
Introduction
Post-transplant lymphoproliferative disorders (PTLD) encompass morphologically heterogeneous entities with variable clinical behavior. [1] PTLDs of B-lineage predominate, while T- and NK-cell PTLDs (T/NK-PTLD) are rare, representing 2-15% of all cases. [2] T/NK-PTLDs usually present late after transplantation, are associated with a poor prognosis, and, in contrast to B-cell PTLDs, only a minority are EBV-related (up to 37%). [2, 3]
The pathogenesis of T/NK-PTLDs and the underlying molecular alterations are presently unknown and their relationship to peripheral T-cell lymphomas (PTCL) arising in immunocompetent hosts remains unclear. Hence, in order to gain insights into the genetic bases of these uncommon PTLDs, we performed targeted next generation sequencing of cancer-associated genes and genome-wide DNA profiling to assess copy number alterations in a relatively large series of well characterized T/NK-PTLDs. We observed frequent mutations of epigenetic modifiers, members of the JAK/STAT signaling cascade, and TP53, and recurrent genomic alterations, similar to those described in PTCL arising in immunocompetent individuals.
Results
Morphology and immunophenotype
All T- and NK-cell lymphomas were classified based on morphologic and immunophenotypic features according to the 2008 WHO guidelines. [4] Immunophenotypic analysis was based on a combination of flow cytometry and immunohistochemistry. Peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS) was the most common subtype (n = 6, 32%), followed by hepatosplenic T-cell lymphoma (HSTCL; n = 3), primary cutaneous anaplastic large cell lymphoma (c-ALCL; n = 3), angioimmunoblastic T-cell lymphoma (AITL; n = 2), extranodal NK/T-cell lymphoma, nasal type (ENKTCL; n = 2), EBV-positive peripheral T-cell lymphoma (EBV+ PTCL; n = 1), lymphomatoid papulosis (LyP; n = 1) and adult T-cell leukemia/lymphoma (ATLL; n = 1) (Table 1, representative cases are illustrated in Figure 1).
PTCL, NOS (n = 6) comprised CD4+ and CD8+ cases with downregulation or loss of CD7 seen in all. One CD8+ T-cell lymphoma expressed CD30 and showed evidence of EBV infection by in situ hybridization for EBER. The three cases of HSTCL were derived from TCR-gamma-delta T-cells. They expressed TIA-1 but not the cytotoxic granule constituents (granzyme-B, perforin) and lacked CD5 expression. The AITL cases were CD4+ and displayed expanded and disrupted follicular dendritic cell meshworks. One case had a concomitant EBV+ clonal B-cell proliferation. The c-ALCL cases showed diffuse dermal infiltrates of large CD30+ T-cells, many exhibiting prominent nucleoli, and lacked ALK expression. Both ENKTCL were EBV+. One was of cytotoxic T-cell origin involving the breast and the other of NK-cell origin involving the nasal vestibule and lung; both displayed cellular atypia, had high proliferation indices and showed angiodestruction and extensive necrosis. The ATLL presented as lymphomatous disease and the neoplastic cells expressed CD4 and CD25.
Table 1: Clinical, morphological and immunohistochemical data. |
|||||||||||||||||||||||||||||||
Case |
Age/ Sex |
Underlying dx |
Graft |
IS therapy |
Time to PTLD (y) |
Diagnosis |
Location |
CD2 |
CD3 |
CD5 |
CD7 |
CD4 |
CD8 |
CD56 |
TIA |
CD30 |
EBER |
Ki-67 |
Other |
B-catenin |
MLH1 |
MSH2 |
PMS2 |
MSH6 |
Gata3 |
Lineage |
MSI status |
TCR |
Treatment |
Status |
Survival (days) |
1 |
75F |
Viral CM |
Heart |
AZA+CS+P |
15.01 |
PTCL, NOS |
BM |
+ |
- |
+ |
- |
- |
+ |
- |
+ |
ND |
- |
30% |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
MSS |
Clonal |
VMP |
Dead |
66 |
|
2 |
29M |
Viral CM |
Heart |
AZA+CS+P |
12.62 |
PTCL, NOS |
Subcutaneous tissue, face |
+ |
+ |
+ |
- |
+ |
- |
- |
ND |
- |
- |
ND |
PD1- |
- |
+ |
+ |
+ |
- |
- |
ND |
MSI-H |
Clonal |
CHOP/RT, gemcitabine |
Dead |
197 |
3 |
57M |
Hypersensitivity pneumonitis |
Left lung |
CNI+MMF+P |
0.47 |
PTCL, NOS |
Subcarinal LN |
+ |
+ |
+/- |
- |
- |
+ |
- |
+ |
- |
- |
80% |
CD10-, CD15-, PD1-, CD25-, MUM1- |
- |
+ |
+ |
+ |
+ |
- |
αβ |
MSS |
Clonal |
EPOCH |
Dead |
98 |
4 |
75M |
Dilated CM |
Heart |
CNI+MMF+P |
2.00 |
PTCL, NOS |
Retroperitoneum |
+ |
+ |
+/- |
+/- |
+ |
- |
- |
- |
- |
- |
40% |
ALK-, BCL2-, MUM1-, PD1- |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
