
INTRODUCTION
Developmental programs control physiological functions at early growth stages, including cell proliferation, differentiation, morphogenesis and tissue homeostasis. Aberrant activation of such programs may disturb the homeostatic balance and thereby trigger tumorigenesis. In this respect, a regulatory network called the Retinal Determination Gene Network (RDGN), originally identified as a fundamental signal for Drosophila melanogaster eye specification, may also be dysregulated in cancer [1, 2]. Functional modification of the key RDGN members, DACH, EYA and SIX, is a potential therapeutic approach to individualized chemotherapy.
RDGN SIGNALING, FROM DROSOPHILA EYE DEVELOPMENT TO HUMAN DISEASE
The RDGN first received attention as a key signaling pathway in Drosophila eye determination [3]. In the last fifteen years, RDGN signaling has been shown to govern the specification of a wide range of tissues, including the retinas of both insects and mammals. This regulatory network mainly includes a structural relative of the Ski/Sno gene family, dachshund; the SIX family members, sine oculis (so) and optix; the tyrosine phosphatase, eyes absent (eya); and the Pax6 homologs, eyeless (ey) and twin of eyeless (toy). These genes are organized in a sophisticated network that controls organogenesis, and mutations of these genes in humans are associated with several clinical disorders [3].
The mammalian DACH1 protein can inhibit target gene expression either directly, by binding to specific DNA sequences within chromatin [4, 5], or indirectly, by combining with other transcription factors (c-Jun, SMADs and SIX) [6-9]. For example, DACH1 can displace FOXM1 and FOXC2 from the cis elements within promoters that have Forkhead family binding sites and thereby attenuate their oncogenic activity [4, 5]. During vertebrate development, DACH1 function is required for organ specification [8, 10-12]. In humans, DACH1 germline mutations have been shown to contribute to bilateral cystic renal dysplasia [13], chronic kidney disease (CKD) [14], familial young-onset diabetes, pre-diabetes and cardiovascular diseases like coronary heart disease (CHD) and coronary arteriosclerosis [15]. DACH1 also inhibits aldosterone secretion in zona glomerulosa cells [16]. However, the functional significance of DACH1 in human diseases still remains a mystery.
As a member of the homeobox gene family, the SIX superfamily has been evolutionarily conserved, and controls embryonic development and tissue specification of the eye, kidney and muscle [2, 3, 17]. During the early stages of development, Six1 activates a diverse range of target genes that determine the proliferation and survival of progenitor cells. Once organ development is complete, Six1 is maintained at low or even undetectable levels in adult tissues [18]. Proteins of the SIX family have two regions of high sequence conservation: the homeoprotein domain (HD) and the SIX domain (SD). SIX proteins recognize specific DNA sequences, and both the HD and the SD contribute to these DNA interactions [19, 20]. Nevertheless, the transcriptional function of SIX depends on an additional cofactor within the complex. For instance, Dach1 functions as a corepressor to inhibit the expression of Six target genes, whereas Eya permits Six to activate downstream signaling [8].
As another component of the conserved RDGN, the EYA family proteins are important transcriptional cofactors. Generally, Eya is recruited to the local chromatin of target genes through Six proteins [8]. Structural analyses have revealed that SIX1 binds to EYA through a single amphipathic helical structure, and that even a single amino acid substitution can abolish SIX1-induced epithelial-mesenchymal transition (EMT) [19]. In humans, mutations in SIX and EYA or disruption of the SIX/EYA complex cause branchio-oto-rena (BOR) syndrome, an autosomal dominant genetic disorder marked by underdeveloped or absent kidneys, deafness, auricular malformations and bronchial arch remnants [19, 21, 22]. The function of SIX/EYA complex is also required for lung morphogenesis [23] and myocardial hypertrophy [24]. Thus, abnormal functioning of the SIX/EYA complex may impact a broad range of human diseases.
