studies, we used a CNR2-specific antagonist (SR144528), and we found that CNR2 blocking has significantly abrogated JWH-015-potential in inhibition of migration and invasion induced by IGF-I in MCF-7 and SUM159 cells (Figure 4A-4B).
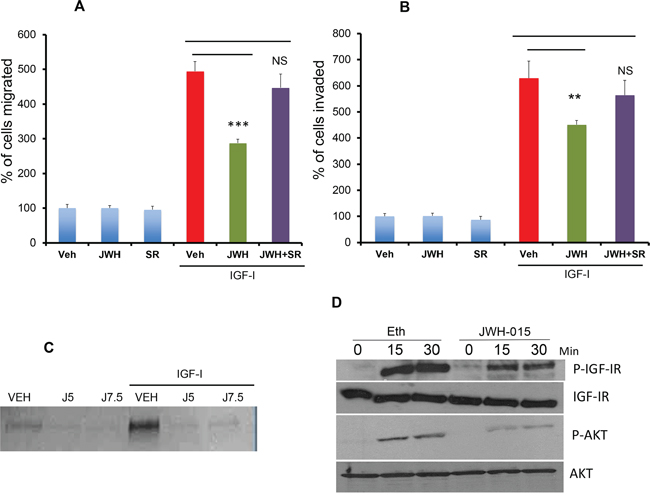
Figure 4: JWH-015 inhibits IGF-I/IGF-IR signaling in ERα+ breast cancer cells. A. MCF-7 cells were treated with JWH-015 for 24 hours and subjected to transwell migration assay in presence or absence of IGF-I (50 ng/ml) and specific CNR2 blocker SR-144528. B. MCF-7 cells were treated with JWH-015 for 24 hours and subjected to transwell invasion assay in presence or absence of IGF-I (50 ng/ml) and specific CNR2 blocker SR-144528. Number of migrated or invaded cells were counted and plotted as % of control. NS = non significant. C. MCF-7 cells were treated with vehicle or JWH-015 5 or 7.5 μM in presence or absence of IGF-I and the collected conditioned media was subjected to gelatin zymography to detect MMP-9. D. MCF-7 cells were treated with vehicle or JWH-015, stimulated with IGF-I (50 ng/ml) for 15 or 30 minutes then cell lysates were used for western blot analysis for the indicated proteins.
These results indicate that CNR2 activation has the ability to inhibit IGF-IR activation and its downstream target. It also inhibits IGF-I-induced secretion of MMP9, which might explain the inhibitory effect of JWH-015 on IGF-I- induced migration and invasion in ERα+ cells.
JWH-015 inhibits EGF/EGFR and IGF-I/IGF-IR axes in ERα- and ERα+ orthotopic mouse models
In order to analyze the anti-tumorigenic potential of CNR2 specific agonist on ERα- cells in vivo, we injected nude mice orthotopically with SUM159 cells. After the tumor became palpable, the mice were treated, through peri-tumoral injection, with either vehicle or JWH-015 (10 mg/Kg) for 4 weeks. The tumors were then harvested for analysis. As shown, JWH-015-treated group has less tumor volume and tumor weight compared to vehicle-treated group (Figure 5A:5C). We analyzed the tumor lysates to find out the ability of CNR2 activation to inhibit EGFR and IGF-IR activation in vivo and found that the JWH-015-treated group has less activation of EGFR, IGF-IR, STAT3, AKT and ERK proteins compared to control group (Figure 5D and Supp. Figure 3A:3E). In order to analyze the anti-tumorigenic potential of CNR2 specific agonist on ERα+ cells in vivo, we injected nude mice orthotopically with MCF-7 cells. After the tumor became palpable, the mice were treated, through peri-tumoral injection, with either vehicle or JWH-015 (10 mg/Kg) for 4 weeks. The tumors were then harvested for analysis. As shown, JWH-015-treated group has less tumor volume and tumor weight compared to vehicle-treated group (Figure 6A:6C). We also analyzed the tumor lysates and found that the JWH-015-treated group has less activation of IGF-IR and AKT proteins compared to the control group (Figure 6D and Supp. Figure 3F-3G). These results suggest that CNR2 activation has the ability to inhibit breast cancer growth through inhibition of EGF/EGFR as well as IGF-I/IGF-IR pathways and their downstream targets.