Clonal |
Cytoxan/Etoposide |
Dead |
29 |
5 |
41F |
Post-partum CM |
Heart |
CNI+P+MMF |
7.01 |
PTCL, NOS |
Retroperitoneum |
+ |
+ |
+ |
- |
+ |
- |
- |
- |
- |
- |
ND |
CD10-, BCL2+, PD1+, MUM1- |
- |
+ |
+ |
+ |
+ |
- |
αβ |
MSS |
PC |
EPOCH, GEMOX, Pralatrexate, Radiation and Auto-SCT |
Alive |
976 |
6 |
32F |
CHF |
Heart |
CS |
14.01 |
EBV+ PTCL |
Breasts, bilateral |
+ |
+ |
+ |
- |
+/- |
+ |
- |
+ |
+ |
+ |
70% |
CD10-, ALK-, MUM1- |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
MSS |
Clonal |
EPOCH, Radiation and gemcitabine |
Alive |
1405 |
7 |
32M |
N/A |
Liver |
CNI+AZA+CS |
5.83 |
HSTCL |
Spleen, liver, BM |
+ |
+ |
- |
+ |
- |
+ |
+ |
+ |
- |
- |
ND |
- |
+ |
+ |
+ |
+ |
γδ |
MSS |
Clonal |
VDMCT |
Dead |
120 |
||
8 |
34M |
Dilated CM |
Heart |
AZA+CS+P |
9.73 |
HSTCL |
Spleen |
ND |
+ |
- |
- |
- |
+ |
ND |
+ |
- |
- |
ND |
- |
+ |
+ |
- |
+ |
- |
γδ |
MSS |
Clonal |
CHOP, ESHAP, MNM |
Dead |
273 |
|
9 |
35M |
GN, NOS |
Kidney |
AZA+CS+P |
10.01 |
HSTCL |
Spleen, liver |
+ |
+ |
- |
ND |
- |
- |
+ |
+ |
ND |
ND |
ND |
PD1- |
- |
- |
+ |
+ |
+ |
70% |
γδ |
MSS |
Clonal |
CHOP |
Dead |
70 |
10 |
57F |
DM |
Kidney |
n/a |
24.43 |
AITL |
Retroperitoneum |
+ |
+ |
+ |
- |
+ |
- |
ND |
ND |
- |
- |
50% |
BCL6+, CD10+, PD1+ |
- |
+ |
+ |
+ |
+ |
30% |
αβ |
MSS |
PC |
RCHOP/Gemcitabine/Pralatrexate |
Dead |
321 |
11 |
44M |
N/A |
Lung |
CNI+CS |
4.08 |
AITL |
LN, lung |
+ |
+ |
+ |
+ |
+ |
- |
- |
ND |
- |
-* |
ND |
BCL6+, PD1+ |
ND |
+ |
+ |
+ |
+ |
ND |
ND |
MSS |
Clonal |
Rituximab |
Dead |
730 |
12 |
83M |
N/A |
Kidney |
CNI+MMF+CS |
6.92 |
Primary cutaneous ALCL, CD30+ |
Skin, pleura |
+ |
- |
- |
- |
+ |
- |
ND |
- |
+ |
- |
ND |
ALK- |
- |
+ |
+ |
+ |
+ |
ND |
ND |
MSS |
ND |
CHOP |
Dead |
120 |
13 |
47M |
Nephrogenic systemic fibrosis |
Kidney |
CNI+MMF |
4.73 |
Primary cutaneous ALCL, CD30+ |
Skin, back |
+ |
- |
- |
- |
+ |
- |
ND |
- |
+ |
- |
80% |
ALK-, BCL2+ |
- |
+ |
+ |
+ |
+ |
80% |
αβ |
MSS |
Clonal |
CHOP/RT |
Dead |
3839 |
14 |
71M |
N/A |
Heart |
CNI+AZA+CS |
18.00 |
Primary cutaneous ALCL, CD30+ |
Skin |
+ |
- |
- |
- |
+ |
- |
ND |
- |
+ |
- |
ND |
ALK- |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
Clonal |
CHOP |
Dead |
210 |
15 |
20F |
Drug induced CM |
Heart |
AZA+CS+P |
5.49 |
ENKTCL |
Right breast |
+ |
+ |
- |
+ |
- |
- |
+ |
+ |
- |
+ |
70% |
CD25+ |
- |
+ |
+ |
+ |
+ |
- |
-- |
MSS |
PC |
N/A |
Dead |
18 |
16 |
71F |
Hypersensitivity pneumonitis |
Lung |
CNI+AZA+P |
7.40 |
ENKTCL |
Nasal vestibule, lung |
+ |
+ |
+ |
+/- |
- |
+ |
- |
+ |
- |
+ |
90% |
ND |
ND |
ND |
ND |
ND |
ND |
αβ |
ND |
Clonal |
None |
Dead |
61 |
|
17 |
43F |
FSGS |
Kidney |
CNI+AZA+P |
6.98 |
ATLL |
Retroperitoneum |
+ |
- |
- |
- |
+ |
- |
- |
ND |
+ |
- |
ND |
ALK-, FoxP3-, CD25+, MUM1+ |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
Clonal |
EPOCH X4 |
Alive |
650 |
18 |
83M |
APCKD |
Kidney |
CNI+MMF |
3.23 |
Lymphomatoid papulosis |
Skin, left forearm |
+ |
- |
- |
- |
- |
- |
- |
ND |
+ |
- |
ND |
CD15+, ALK-, MUM1+ |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
PC |
Reduced IS |
Alive |
2741 |
19 |
70M |
N/A |
Kidney |
CNI+AZA+CS |
1.92 |
PTCL, NOS |
Pleura, pericardium, LN |
+ |
+ |
- |
- |
+ |
- |
- |
- |
- |
- |
ND |
ALK- |
ND |
ND |
ND |
ND |
ND |
ND |
ND |
MSS |
Clonal |
None |
Dead |
60 |
Immunosuppression; MSI: Microsatellite instability; TCR: T-cell receptor clonality; CM: Cardiomyopathy; CHD: Congestive heart failure; GN: Glomerulonephritis; DM: Diabetes Mellitus; FSGS: Focal segmental glomerulosclerosis; APCKD: Autosomal dominant polycystic kidney disease; LN: Lymph node; BM: Bone marrow; AZA: Azathioprine; CS: Corticosteroids; P: Prednisone; CNI: Calcineurin inhibitor; MMF: Mycophenolate mofetil; RT: Radiotherapy; N/A: Not available; ND: Not done; PC: Polyclonal; * EBER+ B-cells were present |
|||||||||||||||||||||||||||||||
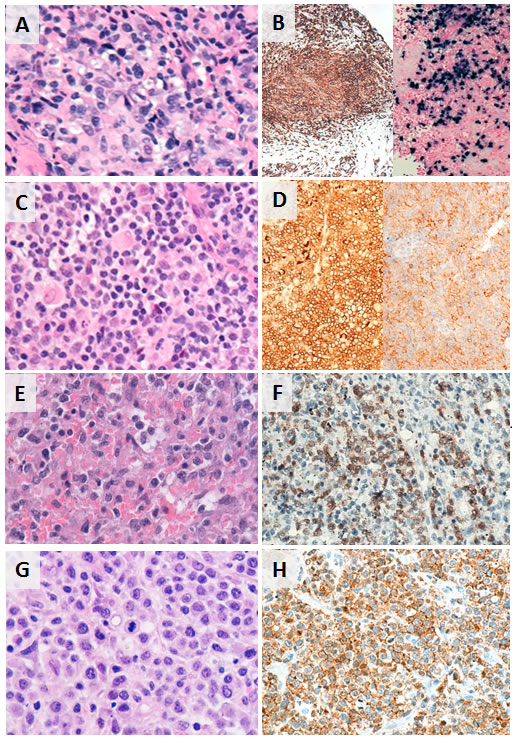
Figure 1: Histopathologic and phenotypic features of selected subtypes of T/NK-PTLD. A. and B. EBV+ PTCL in the breast; CD8+ and EBER+. C. and D. PTCL, NOS presenting as a retroperitoneal mass; CD4+ and PD1+. E. and F. Hepatosplenic T-cell lymphoma in the spleen; TCR-gamma+. G. and H. PTCL, NOS in subcarinal lymph node; CD8+.