Another critical feature that separates EYA proteins from other RDGN members is their tyrosine/threonine-phosphatase activity. The phosphatase activity of Eya bridges Dach and Six, and switches Six-Dach from a repressed to an activated state by displacing a corepressor and recruiting a coactivator [8]. The conserved carboxy-terminal domain of EYA (ED) is essential for protein-protein interactions, for instance, with SIX and DACH. EYA has been classified into the haloacid dehalogenase (HAD) superfamily based on signature motifs in the ED, and functions as an Mg2+-dependent tyrosine phosphatase. The N-terminal region (commonly referred to as ED2), which is characterized by a rich stretch of proline/serine/threonine residues, is mainly responsible for threonine dephosphorylation [2]. This region also provides transactivation domain during Drosophila eye development [25]. Because of this phosphorylation mechanism, the EYA family participates significantly in the cellular response to changes in the microenvironment, but the influences of individual family members vary because of their different phosphatase activities. For example, by dephosphorylating H2AX at tyrosine 142 (pY142), EYA1, EYA2 and EYA3 regulate the formation of γH2AX, which promotes DNA repair and cell survival and thus prevents genotoxic stress-induced apoptosis [26, 27]. In the innate immune response, EYA4 serves as a threonine phosphatase rather than as a tyrosine phosphatase [28]. The phosphatase activity of EYA is essential for the maintenance of tight junctions in lung epithelial cells [29]. However, in breast tumors, EYA2 dephosphorylates estrogen receptor β (ERβ) at Y36, thus reversing its antitumor role and promoting malignant growth and dissemination [30]. Likewise, the tumorigenic role of EYA1 in breast cancer cells relies on its phosphatase function [31]. Thus, the phosphatase activity of EYA may be a good target of cancer therapeutics.
Recent studies have revealed the significance of the RDGN in tumorigenesis and tumor progression [1, 2]. DACH1 is a tumor suppressor, while SIX/EYA promote malignancy, so aberrations in these components of the RDGN signaling machinery enable cells to acquire oncogenic properties. For example, inactivation of DACH1 or hyperactivation of SIX/EYA in breast epithelial cells leads to over-proliferation, tumor formation and invasion into blood vessels, resulting in distant metastases (Figure 1). These activities of the RDGN members will be discussed in detail below.
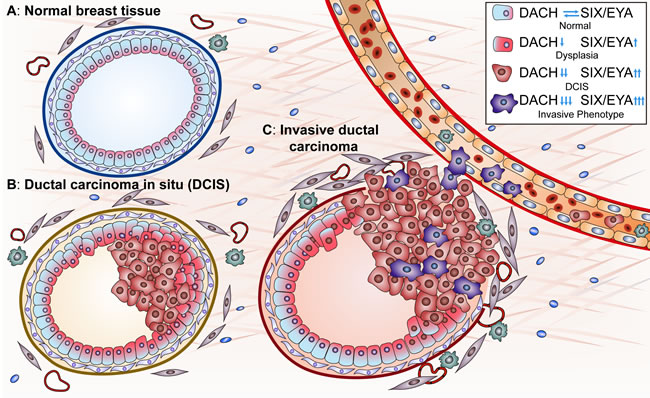
Figure 1: A. RDGN members determine breast cancer initiation and progression. In normal breast epithelial cells, the functional balance of DACH and SIX/EYA maintains homeostasis in luminal cellular proliferation/apoptosis and thereby maintains the luminal structure. Functional loss of DACH or hyperactivity of SIX/EYA drives hyper-proliferation, transformation, and progression to ductal carcinoma in situ. Some malignant cells undergo EMT, acquire cancer stem cell properties, invade through the basal membrane and enter blood vessels, leading to distant metastases. “↑” represents an upregulated function and “↓” represents a downregulated function.