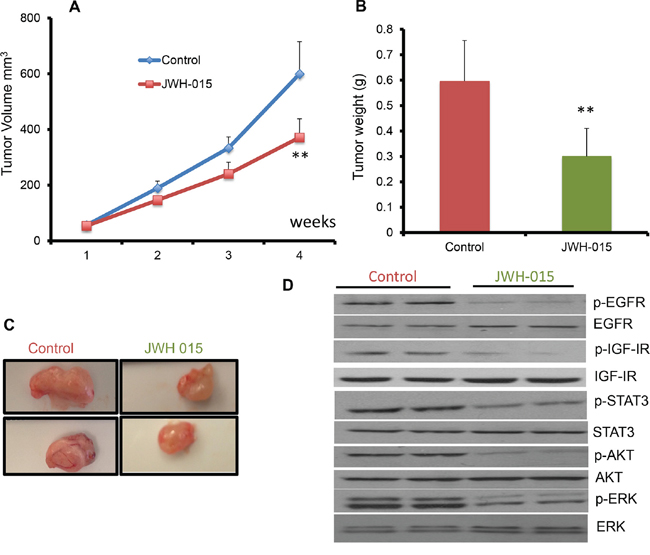
Figure 5: JWH-015 suppresses ERα- breast cancer growth in vivo by inhibiting EGF/EGFR and IGF-I/IGF-IR signaling pathways. A. Tumor volume measurements of orthotopically injected nude mice with SUM159 cells were assessed every week for control and treated groups. B. Tumor weight of vehicle-treated or JWH-015-treated nude mice was determined at the euthanasia day. C. Representative photographs showing tumors dissected from control and treated groups. D. Western blot images of the tumor lysates of the control and treated groups showing the protein expression of phospho-EGFR, phospho-IGF-IR, phospho-STAT3 phospho-ERK and phosphor-AKT (p-EGFR, p-IGF-IR, p-STAT3, p-ERK and p-AKT) and total EGFR, IGF-IR, STAT3, ERK and AKT (EGFR, IGF-IR, STAT3, ERK and AKT).
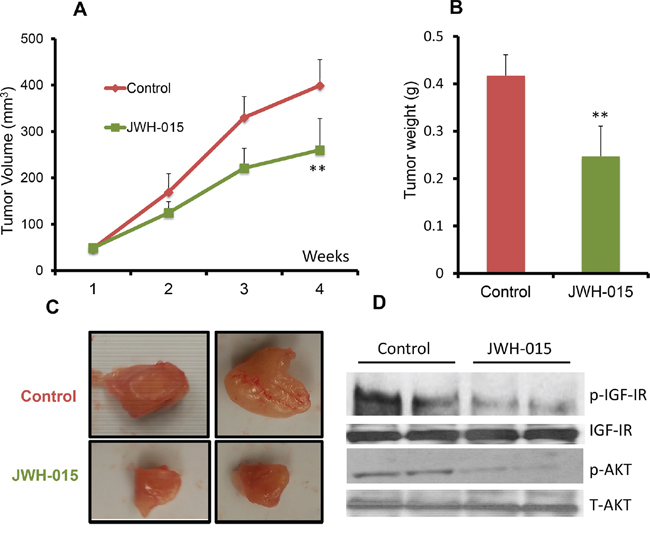
Figure 6: JWH-015 suppresses ERα+ breast cancer growth in vivo. A. Tumor volume measurements of orthotopically injected nude mice with MCF-7 cells were assessed every week for control and treated groups. B. Tumor weight of vehicle-treated or JWH-015-treated nude mice was determined at the euthanasia day. C. Representative photographs showing tumors dissected from control and treated groups. D. Western blot images of the tumor lysates of the control and treated groups showing the protein expression of phospho-IGF-IR and phosphor-AKT (p-IGF-IR and p-AKT) and total IGF-IR and AKT (IGF-IR and AKT).