Clinical characteristics
The detailed clinical features of the 19 T/NK-PTLD are described in Table 1. The median age was 47 (range: 20-83) and 12/19 (63%) patients were male. The most commonly transplanted organ was heart (n = 8), followed by kidney (n = 7), lung (n = 3) and liver (n = 1). Immunosuppressive therapy comprised calcineurin inhibitors (n = 12), corticosteroids (n = 11), azathioprine (n = 10), and mycophenolate mofitil (n = 6); no association between the type of therapy and organ allograft was observed. The median interval between organ transplantation and diagnosis was 7 years (range: 0.5 - 18 y). Extranodal disease was seen in 15/19 (79%) cases and 3 (16%) cases were EBV+. The types of therapies for PTLD and response to treatment varied. The patient with lymphomatoid papulosis (case 18) was treated with reduction in immunosuppression alone, which led to disease regression. Four patients received a combination of chemotherapy and radiation and 12 received chemotherapy alone. Two patients were referred for palliative care. Clinical remission was achieved in 1 patient with c-ALCL (case 13) who presented with localized disease, after treatment with chemotherapy and radiation; he died of disease unrelated causes 12 years post diagnosis. One patient with EBV+ PTCL (case 6) is in remission 4 years after diagnosis having received chemotherapy with etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) and one individual with PTCL, NOS (case 5) achieved remission after chemotherapy, radiation, and allogeneic stem cell transplantation. The patient diagnosed with ATLL, based on positive serology for HTLV-1, was treated with EPOCH and is alive 2 years after diagnosis. Despite aggressive treatment, however, the median overall survival was 6.6 months (range: 18 days to 12 years).
Genome-wide copy number alterations
Copy number alterations were observed in 12/16 cases (75%; complex = 8, simple = 4). Loss of heterozygosity was noted in 7/16 (44%) cases (Figure 3F; Table 2). No recurrent alterations were seen in 5 cases of PTCL, NOS. Two of three HSTCL showed 7q gain and 7p loss, indicative of isochromosome 7q. One of two ENKTCL showed loss of 6q. Copy number alterations in 1 of 2 cases of c-ALCL analyzed included 1p gain and losses at 13q and 16q. No copy number changes were detected in the ATLL and lymphomatoid papulosis.
Table 2: Genomic alterations in T/NK-PTLDs
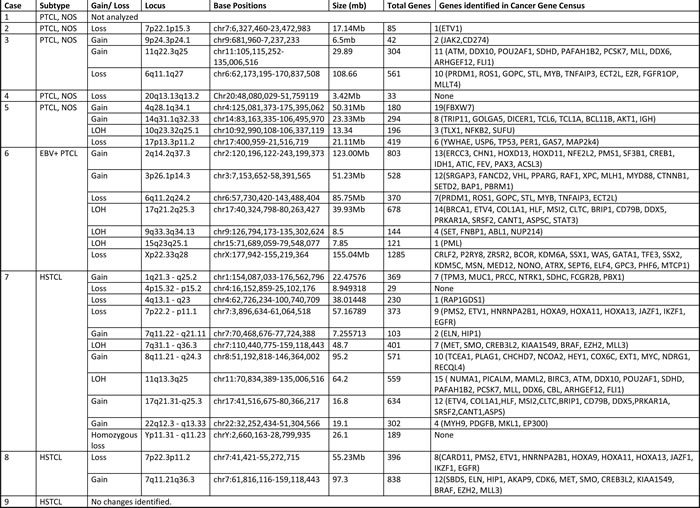
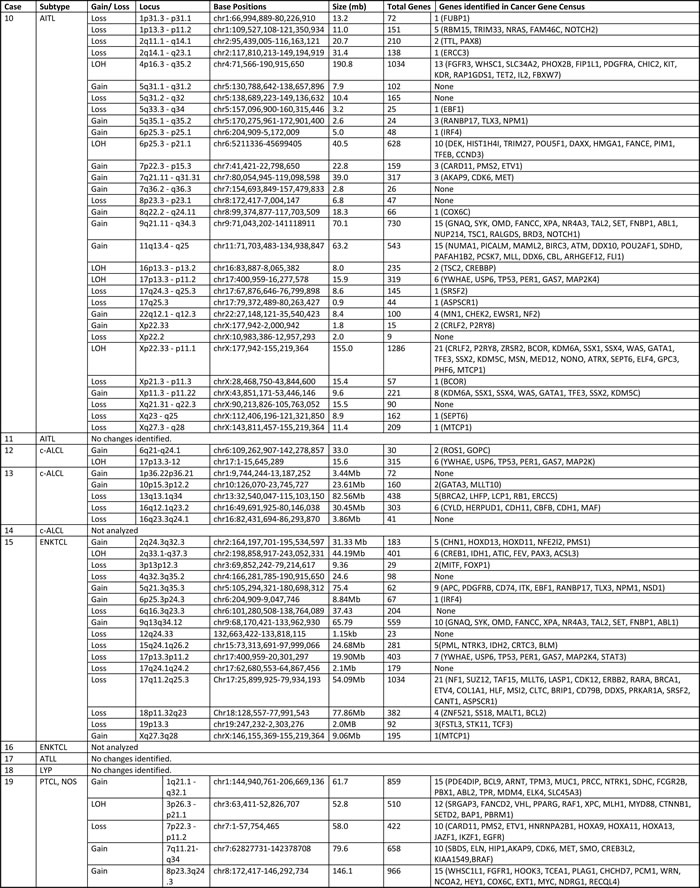
LOH: Loss of heterozygosity
Targeted next generation sequencing
Somatic mutations were detected in 17/18 cases (94%) using targeted sequencing of 465 cancer associated genes. Overall, 377 variants were identified: 286 missense (76%), 31 indels (8%), 16 nonsense (4%), and 44 intronic (12%) (Suppl. Table 1), with an average of 594.8-fold coverage and > 10X coverage for 99% of the coding regions (Suppl. Table 2). The pathogenic variants, focusing on genes recurrently targeted or implicated in hematologic malignancies, are listed in Figure 2.
Heterozygous mutations of epigenetic modifier genes were the most frequently observed alterations (11/18, 61%). TET2 mutations were seen in four cases; missense (n = 2, including R1359G), frameshift (n = 1), and nonsense accompanied by LOH of 4q24 (n = 1, R544X). Mutations in histone and DNA methyltransferases (KMT2D, KMT2C, DNMT3A) and the histone demethylase (KDM6B) occurred in 7/18 (39%) cases. Missense and frameshift mutations in chromatin remodeling complex genes (ARID1B, ARID2) were seen in 5 (28%) cases.
Disruption of TP53 was identified in 6 of 18 (33%) cases (Figure 3A, 3B). Biallelic inactivation was noted in 3 cases (co-occurrence of mutation with 17p LOH) and monoallelic missense mutations in two cases. All missense mutations (n = 4) are classified as non-functional or partially functional in the IARC TP53 database (Version R17). [5] Deletion of 17p encompassing the TP53 locus alone was seen in one case.