THE RDGN IN TUMOR INITIATION AND PROGRESSION
Proliferation and cancer stem cells
Constitutive mitogenic signals drive the misexpression of key cell cycle machinery, leading to the initiation and progression of tumorigenesis [32]. The RDGN maintains a dynamic balance that controls progression through the cell cycle. Suppression of cyclin D1 and cyclin A by DACH1 negatively regulates cell cycle progression [33, 34] and thereby blocks cellular proliferation and tumor growth in various cancers [33-35]. In contrast, EYA1 induces cyclin D1 through its phosphatase activity in breast cancer, and SIX1 directly enhances the transcription of cyclin D1 in rhabdomyosarcoma tumor cells [31, 36]. SIX1 also active cyclin A1 expression to promote proliferation; furthermore, SIX1 alone can transform mammary epithelial cells and promote aggressive tumor formation and peritumoral lymphovascular invasion [37]. DACH1 promotes cell cycle arrest through context-dependent interactions with p53 and activating p53-taget genes [38, 39], whereas SIX1 downregulates p53 by upregulating microRNA-27a-3p and downregulating ribosomal protein L26 (RPL26) [40]. Imbalance between the tumor-restraining effect of DACH1 and the oncogenic functions of SIX/EYA accelerates cell cycle progression and attenuates apoptosis, creating a favorable environment for uncontrolled proliferation.
Cancer stem cells (CSCs), also called tumor initiation cells (TICs), are characterized by self-renewal and differentiation, and contribute to therapeutic resistance. The regulation of breast CSCs by DACH/EYA/SIX is now well documented. A subpopulation of cells with the CD44high/CD24low signature, (where CD44 and CD24 are widely recognized cell surface markers for breast CSCs [41]), can either be negatively regulated by DACH1 as it antagonizes NANOG, KLF4 and SOX2 transcriptional activation [42], or positively enriched by SIX1/EYA1 overexpression [43]. SIX1 also activates TGF-β and MAPK signaling to enhance the accumulation of CSCs [43, 44]. In a bitransgenic mouse model, overexpression of human SIX1 in the adult mouse mammary gland epithelium was found to induce tumors of multiple histological subtypes and to promote stem/progenitor cell characteristics by activating Wnt signaling [45]. Similar to SIX1, EYA1 (through its phosphatase activity) allows the proportion of breast CSCs to increase [31]. The therapeutic potential of the RDGN to limit the population of TICs is substantial.
The RDGN governs EMT and metastasis
EMT, the transformation of epithelial cells into cells with a mesenchymal migratory phenotype, participates in embryonic tissue formation, but also contributes to tumor progression. An interactive network including multiple signaling pathways and post-transcriptional factors facilitates the changes in epithelial cells that drive metastatic seeding [46,47]. TGF-β-driven cell reprogramming is a multistep process that passes first through EMT. The activation of Wnt signaling creates a favorable environment for TGF-β-induced EMT [48]. The cross-talk between RDGN and Wnt/TGF-β signaling promotes EMT (Figure 2).
DACH1 was first shown to inhibit TGF-β signaling by binding to SMAD4 in breast cancer [49]. Subsequently, DACH1 was found to restrain TGF-β/SMAD-induced EMT [50]. DACH1 may also antagonize EMT in part by inactivating Wnt/β-catenin signaling; indeed, DACH1 was recently found to downregulate β-catenin in hepatocellular and gastric cancer [35, 51]. In contrast, SIX1 promotes EMT in a TGF-β signaling-dependent manner, and is a critical enhancer in the switch of TGF-β/SMAD from tumor suppressors to oncogenic proteins [44, 52, 53]. Even a single amino acid mutation in SIX1 disrupts the SIX1-EYA2 complex, thereby abolishing SIX1-dependent TGF-β signaling and EMT progression [19]. In addition, SIX1-enhanced tumorigenesis is coupled to Wnt signaling activation [53]. EYA2 inactivation reverses the activation of TGF-β/SMAD and EMT induced by SIX1 [54].