DISCUSSION
CNR2 has been shown to be expressed in different cancer cells, including breast cancer [6, 33, 34]. Although CNR2 activation has shown significant anti-tumorigenic properties, its functional role in inhibiting breast cancer is not well understood. In this study we showed that CNR2 is expressed in both ERα- and ERα+ breast cancer cells. ERα- breast cancer is characterized by its high proliferative and metastatic potential and poor prognosis. ERα+ breast cancer represents most of all breast cancers cases and is also associated with poor prognosis, low overall survival particularly when associated with lymph node metastasis [35–37]. In the present study, we elucidated the anti-tumor role of CNR2 in ERα+ and ERα- breast cancer cells. Following CNR2 activation, we observed inhibition of breast cancer growth, suppression of EGF/EGFR as well as IGF-I/IGF-IR signaling pathways and their related tumorigenic events in vitro and in vivo.
Since EGFR is upregulated in ERα- breast cancer subtype, EGFR inhibitors have been used as a possible therapeutic option. However, EGFR inhibitors usually gain rapid resistance and they eventually fail [38]. Therefore, it is crucial to identify other small molecules that can achieve the interruption of EGF/EGFR axis by different, possibly indirect, ways. In our present study, we showed that CNR2 activation by CNR2 specific agonist (JWH-015) inhibits EGF/EGFR signaling transduction pathway’s activation in ERα- cells in vitro and in vivo. This pathway involves important key molecules STAT3, AKT and ERK and NF-kB, which are important in cancer cell survival, proliferation, migration and invasion. These results explain the ability of JWH-015 to inhibit EGF-induced migration and invasion as well as inhibition of phosphorylation of EGFR, AKT and ERK proteins in ERα- cells in vitro and in vivo. In our study, we found that JWH-015 also inhibited EGF-induced NF-kB activation in ERα- breast cancer cells. NF-kB is highly active in ERα- breast cancer cells and it enhances breast cancer cell’s migration, invasion and metastasis through different mechanisms [39]. It promotes the expression of several pro-tumorigenic genes such as CXCR4, which is crucial for cancer cell migration and invasion [30, 40]. Furthermore, NF-kB activation is important for epithelial to mesenchymal transition (EMT) process [41]. Our studies are in accordance with previous studies, which showed that CNR2 agonists inhibit the tumor growth and metastasis through induction of apoptosis and inhibition of EGFR activation in non-small cell lung cancer (NSCLC) [5]. Our findings might explain the observation, which shows that higher CNR2 expression is significantly correlated with better prognosis in ERα- patients.
A common problem for ERα+ breast cancer is the resistance to the anti-estrogen therapy [42, 43]. After gaining resistance to the estrogen deprivation, ERα+ cells show upregulation and activation of IGF-I/IGF-IR and PI3K/AKT signaling [44]. Furthermore, IGF-IR inhibitors also gain rapid resistance [45, 46]. It is urgently needed to find other options for targeting IGF-I/IGF-IR in ERα+ and ERα- cells rather than conventional direct therapies. Therefore, we studied the effect of CNR2 agonist on IGF-I-induced tumorigenic events. Our results showed that JWH-015 inhibits phosphorylation and activation of IGF-IR and its downstream target AKT in vitro and in vivo. This might explain its ability to inhibit IGF-I-induced migration in ERα+ and ERα- cells. The inhibition of IGF-I-induced secretion of MMP-9 by JWH-015 might also explain the ability of CNR2 agonist to inhibit IGF-I-induced invasion in ERα+ cells. These findings might explain our observation that show that higher CNR2 expression is significantly correlated with better prognosis in ERα- as well as ERα+ patients.