Alterations of JAK/STAT pathway genes were seen in 5 of 18 cases (28%). STAT3 and STAT5B mutations were mutually exclusive (Figure 3C-3E). STAT5B mutations were present in one case each of HSTCL and PTCL, NOS. STAT3 mutations were present in one case each of c-ALCL, ENKTCL, and the EBV+ PTCL. LOH of the STAT3 locus on 17q was observed in the two EBV+ PTLDs with STAT3 mutations (EBV+ PTCL and ENKTCL), suggesting biallelic gain of function. These two cases also had loss of 6q encompassing FOXO3, PRDM1 and PTPRK. A JAK3 variant (S568P) adjacent to a previously described hotspot was also present in the STAT3-mutated EBV+ PTCL. [6]
Subtype-specific genetic alterations were also identified. One of the two cases of angioimmunoblastic T-cell lymphoma demonstrated known pathogenic mutations in RHOA and FYN, proteins involved in T-cell motility and proliferation and TCR signaling. [7]
Importantly, mutations in genes not previously implicated in PTCL pathogenesis were also observed. Missense mutations in MED12 were seen in 3 cases (1 PTCL, NOS and 2 HSTCL), including two novel and potentially damaging variants (R516H and R816Q) and one exon 2 mutation (L36P). Alterations of TBX3 were observed in 3 of 5 PTCL, NOS. A missense mutation in MTOR (W1456L) involving the FAT domain was observed in the EBV+ PTCL. A three base pair insertion mutation was observed in FBXW7 in one PTCL, NOS, which was confirmed by Sanger sequencing (Suppl. Figure 1).
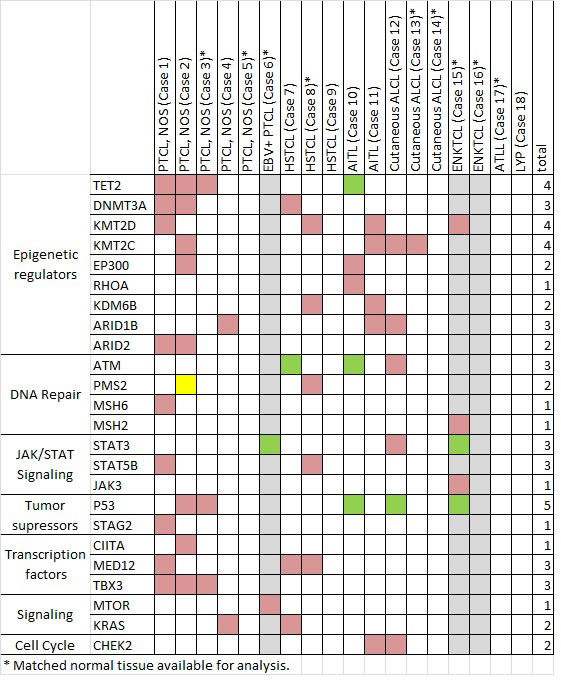
Figure 2: Selected mutations identified by next-generation sequencing in 18 cases of T/NK-PTLD. Total number of subjects with mutations is listed at right. A green box indicates mutation co-occurring with genomic loss or LOH, yellow box indicates MSI-H status as a consequence of mutation and grey columns indicate 3 EBV+ PTLD.
Microsatellite instability
Variants in mismatch repair (MMR) genes were detected in 4 of 18 PTLD (cases 1, 2, 8, and 15, Figure 2). However only one (6%) PTLD (case 2) with a heterozygous PMS2 mutation and PMS2 loss by immunohistochemistry showed evidence of high level microsatellite instability (MSI-H); alterations at 5/5 mononucleotide repeats accounting for 7% (1/14) of all cases analyzed with a panel of microsatellite markers.. The remaining 3 cases showed normal immunohistochemical expression of the MMR gene products.
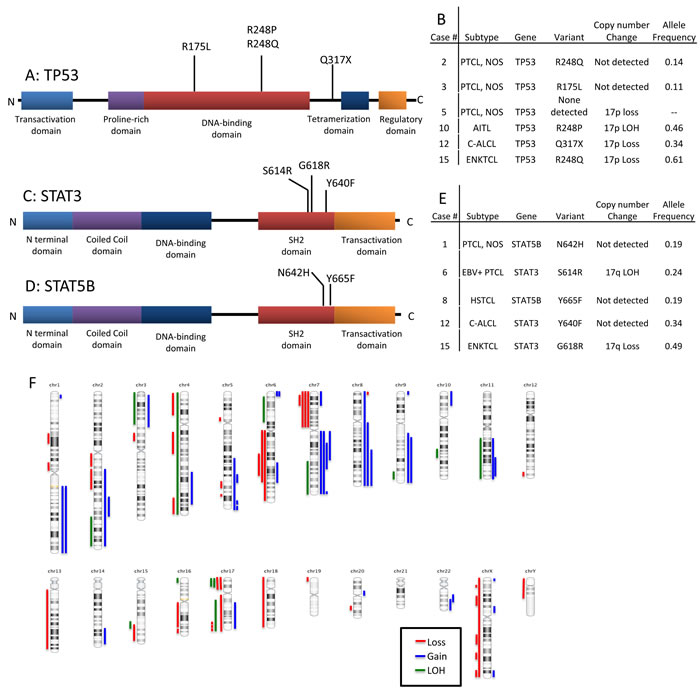
Figure 3: Schematic representation of the types and locations of TP53, STAT3 and STAT5b mutations. A. TP53 mutations. B. Table showing T/NK-PTLD subtypes with TP53 mutation and 17p copy number changes. C. STAT3 mutations in the SH2 domain. D. STAT5b mutations in the SH2 domain. E. Table showing T/NK-PTLD subtypes with STAT3 or 5b mutations, and 17q copy number changes. F. Ideogram of copy number changes detected in 16 cases of T/NK-PTLD. Gains are indicated in blue, losses in red and LOH in green.
Discussion
Our series of T/NK-PTLD comprised a spectrum of PTCL subtypes that have been reported to occur post organ transplantation and are recognized as monomorphic PTLDs in the 2008 WHO classification [3, 4]. Epidemiologic data drawn from national registries have shown a higher frequency of PTCL, NOS, ALCL, HSTCL, and ENKTCL in transplant recipients. [8] Data regarding other PTCL subtypes in the setting of immunosuppression (e.g. AITL, ATLL) are limited. [9, 10] Cases of ATLL, however, have been reported in recipients of solid organ allografts in multiple series. [11, 12] In fact, immunosuppression has been proposed as a risk factor for neoplastic transformation in HTLV-1 carriers post organ transplantation. [13] Our observations support the broad definition of T/NK-PTLD in the current WHO classification and suggest inclusion of more detailed phenotypic information in PTLD databases, which will allow precise classification of PTCL and help determine the role of immune deregulation in the pathogenesis of different disease subtypes.
The clinicopathological features of T/NK-PTLD are distinct from B-cell PTLDs. In contrast to B-cell PTLDs, which are EBV-driven in 55-65% of cases, only 3 of 19 (16%) T/NK-PTLD in our series showed evidence of EBV infection. [14] Consistent with prior reports, disease presentation was late after transplant, with a median of 7 years between onset of immunosuppression and the development of lymphoma. The time of occurrence of B-cell PTLDs has an apparent bimodal distribution with the largest peak occurring in the first year after transplantation and a smaller peak observed five years after transplantation. [8] While a subset of B-cell PTLDs respond to reduction in immunosuppression, the majority of the T/NK-cell PTLDs do not, and they are associated with poor outcomes despite aggressive treatment. [15] Disease biology, however, appears to mirror the subtype specific differences observed in T-cell lymphomas of immunocompetent individuals. Lymphomatoid papulosis followed a fairly indolent course with survival over 10 years, while ENKTCL and HSTCL were rapidly fatal.