The RDGN also governs EMT by regulating the expression of EMT-associated genes. DACH1 was found to abolish YB-1-induced SNAI1 translation and thus reverse EMT in basal-like breast cancer, an aggressive subtype [55]. By downregulating SNAI1, DACH1 increases E-cadherin expression and induces morphological changes that enhance epithelial properties [56]. SIX1 suppresses miR-200-family expression, a negative feedback loop in ZEB1/ZEB2 regulation, which in turn promotes EMT and carcinogenesis [57]. The role of EYA in EMT remains to be understood. The tyrosine-phosphatase activity of EYA3 was shown to be critical for cell motility and invasiveness rather than proliferation, indicating that EYA3 promotes metastatic behavior in cancer cells [58]. However, our previous study also indicated that transactivation of cyclin D1 through the tyrosine-phosphatase activity of EYA1 is required for tumor growth and cellular proliferation [31]. Some of these inconsistent results are due to the fact that different researchers have tested the phosphatase activities of the EYAs in different breast cancer cell lines. Although the EYA family members may behave distinctly in different studies, their underlying mechanisms may overlap. Whether they share similar functions during cancer development needs to be further examined.
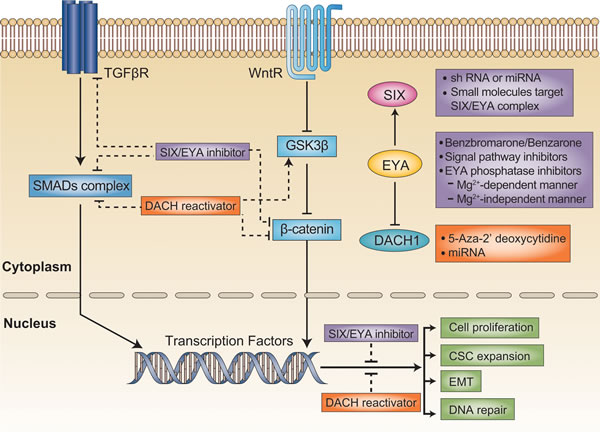
Figure 2: Targeting the DACH/SIX/EYA pathway in cancer treatment. TGFβ and Wnt signaling are both critical for tumor initiation and progression. Generally, SIX/EYA activates these two signaling pathways, whereas DACH1 negatively regulates them. The function of SIX could be blocked by the silencing of SIX expression with shRNA, or by the disruption of the SIX/EYA complex with small molecules. EYA phosphatase activity could be blocked by biochemical inhibitors. On the other hand, DACH1 expression could be reactivated with demethylating agents or miRNA. Thus, targeting RDGN members is a promising therapeutic strategy that could restrain malignant behavior in tumors.
The regulation of the tumor microenvironment by RDGN signaling
In response to oncogenic signals from solid tumors or evolving genetic/epigenetic alternations, the tumor microenvironment continually changes to support malignant behavior. Numerous components participate in this process, but cancer-related inflammation and dysregulation of innate immunity especially disturb the physiological homeostasis. It has been indicated that DACH1 binds the endogenous IL-8 promoter to block breast cancer cellular migration in vitro and metastasis in vivo [59]. Dach1 gene deletion in mice was shown to dramatically increase IL-8 and IL-6 abundance by nearly 1000-fold and thus promote prostate cancer cellular migration [60]. CXCL5 is another downstream target whereby DACH1 inhibits lung tumorigenesis and metastasis [61]: endogenous DACH1 negatively regulates CXCL signaling to inhibit cytokine secretion and restrain malignant cell growth and migration. DACH1 also was found to repress bFGF-induced tumor initiation in glioma cells [62]. In contrast, as an enhancer of the innate immune response, EYA4 induces IFN-β and CXCL10 expression by phosphorylating IRF3 and activating NF-κB to against the undigested DNA from apoptotic cells [28]. While the involvement of SIX1 in cytokine secretion and immune regulation is yet to be established, the current evidence suggests that an imbalance in the RDGN would create a favorable microenvironment to enhance the survival of cancer cells and their escape from the primary tumor.
Tumor-associated angiogenesis is another crucial step early in the development and growth of most solid neoplasms, and is also necessary for the hematogenous dissemination of cancer cells. Indeed, EYAs have been detected in vascular endothelial cells and enhance sprouting angiogenesis, mainly by their tyrosine-phosphatase activity [63]. This may explain why transgenic Eya knockout mice are characterized by serious defection in the smooth muscle component of the bronchi and pulmonary vessels [64]. Although EYAs promote vascular development, it remains unclear whether they function similarly in endothelial cells and vascular smooth muscle cells.