In this study, we showed that CNR2 activation inhibits the activation of EGFR and IGF-IR receptor tyrosine kinases (RTKs), however the mechanism by which CNR2 inhibits these RTKs activation is not known. Importantly, Pérez-Gómez et al; have recently reported that CNR2 cross talks with HER-2 through forming a heterodimer. In addition, CNR2 has been shown to cross talks with other G-protein coupled receptors (GPCR) such as CXCR4 through forming heterodimers and thereby inhibiting its activity [5, 47]. Therefore, there is possibility of similar direct interaction between CNR2 with EGFR and IGF-IR. There is also a probability that the cross-talk between CNR2 and these RTKs occurs through indirect way. Previous studies showed that MMP inhibitor inhibits EGFR ligand shedding and secretion that resulted in less activation of EGFR [48]. Since we have shown that JWH-015 inhibits MMP secretion, this may lead to less shedding and secretion of EGFR ligands and eventually less activation of EGFR and its downstream signaling.
Overall, we show in this study that CNR2 is an important therapeutic target in ERα- and ERα+ breast cancer subtypes and its higher expression is associated with better prognosis. In addition, we show, for the first time, that CNR2 activation inhibits the activation of EGFR/IGF-IR signaling pathways and EGF/IGF-I-induced tumorigenic events in ERα- and ERα+ breast cancer subtypes in vitro and in vivo (Figure 7). Our data support the use of CNR2 agonists as an adjuvant therapy for ERα- and ERα+ breast cancer patients.
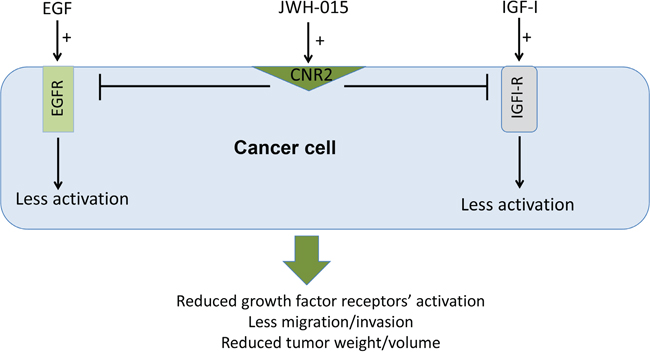
Figure 7: Schematic representation of the anti-tumor role of CNR2 activation in breast cancer. Schematic representation of the direct anti-tumor role of CNR2 activation showing the possible cross-talk between CNR2 receptor and EGFR and IGF-IR.
MATERIALS AND METHODS
Reagents and antibodies
Cell culture reagents were obtained from Gibco Laboratories (Grand Island, NY). The following reagents and antibodies used in this study were purchased from different sources: JWH-015 (Tocris Bioscience) is a CNR2 agonist that is used in the whole study, human EGF and IGF-I proteins (Peprotech). Antibodies: CNR2 (Abcam), p-AKT (serine 473), p-STAT3 (serine 727) and p-IGF-IR (Y1135/1136) (Cell Signaling); p-ERK/ERK, p-EGFR (Tyr 1173), GAPDH, STAT3 and AKT (Santa Cruz).
Cell culture
SUM159 cells were obtained from Dr. Sarmila Majumder (The Ohio State University). MDA-MB 231, T47D and MCF7 cells were purchased from ATCC. SCP2 cells were kindly obtained from Dr. Joan Massagué [49]. Cells were cultured in DMEM containing 10% heat-inactivated fetal bovine serum (FBS), 5-units/mL penicillin, and 5-mg/mL streptomycin.
Western blot analysis
Cells were plated in 100 cm2 dishes and lysed in lysis buffer (RIPA). Tumor samples were homogenized and lysed in RIPA buffer for further analysis. 50 mg of proteins were loaded on 4–12% SDS–polyacrylamide gels (Invitrogen) then transferred to nitrocellulose membranes (BioRad) and blocked with 5% milk. Membranes were incubated overnight with primary antibody, washed, and incubated for 1 h at RT with horseradish peroxidase conjugated secondary antibody. The membranes were then washed and stained using a chemiluminescence system (ECL Amersham Biosciences) and exposed to X-ray film (Kodak).