The genomic alterations detected in our cohort were similar to those described in PTCLs occurring in immunocompetent hosts. [16, 17] Changes consistent with isochromosome 7q, a characteristic feature of HSTCL, were identified in 2 cases of HSTCL. Loss of 13q, encompassing ING1 and 16q, encompassing the CTCF locus, seen in one c-ALCL, have been reported in non-transplant associated c-ALCL. [18] One of two ENKTCL and the EBV+ PTCL showed loss of 6q. This recurrent abnormality, which is observed in 29-52% of ENKTCL arising in immunocompetent individuals, encompasses loci of the FOXO3 and PRDM1 genes, which have been shown to function as tumor suppressors in ENKTCL and subtypes of PTCL. [16, 19, 20] In contrast to EBV+ B-PTLD, however, we observed a higher frequency of complex copy number changes in EBV+ T-PTLD. [21]
Mutations of epigenetic modifier genes were the most frequent alterations (61%) in T/NK-PTLD and were most commonly observed in cases of PTCL, NOS and AITL. A similar mutational spectrum is frequently observed in PTCL occurring in immunocompetent individuals. [7, 22, 23] Loss of function mutations in KMT2D/MLL2, a lysine specific histone methyltransferase targeting H3K4 residues that is considered to function as a tumor suppressor, have been reported in PTCL arising in immunocompetent individuals and were seen in a variety of T/NK-PTLD. [24] Missense mutations in TET2, a methylcytosine dioxygenase that plays an important role in active DNA demethylation, are frequent in both PTCL, NOS and AITL (38% and 33-76%, respectively). [23, 25, 26] Loss of function mutations in DNMT3A, a DNA methyltransferase that is required for genome-wide de novo methylation, are frequent in a variety of malignancies and have been reported in up to 38% of PTCL, NOS and 33%-37% AITL. [23, 26, 27] In a murine model, the combination of TET2 loss and DNMT3A mutation appeared to cooperate in the development of T-cell lymphomas exhibiting features of AITL. [28] Similar to observations in PTCL, NOS and AITL occurring in immunocompetent individuals, concomitant TET2 and DNMT3A mutations were noted in two cases of PTCL, NOS in our series. [29] Mutations in chromatin modifier genes, including ARID1B, and ARID2, were also common, a finding in line with prior studies of PTCL occurring in immunocompetent hosts. [24]
The TP53 pathway is frequently dysregulated in a variety of neoplasms, most commonly by TP53 mutation or loss, and these lesions are associated with aggressive disease. [30] TP53 abnormalities are known recurrent changes in a variety of PTCL, albeit at a low frequency (8-9%) and their prognostic significance is unclear in these neoplasms. [31-33] The higher frequency of TP53 aberrations in our series (33%) could reflect the small sample size and future larger studies are warranted to determine if the frequency of this genetic lesion is higher in PTCL arising in states of immune dysregulation.
Alterations of the JAK/STAT pathway are frequent in diverse subtypes of PTCL of immunocompetent hosts, including ALCL, HSTCL, T-PLL, ENKTCL, and also in LGL. [6, 34-37] Genetic aberrations activating the JAK-STAT pathway were identified in 28% of T/NK-PTLD, including PTCL, NOS, HSTCL, c-ALCL, and ENKTCL, and an EBV+ PTCL. As previously observed in PTCL, STAT3 and STAT5b alterations were mutually exclusive in T/NK-cell PTLD as well. [38] We observed an association between EBV infection and STAT3 activation. STAT3 mutations and LOH of the STAT3 locus were seen in 2 of 3 EBV+ T/NK-PTLDs with concomitant loss of 6q encompassing PTPRK. Loss of PTPRK has been associated with constitutive STAT3 activation in ENKTCL. [39] Activation of STAT3 by EBV, even in the absence of genetic lesions, has been shown to induce lymphoproliferative disorders of T- and NK-cells. [40] EBNA-2 is a coactivator of STAT3 transcription and LMP1 is known to constitutively activate STAT3 in B-cells. [41, 42] Of interest, IFN-gamma induced by LMP1 has been shown to enhance STAT1 expression in EBV+ B-PTLD. [43] Whether analogous mechanisms are operational in EBV-associated T/NK-PTLD is not yet known. Nonetheless, our findings suggest that targeting the JAK-STAT pathway with currently available drugs or more selective agents might be of therapeutic benefit in a subset of T/NK-PTLD.
Mutations in genes not previously described in PTCL were also observed. Novel and potentially damaging variants in MED12 were observed in two cases and a MED12 exon 2 mutation (L36P), recently described in chronic lymphocytic leukemia, was seen in one case. [44] MED12 is part of the CDK8 subcomplex, which includes CDK8 kinase and cyclin C, of Mediator, a component of the preinitiation complex, with integral roles in transcriptional regulation. [45] The MED12 protein has been shown to regulate JAK/STAT signaling in mouse embryonic fibroblasts [46]. The role of MED12 in normal and neoplastic T-cells, however, is unclear. The functional consequence of mutations of the transcription factor T-box 3 (TBX3), a transcriptional repressor, important for myriad developmental processes, [47] noted in three cases, is also not known at present. A novel missense mutation in MTOR was detected in an EBV+ PTCL. Similar mutations, which have been shown to activate both mTORC1 and 2 signaling in other neoplasms, may contribute to T-cell growth, survival and proliferation, in part by metabolic reprogramming of lymphocytes. [48] Activation of the mTOR pathway has been reported in T-cell lymphomas, but the presence of mTOR mutations has not been systematically investigated in these malignancies. Of interest, rapamycin analogues, which disrupt the mTORC signaling cascade have shown promise in early phase clinical trials of PTCL patients. [49-51]
Azathioprine use has been associated with an increased risk of hematological malignancies in allograft recipients and individuals with autoimmune diseases. [52] Thiopurines have been implicated in MMR pathway defects possibly due to epigenetic silencing of the MMR enzymes. [53] Borie et al. reported microsatellite instability (MSI-H) in 9% of immunosuppression-related lymphomas, including 33% (6/18 cases) of PTCL post solid organ transplantation. [52] Only 1 of 10 patients in our series treated with an azathioprine-based immunosuppressive regimen developed a MSI-H tumor. Although the observed frequency of MSI-H was lower in our series (1/14 cases; 7%) than a prior study, this difference was not statisically significant (p = 0.1).
In summary, our study represents the first survey of the genetic landscape of T/NK-PTLDs and adds to the growing list of genes altered in T-cell neoplasms that affect a variety of T-cell functions, including activation, proliferation, epigenetic regulation and metabolism. The observed clinicopathologic features and genetic aberrations appear similar to those reported for PTCL subtypes occurring in “immunocompetent” individuals, suggesting shared pathogenetic mechanisms. Our findings raise the possibility of novel therapeutic options for these aggressive lymphomas that need to be explored further in pre-clinical studies.