To invade distant organs, cancer cells must either enter the blood or lymphatic vasculature. The changes in lymphatic vessels can be detected even in early tumor initiation; for instance, the expression of vascular endothelial growth factor C (VEGF-C), a key promoter of lymphangiogenesis, is enhanced at this stage. SIX1 promotes lymphangiogenesis and lymphatic metastasis by upregulating VEGF-C expression [65, 66]. SIX1 transcriptionally activates VEGF-C [65], and overexpression of SIX1 in tumor cells may abrogate the inhibitory effect of TGF-β on lymphangiogenesis by upregulating VEGF-C [66]. Considering that EYA2 is an indispensable coactivator in SIX1-induced TGF-β signal activation [19], EYA2 may participate in promoting lymphangiogenesis in a SIX1-dependent manner.
POTENTIAL STRATEGY OF TARGETING THE RDGN PATHWAY FOR CLINICAL BENEFIT
Successful cancer drugs can be designed against specific molecular targets involved in proliferation, invasion, metastasis and TIC formation [67, 68]. However, discovering novel drug targets is challenging because the inhibition or activation of target genes expressed in normal tissue may result in nonspecific toxicity. Targeting RDGN members like SIX/EYA, which are silent or expressed at low levels in adult tissue but are overexpressed in tumors, is a tremendous opportunity for such anticancer therapy. The tyrosine-phosphatase activity of EYA is significant because it promotes DNA damage repair [26] and tumor growth [31], which may promote relapse from chemotherapy and ionizing radiation. Thus, a combination therapy including highly selective EYA inhibitors would reduce the incidence of chemotherapy resistance. Alternatively, reactivation of DACH1 expression could restrain tumor growth and metastasis [59]. Therefore, therapies co-targeting RDGN members may be more effective and durable cancer treatments than existing treatment options (Figure 2).
DACH1
Reduced expression of DACH1 tightly correlates with poor prognosis in breast cancer, as was first demonstrated in a tissue microarray analysis of over 2,100 samples, and was subsequently supported in an artificial neural network (ANN) approach [33, 69]. Reduced DACH1 expression also correlates with poor prognosis in prostate cancer and lung and hepatocellular carcinoma patients [35, 59, 70]. Using Dach1 gene deletion, Dach1 was shown to inhibit prostate cellular proliferation in vivo [60]. Still, whereas a number of studies have demonstrated that DACH1 profoundly improves the prognosis of cancer patients as a tumor suppressor, a few studies have also observed an oncogenic role of DACH1 in endometrial cancer and mixed lineage leukemia (MLL) [2, 24, 71]. These seemingly disparate findings might indicate that the exact function of DACH1 depends on its communication with specific signaling pathways, especially in the context of organ heterogeneity.
Various studies have explored the potential of restoring DACH1 expression in cancer cells. Epigenetic silencing of the DACH1 promoter region is often responsible for its inactivation and the shift towards a malignant phenotypic [50, 51, 72]. Meanwhile, restoration of DACH1 expression sensitizes gastric and colorectal cancer cells to docetaxel [50, 51] and enhances chemosensitivity to 5-fluorouracil (5-FU) in hepatocellular carcinoma [62]. The next logical step would be to search for a compound that can induce the expression of DACH1 in tumor tissues. Intriguingly, 5-Aza-2’-deoxycytidine (decitabine), a potent demethylating agent, reactivates DACH1 in gastric cancer, suggesting that low-dose decitabine combined with traditional chemotherapy may improve the efficiency of treatments against tumors in which DACH1 expression is silenced [50]. Additionally, ectopic expression of miR-217 directly inhibits DACH1 by binding to its 3’UTR, which not only may exhibit the tumorigenic role of miR-217 in breast cancer, but also suggests an approach for recovering DACH1 expression by diminishing the activation of a particular miRNA [73].