Chemotactic assays
Chemotactic assays were performed using transwell chambers (Costar 8.0 mm pore size) as described previously [5, 6, 50]. Briefly, serum starved cells were pretreated with JWH-015 or vehicle for 24 h. Top chambers were loaded with cells in serum free medium (SFM) and bottom chambers contained SFM in the presence or absence of chemo-attractants. Cells that migrated or invaded across the membrane were counted by fixing in 37% formaldehyde and 25% glutaraldehyde in PBS and stained with 0.1% crystal violet in PBS for 30 minutes. Migrated/invaded cells per membrane were detected by light microscopy, counted in 5 fields and the percentage of migration determined.
Gelatin zymography
Gelatin zymography for collected conditioned media was performed as described earlier [51].
Luciferase reporter assay
We used NF-kB luciferase reporter assay (Promega) to determine NF-kB activity as described previously [50, 51]. Briefly, NF-kB luciferase constructs containing either wild type or NF-kB vector were transfected in the pretreated cells using lipofectamine 2000 (Invitrogen). For internal control, we co-transfected cells with Renilla luciferase vector. 24 h after transfection, EGF (100 ng/ml) was added and then incubated for another 24 h. Cells were lysed and luciferase assay was performed according to manufacturer’s protocol.
Mouse models
Female nude (NU/NU) mice were purchased from (Charles River Laboratories Inc.). Tumors were formed by orthotopically injecting SUM159 (5 x 106) cells or MCF-7 (5 x 106) in the 4th mammary glands of female nude mice (n=6 mice per group). Mice were randomized when tumors became palpable, and then injected peri-tumorally with JWH-015 (10 mg/kg) or vehicle on alternate days for 4 weeks. Mice with MCF7 cell were subcutaneously injected once a week with 2.5 μg β-estradiol 17-valerate in 50 μL of sesame oil. Tumors were measured every week with external calipers and tumor volume was calculated according to the formula tumor volume = (length x width2 x 0.52) [1, 52]. The mice did not experience any undesirable effects following administration of vehicle or JWH-015 during the whole period of the experiment.
Statistical analysis
Results were represented as mean ± SD. Student’s t test was used. P<0.05 was considered to be statistically significant. For all graphs, * indicates P<0.05, ** indicates P<0.01 and *** indicates P<0.001.
ACKNOWLEDGMENTS
This study has been funded in part by NIH (CA109527 and CA153490), and Pelotonia Idea Grant to RKG. Mohamad Elbaz is supported by fellowship from Egyptian government.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Nasser MW, Qamri Z, Deol YS, Smith D, Shilo K, Zou X, Ganju RK. Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. PloS one. 2011; 6:e23901.
2. Pertwee R. The pharmacology of cannabinoid receptors and their ligands: an overview. International journal of obesity. 2006; 30:S13-S18.
3. Guindon J, Hohmann AG. The endocannabinoid system and cancer: therapeutic implication. British journal of pharmacology. 2011; 163:1447-1463.
4. Blázquez C, Carracedo A, Barrado L, Real PJ, Fernández-Luna JL, Velasco G, Malumbres M, Guzmán M. Cannabinoid receptors as novel targets for the treatment of melanoma. The FASEB journal. 2006; 20:2633-2635.
5. Preet A, Qamri Z, Nasser MW, Prasad A, Shilo K, Zou X, Groopman JE, Ganju RK. Cannabinoid Receptors, CB1 and CB2, as Novel Targets for Inhibition of Non–Small Cell Lung Cancer Growth and Metastasis. Cancer Prevention Research. 2011; 4:65-75.
6. Qamri Z, Preet A, Nasser MW, Bass CE, Leone G, Barsky SH, Ganju RK. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Molecular cancer therapeutics. 2009; 8:3117-3129.
7. Elbaz M, Nasser MW, Ravi J, Wani NA, Ahirwar DK, Zhao H, Oghumu S, Satoskar AR, Shilo K, Carson WE. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Molecular oncology. 2015; 9:906-919.
8. Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Fur G, Casellas P. Cannabinoid-receptor expression in human leukocytes. European Journal of Biochemistry. 1993; 214:173-180.
9. Rayman N, Lam KH, Laman JD, Simons PJ, Löwenberg B, Sonneveld P, Delwel R. Distinct expression profiles of the peripheral cannabinoid receptor in lymphoid tissues depending on receptor activation status. The Journal of Immunology. 2004; 172:2111-2117.
10. Ziring D, Wei B, Velazquez P, Schrage M, Buckley NE, Braun J. Formation of B and T cell subsets require the cannabinoid receptor CB2. Immunogenetics. 2006; 58:714-725.
11. Park HS, Jang MH, Kim EJ, Kim HJ, Lee HJ, Kim YJ, Kim JH, Kang E, Kim SW, Kim IA. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Modern Pathology. 2014; 27:1212-1222.
12. Nogi H, Kobayashi T, Suzuki M, Tabei I, Kawase K, Toriumi Y, Fukushima H, Uchida K. EGFR as paradoxical predictor of chemosensitivity and outcome among triple-negative breast cancer. Oncology reports. 2009; 21:413-417.
13. Dey N, R Smith B, Leyland-Jones B. Targeting basal-like breast cancers. Current Drug Targets. 2012; 13:1510-1524.
14. Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer. 2007; 109:1721-1728.
15. Gazinska P, Grigoriadis A, Brown JP, Millis RR, Mera A, Gillett CE, Holmberg LH, Tutt AN, Pinder SE. Comparison of basal-like triple-negative breast cancer defined by morphology, immunohistochemistry and transcriptional profiles. Modern Pathology. 2013; 26:955-966.
16. Bayraktar S, Glück S. Molecularly targeted therapies for metastatic triple-negative breast cancer. Breast cancer research and treatment. 2013; 138:21-35.
17. Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011; 16:1-11.
18. Liedtke C, Mazouni C, Hess KR, André F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. Journal of Clinical Oncology. 2008; 26:1275-1281.
19. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research. 2007; 13:4429-4434.
20. Pogoda K, Niwińska A, Murawska M, Pieńkowski T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Medical Oncology. 2013; 30:1-8.
21. Bhargava R, Beriwal S, McManus K, Dabbs DJ. Insulin-like growth factor receptor-1 (IGF-1R) expression in normal breast, proliferative breast lesions, and breast carcinoma. Applied Immunohistochemistry & Molecular Morphology. 2011; 19:218-225.
22. Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, Carboni JM, Gottardis MM, Huang F, Chang JC. High IGF-IR Activity in Triple-Negative Breast Cancer Cell Lines and Tumorgrafts Correlates with Sensitivity to Anti–IGF-IR Therapy. Clinical Cancer Research. 2011; 17:2314-2327.
23. Hankinson SE. (2008). Circulating levels of sex steroids and prolactin in premenopausal women and risk of breast cancer. Hormonal Carcinogenesis V: Springer), pp. 161-169.
24. Walsh LA, Damjanovski S. IGF-1 increases invasive potential of MCF 7 breast cancer cells and induces activation of latent TGF-b1 resulting in epithelial to mesenchymal transition. Cell Commun Signal. 2011; 9:10.
25. Davison Z, de Blacquière GE, Westley BR, May FE. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: implications for therapy. Neoplasia. 2011; 13:504-515.
26. Aaltonen KE, Rosendahl AH, Olsson H, Malmström P, Hartman L, Fernö M. Association between insulin-like growth factor-1 receptor (IGF1R) negativity and poor prognosis in a cohort of women with primary breast cancer. BMC cancer. 2014; 14:794.
27. Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Research. 2008; 68:10238-10246.
28. Grigoriadis A, Mackay A, Noel E, Wu PJ, Natrajan R, Frankum J, Reis-Filho JS, Tutt A. Molecular characterisation of cell line models for triple-negative breast cancers. BMC genomics. 2012; 13:619.