Materials and Methods
Case selection
The archives of the Departments of Pathology, Columbia University Medical Center, New York and University Hospitals of KU, Leuven were searched for cases of T/NK-PTLD. Nineteen cases were identified (Columbia University Medical Center, n = 14; University Hospitals of KU, Leuven, n = 5) that had adequate formalin-fixed, paraffin-embedded (FFPE) tissue for analysis ( > 50% tumor, except for one case of lymphomatoid papulosis with a tumor burden of 20%). We defined T/NK-PTLD as T- or NK-cell lymphomas occurring in individuals after organ transplantation, in accordance with the WHO 2008 guidelines. Morphologic and phenotypic data were reviewed and the lymphomas were classified according to current WHO criteria. [1] Patient demographic and clinical data were collected from the electronic medical record. This study was approved by the Institutional Review Board of Columbia University and performed in accordance with the Declaration of Helsinki.
Whole genome copy number and LOH analysis
Genome-wide copy number and LOH analysis was performed for 16 cases using the Affymetrix OncoScan FFPE assay (Affymetrix, CA, USA). Briefly, the Oncoscan assay has been optimized to work with DNA from FFPE tissue, utilizing molecular inversion probes to identify changes in copy number and loss of heterozygosity. [54] DNA extraction, sample preparation, hybridization, and scanning were performed according to the manufacturer’s specifications. Analysis was performed using the Affymetrix Chromosome Analysis Suite 2.0 (ChAS) and Nexus Copy Number 7.5 software (Biodiscovery, Inc. CA, USA).
All copy number alterations and regions of LOH called by the software were verified manually to determine erroneous calls and identify clonal gains and losses undetected by the software(s). Analysis was restricted to gains and losses > 1 Mb in length. Genomic alterations are reported based on the NCBI build 37 (hg19) of the human genome and cancer-associated genes were curated from the Cancer Gene Census (COSMIC v61 Release;http://www.sanger.ac.uk/genetics/CGP/Census/).
Next generation sequencing analysis
Targeted next generation sequencing of 18 tumors and 9 matched normal samples was performed using a panel comprising 465 cancer-associated genes (Suppl. Table 3). Fifty to 250 ng of DNA, extracted using the Qiamp mini kit or the Qiamp FFPE kit (Qiagen, Germantown, USA) was fragmented to a median of 150-200bp, by sonication. Following end-repair and 3’ adenylation of the fragments, and ligation of double-stranded sequencing and indexing adaptors to both ends, target capture and enrichment was performed with the Sure Select Hybrid Capture system (Agilent Technologies, Santa Clara, USA), using custom designed probes. Libraries were then quantified using qPCR, diluted to 2nM and pooled, prior to cluster generation and analysis on Illumina HiSeq2500, using Illumina TruSeq v3 chemistry (San Diego, USA) and 100bp paired-end reads (up to 9 indexed samples per run). Fastq files (more than 70% of reads with > Q30) were demultiplexed with CASAVA, and samples with at least 6Gb of sequencing data were used for mapping and variant calling using NextGene Software (Softgenetics, State College, USA). Reads were aligned to the hg19 (GRCh37) reference genome. At least 3 variant reads were required to call a variant. All calls with ambiguous alignments were excluded, including calls corresponding to regions with large indels. Candidate variants at very low allele frequency ( < 10% of variant reads) were eliminated.
After annotation, the variants were cross referenced with those in the 1000 Genomes Project, OMIM, dbSNP, and the Exome Variant Server. Variants with an allele prevalence > 1% in the 1000 genomes project were excluded. Common variants present in our departmental database of variants identified in prior constitutional exome analysis, non-pathogenic variants reported in dbSNP, and low quality calls were filtered out. The remaining variants were submitted for manual curation and variant prioritization with visual review of alignments. Synonymous variants and intronic variants greater than 2 bp from the coding sequence were excluded. Variants were manually cross-referenced with the Catalog of Somatic Variants in Cancer (COSMIC) and those that were not known “hot spot” mutations or had not been previously reported as potential driver variants were analyzed by PROVEAN and SIFT algorithms. For cases with matched normal specimens, variants present in the normal samples were excluded.
Assessment of microsatellite instability
Microsatellite instability (MSI) testing was performed in 14 cases using a fluorescent PCR-based assay (MSI Analysis system, Version 1.2, Promega, Madison, WI). Briefly, this test assessed 5 mononucleotide repeats (BAT25, BAT26, NR-21, NR-24, and MONO-27) and 2 pentanucleotide repeats (PentaC and PentaD) on genomic DNA and matched normal, where available. The fluorescently labeled PCR products were analyzed by capillary gel electrophoresis. MSI was determined if the tumor alleles showed a size difference ≥3 bp. Tumors with 2 or more microsatellite unstable markers were classified as MSI-H; the remainder were classified as microsatellite stable (MSS). Immunohistochemistry was performed for MLH1 (clone M1, predilute, Ventana), MSH6 (clone 44, predilute, Ventana), MSH2 (clone G219-1129, predilute, Cell Marque) and PMS2 (clone EPR3947, predilute, Cell Marque) using an autostainer (Benchmark UltraTM Ventana, Tuscon, AZ) after heat induced antigen retrieval (EDTA, pH 7.3). The ultraViewTM Universal DAB Detection kit (Ventana) was used for visualization. Absence of nuclear staining was evaluated in lesional tissue; adjacent stromal cells were used as an internal control.
Methods for DNA extraction, immunohistochemistry, flow cytometry, and Sanger sequencing are described in the supplemental methods.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Grant Support
Thomas Tousseyn holds a Mandate for Fundamental and Translational Research from the “Stichting tegen Kanker” (2014-083). This study was funded by an intramural research grant provided by the Department of Pathology and Cell Biology, Columbia University Medical Center.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Swerdlow SH WS, Chadburn A, Ferry JA. (2008). “Post-transplant lymphoproliferative disorders”. In: Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, ed. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (Lyon: IARC), pp. 343-349.
2. Swerdlow SH. T-cell and NK-cell posttransplantation lymphoproliferative disorders. American journal of clinical pathology. 2007; 127:887-895.
3. Herreman A, Dierickx D, Morscio J, Camps J, Bittoun E, Verhoef G, De Wolf-Peeters C, Sagaert X and Tousseyn T. Clinicopathological characteristics of posttransplant lymphoproliferative disorders of T-cell origin: single-center series of nine cases and meta-analysis of 147 reported cases. Leukemia & lymphoma. 2013; 54:2190-2199.
4. (2008). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. (Lyon: IARC).
5. Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P and Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Human mutation. 2007; 28:622-629.
6. Lee S, Park HY, Kang SY, Kim SJ, Hwang J, Lee S, Kwak SH, Park KS, Yoo HY, Kim WS, Kim JI and Ko YH. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget. 2015; 6:17764-17776. doi: 10.18632/oncotarget.3776.
7. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, Jiang X, Lossos IS, Nicolas C, Balbin M, Bastard C, Bhagat G, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nature genetics. 2014; 46:166-170.
8. Clarke CA, Morton LM, Lynch C, Pfeiffer RM, Hall EC, Gibson TM, Weisenburger DD, Martinez-Maza O, Hussain SK, Yang J, Chang ET and Engels EA. Risk of lymphoma subtypes after solid organ transplantation in the United States. British journal of cancer. 2013; 109:280-288.
9. Hoshida Y, Li T, Dong Z, Tomita Y, Yamauchi A, Hanai J and Aozasa K. Lymphoproliferative disorders in renal transplant patients in Japan. International journal of cancer. 2001; 91:869-875.
10. Kraus TS, Twist CJ and Tan BT. Angioimmunoblastic T cell lymphoma: an unusual presentation of posttransplant lymphoproliferative disorder in a pediatric patient. Acta Haematologica. 2014; 131:95-101.
11. Yoshizumi T, Takada Y, Shirabe K, Kaido T, Hidaka M, Honda M, Ito T, Shinoda M, Ohdan H, Kawagishi N, Sugawara Y, Ogura Y, Kasahara M, Kubo S, Taketomi A, Yamashita N, et al. Impact of human T-cell leukemia virus type 1 on living donor liver transplantation: a multi-center study in Japan. Journal of hepato-biliary-pancreatic sciences. 2016; doi: 10.1002/jhbp.345.
12. Draoua HY, Tsao L, Mancini DM, Addonizio LJ, Bhagat G and Alobeid B. T-cell post-transplantation lymphoproliferative disorders after cardiac transplantation: a single institutional experience. British Journal of Haematology. 2004; 127:429-432.
13. Jenks PJ, Barrett WY, Raftery MJ, Kelsey SM, van der Walt JD, Kon SP and Breuer J. Development of human T-cell lymphotropic virus type I-associated adult T-cell leukemia/lymphoma during immunosuppressive treatment following renal transplantation. Clinical infectious diseases. 1995; 21:992-993.
14. Johnson LR, Nalesnik MA and Swerdlow SH. Impact of Epstein-Barr virus in monomorphic B-cell posttransplant lymphoproliferative disorders: a histogenetic study. The American journal of surgical pathology. 2006; 30:1604-1612.
15. Leblond V, Dhedin N, Mamzer Bruneel MF, Choquet S, Hermine O, Porcher R, Nguyen Quoc S, Davi F, Charlotte F, Dorent R, Barrou B, Vernant JP, Raphael M and Levy V. Identification of prognostic factors in 61 patients with posttransplantation lymphoproliferative disorders. Journal of clinical oncology. 2001; 19:772-778.
16. Hartmann S, Gesk S, Scholtysik R, Kreuz M, Bug S, Vater I, Doring C, Cogliatti S, Parrens M, Merlio JP, Kwiecinska A, Porwit A, Piccaluga PP, Pileri S, Hoefler G, Kuppers R, et al. High resolution SNP array genomic profiling of peripheral T cell lymphomas, not otherwise specified, identifies a subgroup with chromosomal aberrations affecting the REL locus. British journal of haematology. 2010; 148:402-412.
17. Nakagawa M, Nakagawa-Oshiro A, Karnan S, Tagawa H, Utsunomiya A, Nakamura S, Takeuchi I, Ohshima K and Seto M. Array comparative genomic hybridization analysis of PTCL-U reveals a distinct subgroup with genetic alterations similar to lymphoma-type adult T-cell leukemia/lymphoma. Clinical cancer research. 2009; 15:30-38.
18. Sanchez-Schmidt JM, Salgado R, Servitje O, Gallardo F, Ortiz-Romero PL, Karpova MB, Zipser MC, Garcia-Muret MP, Estrach T, Rodriguez-Pinilla SM, Climent F, Suela J, Ferreira BI, Cigudosa JC, Salido M, Barranco C, et al. Primary cutaneous CD30+ anaplastic large-cell lymphomas show a heterogeneous genomic profile: an oligonucleotide arrayCGH approach. The Journal of investigative dermatology. 2011; 131:269-271.
19. Zettl A, Rudiger T, Konrad MA, Chott A, Simonitsch-Klupp I, Sonnen R, Muller-Hermelink HK and Ott G. Genomic profiling of peripheral T-cell lymphoma, unspecified, and anaplastic large T-cell lymphoma delineates novel recurrent chromosomal alterations. The American journal of pathology. 2004; 164:1837-1848.
20. Boi M, Rinaldi A, Kwee I, Bonetti P, Todaro M, Tabbo F, Piva R, Rancoita PM, Matolcsy A, Timar B, Tousseyn T, Rodriguez-Pinilla SM, Piris MA, Bea S, Campo E, Bhagat G, et al. PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood. 2013; 122:2683-2693.
21. Rinaldi A, Capello D, Scandurra M, Greiner TC, Chan WC, Bhagat G, Rossi D, Morra E, Paulli M, Rambaldi A, Rancoita PM, Inghirami G, Ponzoni M, Moreno SM, Piris MA, Mian M, et al. Single nucleotide polymorphism-arrays provide new insights in the pathogenesis of post-transplant diffuse large B-cell lymphoma. British Journal of Haematology. 2010; 149:569-577.
22. Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, Chan LC, Chan WC, Gaulard P and Mak TW. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012; 119:1901-1903.
23. Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, Gascoyne RD, Wu X, Wang J, Muhammad Z, Jiang B, Rohr J, Cannon A, Steidl C, Fu K, Li Y, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015; 126:1741-1752.
24. Schatz JH, Horwitz SM, Teruya-Feldstein J, Lunning MA, Viale A, Huberman K, Socci ND, Lailler N, Heguy A, Dolgalev I, Migliacci JC, Pirun M, Palomba ML, Weinstock DM and Wendel HG. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia. 2015; 29:237-241.
25. Lemonnier F, Couronne L, Parrens M, Jais JP, Travert M, Lamant L, Tournillac O, Rousset T, Fabiani B, Cairns RA, Mak T, Bastard C, Bernard OA, de Leval L and Gaulard P. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012; 120:1466-1469.
26. Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, Kim S, van Bodegom D, Bolla S, Schatz JH, Teruya-Feldstein J, Hochberg E, Louissaint A, Dorfman D, Stevenson K, Rodig SJ, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014; 123:1293-1296.
27. Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK and Ley TJ. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer cell. 2014; 25:442-454.
28. Scourzic L, Couronne L, Pedersen MT, Della Valle V, Diop M, Mylonas E, Calvo J, Mouly E, Lopez CK, Martin N, Fontenay M, Bender A, Guibert S, Dubreuil P, Dessen P, Droin N, et al. DNMT3A mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia. 2016; doi: 10.1038/leu.2016.29.
29. Couronne L, Bastard C and Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. The New England journal of medicine. 2012; 366:95-96.
30. Greenblatt MS, Bennett WP, Hollstein M and Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer research. 1994; 54:4855-4878.