SIX1
The current studies have defined an oncogenic role for SIX family members in the development of diverse tumor types. SIX1 combined with specific transcription factors increases a particular subpopulation of cancer stem/progenitor cells. Furthermore, SIX family members are directly responsible for the malignant behavior of cancer cells [2, 37, 53]. Indeed, SIX1 has been highlighted as an independent prognostic marker in colorectal cancer, and its profile can be used to stratify patients into different risk groups and guide individualized regimens [74]. Accumulation of SIX1 reduces paclitaxel sensitivity in patients undergoing breast cancer chemotherapy [75], and diminishes the therapeutic response to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in ovarian carcinoma [76]. Intriguingly, overexpression of SIX1 reduces p53 abundance and induces resistance to molecular therapy targeting the murine double mimute2 (MDM2)-p53 interaction. Therefore, the expression of SIX1 could be used as a molecular marker to predict which patients would benefit from MDM2 antagonists [40].
Perhaps the most critical question is how to inhibit the transcription of SIX1 in a controllable and predictable manner. Silencing SIX1 expression using short hairpin RNA (shRNA) or miRNA is now viewed as a potential cancer treatment strategy. For instance, miRNA-185 directly reduces SIX1 abundance and thereby sensitizes SIX1-overexpressing cancer cells to TRAIL-induced apoptosis [77]. miR-30b blocks the translation of SIX1 by binding to its 3’-UTR, thereby delaying the progression of colorectal cancer [78]. Epigenetic modifications, including histone acetylation and methylation, can also restrain the oncogenic role of SIX1 [79, 80]. Since DNA methylation inhibitors (e.g., Dacogen, Vidaza) and broad-spectrum HDAC inhibitors (e.g., Vorinostat, Romidepsin) have been successfully developed, inhibiting epigenetic modifications may be an attractive approach for the multi-modal treatment of advanced cancer [81]. However, it is not known whether these small molecule inhibitors of epigenetic enzymes can block the function of SIX1 in vivo.
Although structural and biochemical evidence has confirmed that SIX1 is a DNA-binding transcription factor, the recruitment of additional corepressors or coactivators is necessary for SIX1 signaling [18]. In vertebrates, SIX1 requires EYA for its oncogenic function [19]. Thus, another way to inhibit the aberrant function of SIX1 is by destroying the SIX1/EYA transcriptional complex with a small molecule inhibitor. Since the successful discovery of small molecules like Nutlin-3, which releases p53 from the binding pocket in the MDM2 protein [82], there has been greater hope that effective inhibitors of the SIX1/EYA complex could be generated.
EYA
EYA proteins facilitate tumor progression and are independent prognostic factors in breast and ovarian cancer [31, 83], but not in pancreatic cancer, in which epigenetic silencing of EYA2 increases the cancer invasion capacity [84]. The distinct features of the EYA family, whose members can function as either transcriptional coactivators or tyrosine phosphatases, suggest that EYA expression could be inactivated in different ways. EYA activity could be regulated by multiple signaling pathways. Dr. Li and her coworkers demonstrated that hyperactivation of the canonical Wnt and PI3K/Akt signaling pathways reduces EYA1 ubiquitination and thus prevents its premature degradation [85]. In particular, PI3K/Akt signaling was shown to repress the SUMOylation of EYA1 in a phosphorylation-dependent manner and thus enhance the transcriptional activity of EYA1 [86]. Since the knockdown of SUMO-activating enzyme subunit 2 (SAE2) restrains tumor growth and enhances chemosensitivity, it may be possible to suppress EYA expression by promoting its SUMOylation [87]. Meanwhile, combination regimens with specific pathway blockers (PI3K: LY294002; Akt: Perifosine; Wnt: XAV939) may be effective treatments for EYA1-dependent breast cancer.