29. Rondón-Lagos M, Verdun Di Cantogno L, Marchiò C, Rangel N, Payan-Gomez C, Gugliotta P, Botta C, Bussolati G, Ramírez-Clavijo SR, Pasini B. Differences and homologies of chromosomal alterations within and between breast cancer cell lines: a clustering analysis. Mol Cytogenet. 2014; 7:8.
30. Helbig G, Christopherson KW, 2nd, Bhat-Nakshatri P, Kumar S, Kishimoto H, Miller KD, Broxmeyer HE, Nakshatri H. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003; 278:21631-21638.
31. Singel SM, Batten K, Cornelius C, Jia G, Fasciani G, Barron SL, Wright WE, Shay JW. Receptor-interacting protein kinase 2 promotes triple-negative breast cancer cell migration and invasion via activation of nuclear factor-kappaB and c-Jun N-terminal kinase pathways. Breast Cancer Research. 2014; 16:R28.
32. Biswas DK, Iglehart JD. Linkage between EGFR family receptors and nuclear factor kappaB (NF-κB) signaling in breast cancer. Journal of cellular physiology. 2006; 209:645-652.
33. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990; 561-564.
34. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993; 61-65.
35. Schnitt SJ, Collins LC. (2009). Biopsy interpretation of the breast: Lippincott Williams & Wilkins).
36. Turner NC, Jones AL. Management of breast cancer: part II. BMJ: British Medical Journal. 2008; 337:164-169.
37. Lønning PE. Poor-prognosis estrogen receptor-positive disease: present and future clinical solutions. Therapeutic advances in medical oncology. 2012:1758834012439338.
38. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nature reviews Clinical oncology. 2010; 7:493-507.
39. Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Sledge G. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Molecular and cellular biology. 1997; 17:3629-3639.
40. Chen YJ, Yeh MH, Yu MC, Wei YL, Chen WS, Chen JY, Shih CY, Tu CY, Chen CH, Hsia TC. Lapatinib–induced NF-kappaB activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Research. 2013; 15:1-14.
41. Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. Journal of Clinical Investigation. 2004; 114:569-581.
42. Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer a southwest oncology group study. Clinical Cancer Research. 2004; 10:5670-5676.
43. De Laurentiis M, Arpino G, Massarelli E, Ruggiero A, Carlomagno C, Ciardiello F, Tortora G, D'Agostino D, Caputo F, Cancello G. A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clinical Cancer Research. 2005; 11:4741-4748.
44. Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res. 2013; 15:R55.
45. Pitts TM, Tan AC, Kulikowski GN, Tentler JJ, Brown AM, Flanigan SA, Leong S, Coldren CD, Hirsch FR, Varella-Garcia M. Development of an integrated genomic classifier for a novel agent in colorectal cancer: approach to individualized therapy in early development. Clinical Cancer Research. 2010; 16:3193-3204.
46. Ekyalongo RC, Mukohara T, Kataoka Y, Funakoshi Y, Tomioka H, Kiyota N, Fujiwara Y, Minami H. Mechanisms of acquired resistance to insulin-like growth factor 1 receptor inhibitor in MCF-7 breast cancer cell line. Investigational new drugs. 2013; 31:293-303.
47. Coke CJ, Scarlett KA, Chetram MA, Jones KJ, Sandifer BJ, Davis AS, Marcus AI, Hinton CV. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. Journal of Biological Chemistry. 2016; 291:9991-10005.
48. Xu KP, Ding Y, Ling J, Dong Z, Fu-Shin XY. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Investigative ophthalmology & visual science. 2004; 45:813-820.
49. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massagué J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. The Journal of clinical investigation. 2005; 115:44-55.
50. Ravi J, Sneh A, Shilo K, Nasser MW, Ganju RK. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget. 2014; 5:2475-2486. doi: 10.18632/oncotarget.1723.
51. Nasser MW, Qamri Z, Deol YS, Ravi J, Powell CA, Trikha P, Schwendener RA, Bai XF, Shilo K, Zou X. S100A7 enhances mammary tumorigenesis through upregulation of inflammatory pathways. Cancer Research. 2012; 72:604-615.