31. Matsushima AY, Cesarman E, Chadburn A and Knowles DM. Post-thymic T cell lymphomas frequently overexpress p53 protein but infrequently exhibit p53 gene mutations. The American journal of pathology. 1994; 144:573-584.
32. Rassidakis GZ, Thomaides A, Wang S, Jiang Y, Fourtouna A, Lai R and Medeiros LJ. p53 gene mutations are uncommon but p53 is commonly expressed in anaplastic large-cell lymphoma. Leukemia. 2005; 19:1663-1669.
33. Vasmatzis G, Johnson SH, Knudson RA, Ketterling RP, Braggio E, Fonseca R, Viswanatha DS, Law ME, Kip NS, Ozsan N, Grebe SK, Frederick LA, Eckloff BW, Thompson EA, Kadin ME, Milosevic D, et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood. 2012; 120:2280-2289.
34. Nicolae A, Xi L, Pittaluga S, Abdullaev Z, Pack SD, Chen J, Waldmann TA, Jaffe ES and Raffeld M. Frequent STAT5B mutations in gammadelta hepatosplenic T-cell lymphomas. Leukemia. 2014; 28:2244-2248.
35. Lopez C, Bergmann AK, Paul U, Murga Penas EM, Nagel I, Betts MJ, Johansson P, Ritgen M, Baumann T, Aymerich M, Jayne S, Russell RB, Campo E, Dyer MJ, Durig J and Siebert R. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. British journal of haematology. 2016; 173:265-273.
36. Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, Lagstrom S, Clemente MJ, Olson T, Jalkanen SE, Majumder MM, Almusa H, Edgren H, Lepisto M, Mattila P, Guinta K, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. The New England journal of medicine. 2012; 366:1905-1913.
37. Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E, Todaro M, Boi M, Acquaviva A, Ficarra E, Novero D, Rinaldi A, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer cell. 2015; 27:516-532.
38. Kucuk C, Jiang B, Hu X, Zhang W, Chan JK, Xiao W, Lack N, Alkan C, Williams JC, Avery KN, Kavak P, Scuto A, Sen E, Gaulard P, Staudt L, Iqbal J, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nature communications. 2015; 6:6025.
39. Chen YW, Guo T, Shen L, Wong KY, Tao Q, Choi WW, Au-Yeung RK, Chan YP, Wong ML, Tang JC, Liu WP, Li GD, Shimizu N, Loong F, Tse E, Kwong YL, et al. Receptor-type tyrosine-protein phosphatase kappa directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood. 2015; 125:1589-1600.
40. Honami Komatsu K-II, Haruna Shibayama, Tomotaka Yada, Momoko Yamada, Kouhei Yamamoto, MD, PhD, Takatoshi Koyama, MD, PhD, Shigeyoshi Fujiwara, MD, PhD, Osamu Miura, MD, PhD and Ayako Arai, MD, PhD. (2014). STAT3 Is Activated By EBV in T or NK Cells Leading to Development of EBV-T/NK-Lymphoproliferative Disorders American Society of Hematology. (San Francisco).
41. Shair KH, Bendt KM, Edwards RH, Bedford EC, Nielsen JN and Raab-Traub N. EBV latent membrane protein 1 activates Akt, NFkappaB, and Stat3 in B cell lymphomas. PLoS pathogens. 2007; 3:e166.
42. Muromoto R, Ikeda O, Okabe K, Togi S, Kamitani S, Fujimuro M, Harada S, Oritani K and Matsuda T. Epstein-Barr virus-derived EBNA2 regulates STAT3 activation. Biochemical and biophysical research communications. 2009; 378:439-443.
43. Vaysberg M, Lambert SL, Krams SM and Martinez OM. Activation of the JAK/STAT pathway in Epstein Barr virus+-associated posttransplant lymphoproliferative disease: role of interferon-gamma. American journal of transplantation. 2009; 9:2292-2302.
44. Kampjarvi K, Jarvinen TM, Heikkinen T, Ruppert AS, Senter L, Hoag KW, Dufva O, Kontro M, Rassenti L, Hertlein E, Kipps TJ, Porkka K, Byrd JC, de la Chapelle A and Vahteristo P. Somatic MED12 mutations are associated with poor prognosis markers in chronic lymphocytic leukemia. Oncotarget. 2015; 6:1884-1888. doi: 10.18632/oncotarget.2753.
45. Turunen M, Spaeth JM, Keskitalo S, Park MJ, Kivioja T, Clark AD, Makinen N, Gao F, Palin K, Nurkkala H, Vaharautio A, Aavikko M, Kampjarvi K, Vahteristo P, Kim CA, Aaltonen LA, et al. Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell reports. 2014; 7:654-660.
46. Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, Dolken L, Strobl B, Muller M, Taatjes DJ and Kovarik P. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013; 38:250-262.
47. Wansleben S, Peres J, Hare S, Goding CR and Prince S. T-box transcription factors in cancer biology. Biochimica et biophysica acta. 2014; 1846:380-391.
48. Ghosh AP, Marshall CB, Coric T, Shim EH, Kirkman R, Ballestas ME, Ikura M, Bjornsti MA and Sudarshan S. Point mutations of the mTOR-RHEB pathway in renal cell carcinoma. Oncotarget. 2015; 6:17895-17910. doi: 10.18632/oncotarget.4963.
49. Yun S, Vincelette ND, Knorr KL, Almada LL, Schneider PA, Peterson KL, Flatten KS, Dai H, Pratz KW, Hess AD, Smith BD, Karp JE, Wahner Hendrickson AE, Fernandez-Zapico ME and Kaufmann SH. 4EBP1/c-MYC/PUMA and NFkappaB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood. 2016.
50. Kim SJ, Shin DY, Kim JS, Yoon DH, Lee WS, Lee H, Do YR, Kang HJ, Eom HS, Ko YH, Lee SH, Yoo HY, Hong M, Suh C and Kim WS. A phase II study of everolimus (RAD001), an mTOR inhibitor plus CHOP for newly diagnosed peripheral T-cell lymphomas. Annals of oncology. 2016; 27:712-718.
51. Witzig TE, Reeder C, Han JJ, LaPlant B, Stenson M, Tun HW, Macon W, Ansell SM, Habermann TM, Inwards DJ, Micallef IN, Johnston PB, Porrata LF, Colgan JP, Markovic S, Nowakowski GS, et al. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma. Blood. 2015; 126:328-335.
52. Borie C, Colas C, Dartigues P, Lazure T, Rince P, Buhard O, Folliot P, Chalastanis A, Muleris M, Hamelin R, Mercier D, Oliveira C, Seruca R, Chadburn A, Leblond V, Barete S, et al. The mechanisms underlying MMR deficiency in immunodeficiency-related non-Hodgkin lymphomas are different from those in other sporadic microsatellite instable neoplasms. International journal of cancer. 2009; 125:2360-2366.
53. Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. British medical bulletin. 2006; 79-80:153-170.
54. Wang Y, Cottman M and Schiffman JD. Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer genetics. 2012; 205:341-355.