Several lines of evidence strongly demonstrate that the function of EYA depends on its phosphatase activity; therefore, inhibition of this activity may be another promising treatment strategy. Designing phosphatase inhibitors is challenging, because it requires not only detailed information about the three-dimensional structure of the protein, but also the identification of a unique catalytic property that can be specifically inhibited. In the EYA X-ray crystal structure, aspartic acid appears to be involved in a metal-dependent reaction, rather than the more common cysteine and arginine residues. Thus, EYA is distinct from the classical tyrosine phosphatases in the HAD family, offering a great chance to identify selective EYA phosphatase inhibitors [27, 88, 89]. Seven novel classes of compounds, which bind to the active site of EYA and chelate the active site Mg2+ ion, were discovered through structure-based virtual screening technology [90]. Using a structure-based de novo design, Dr. Kim’s group further identified 29 inhibitors of EYA2 phosphatase with moderate inhibitory activities (IC50 values from 6 to 50 μM) [91]. While the tyrosine-phosphatase activity of EYA is critical for regulating the chromatin structure, the threonine-phosphatase activity otherwise determines the involvement of EYA in the innate immune response, and could be inhibited by okadaic acid [28]. However, whether the threonine-phosphatase activity of the EYAs promotes cancer initiation and progression remains to be determined.
Another well-recognized inhibitor of the EYA family is Benzbromarone and its derivative, Benzarone, which selectively blocks both EYA2 and EYA3 activity in a non-competitive way [63, 92]. Pharmacological studies and clinical evaluation in gout treatment have provided comprehensive insights into the safety, metabolism, pharmacokinetics and both chronic and acute toxicity of Benzbromarone and Benzarone. Another advantage of Benzbromarone and its derivatives is the anti-angiogenic effect, which may thereby reduce the neovascular-based dissemination of cancer cells [87]. The major metabolite of Benzbromarone, 6-hydroxy Benzbromarone, is a more powerful inhibitor of EYA tyrosine-phosphatase activity, and may block tumor growth by inhibiting angiogenesis [92]. As a result of these studies, there is now a discussion of whether luminal B-type breast cancer patients could benefit from Benzbromarone-based combination therapy due to its suppression of EYA [31].
Recently, a fluorescent high-throughput-screening phosphatase assay identified a series of N-arylidenebenzohydrazide-containing compounds that inhibit the catalytic site of EYA2 compared with other cellular phosphatases in HAD family [93]. These compounds selectively inhibit the phosphatase activity of EYA2 rather than EYA3 [94]. In contrast with those metal-chelating compounds and Benzbromarone, which physiologically perturbs metal ions, N-arylidenebenzohydrazide-containing compounds bind to the allosteric site in a Mg2+-independent manner [89]. Since EYA2-induced migration could be reversed by these compounds, EYA2 phosphatase-specific anti-cancer drugs appear promising. With great technological advancements, like the establishment of high-throughput material screening platforms, it will be possible to find a specific EYA inhibitor for clinical application.
CONCLUSION AND PERSPECTIVES
The RDGN could directly or indirectly determine the biological features and clinical outcomes of various cancer types, so RDGN genes are promising biomarkers. Evaluation of profiles of RDGN members could guide the design of individualized treatment regimens [95]. At the core of this research area are the questions: Which particular cancers are driven by the RDGN? And then, how will it be possible to specifically reactivate the expression of DACH1 or efficiently block the functions of SIX1/EYA? At this time, only a few drugs and small molecules have been shown to counteract the aberrant expression of RDGN members in vitro. Therapeutic agents targeting the RDGN are worthy of development in light of the profound biological effects of these gene products in cancer progression and metastasis.
ACKNOWLEDGMENTs
This work was supported by the National Science Foundation of China (No. 81572608, 81172422, 81502209) and the National Science Foundation of Hubei Province No. WJ2015MA003. This work was supported in part by NIH grants (1R01CA137494, R01CA132115, to R.G.P.), a grant from the Breast Cancer Research Foundation (R.G.P.) and the Dr. Ralph and Marian C. Falk Medical Research Trust (R.G.P.), and a grant from the Pennsylvania Department of Health (R.G.P.).
conflicts of interest
All authors declare no conflicts of interest.
References
1. Blevins MA, Towers CG, Patrick AN, Zhao R, Ford HL. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opin Ther Targets. 2015; 19:213-225.
2. Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H, Zhang C, Yin T, Wu K. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. Int J Cancer. 2016; 138:1067-1075.
3. Kumar JP. The molecular circuitry governing retinal determination. Biochim Biophys Acta. 2009; 1789:306-314.
4. Zhou J, Wang C, Wang Z, Dampier W, Wu K, Casimiro MC, Chepelev I, Popov VM, Quong A, Tozeren A, Zhao K, Lisanti MP, Pestell RG. Attenuation of Forkhead signaling by the retinal determination factor DACH1. Proc Natl Acad Sci U S A. 2010; 107:6864-6869.
5. Zhou J, Liu Y, Zhang W, Popov VM, Wang M, Pattabiraman N, Sune C, Cvekl A, Wu K, Jiang J, Wang C, Pestell RG. Transcription elongation regulator 1 is a co-integrator of the cell fate determination factor Dachshund homolog 1. J Biol Chem. 2010; 285:40342-40350.
6. Popov VM, Zhou J, Shirley LA, Quong J, Yeow WS, Wright JA, Wu K, Rui H, Vadlamudi RK, Jiang J, Kumar R, Wang C, Pestell RG. The cell fate determination factor DACH1 is expressed in estrogen receptor-alpha-positive breast cancer and represses estrogen receptor-alpha signaling. Cancer Res. 2009; 69:5752-5760.
7. Wu K, Liu M, Li A, Donninger H, Rao M, Jiao X, Lisanti MP, Cvekl A, Birrer M, Pestell RG. Cell fate determination factor DACH1 inhibits c-Jun-induced contact-independent growth. Mol Biol Cell. 2007; 18:755-767.
8. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, Rosenfeld MG. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003; 426:247-254.
9. Ikeda K, Watanabe Y, Ohto H, Kawakami K. Molecular interaction and synergistic activation of a promoter by Six, Eya, and Dach proteins mediated through CREB binding protein. Mol Cell Biol. 2002; 22:6759-6766.
10. Horner A, Shum L, Ayres JA, Nonaka K, Nuckolls GH. Fibroblast growth factor signaling regulates Dach1 expression during skeletal development. Dev Dyn. 2002; 225:35-45.
11. Klassen H, Kiilgaard JF, Zahir T, Ziaeian B, Kirov I, Scherfig E, Warfvinge K, Young MJ. Progenitor cells from the porcine neural retina express photoreceptor markers after transplantation to the subretinal space of allorecipients. Stem Cells. 2007; 25:1222-1230.
12. Kalousova A, Mavropoulos A, Adams BA, Nekrep N, Li Z, Krauss S, Stainier DY, German MS. Dachshund homologues play a conserved role in islet cell development. Dev Biol. 2010; 348:143-152.
13. Schild R, Knüppel T, Konrad M, Bergmann C, Trautmann A, Kemper MJ, Wu K, Yaklichkin S, Wang J, Pestell R, Müller-Wiefel DE, Schaefer F, Weber S. Double homozygous missense mutations in DACH1 and BMP4 in a patient with bilateral cystic renal dysplasia. Nephrol Dial Transplant. 2013; 28:227-232.
14. Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, O’Connell JR, Li M, Schmidt H, Tanaka T, Isaacs A, Ketkar S, et al. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010; 42:376-384.
15. Ma RCW, Lee HM, Lam VKL, Tam CHT, Ho JSK, Zhao H-L, Guan J, Kong APS, Lau E, Zhang G, Luk A, Wang Y, Tsui SKW, Chan TF, Hu C, Jia WP, et al. Familial young-onset diabetes, pre-diabetes and cardiovascular disease are associated with genetic variants of DACH1 in Chinese. PloS One. 2014; 9: e84770.
16. Zhou J, Shaikh LH, Neogi SG, McFarlane I, Zhao W, Figg N, Brighton CA, Maniero C, Teo AED, Azizan EAB, Brown MJ. DACH1, a zona glomerulosa selective gene in the human adrenal, activates transforming growth factor-β signaling and suppresses aldosterone secretion. Hypertension. 2015; 65:1103-1110.
17. Wu W, Ren Z, Li P, Yu D, Chen J, Huang R, Liu H. Six1: A critical transcription factor in tumorigenesis. Int J Cancer. 2015;136:1245-1253.
18. Zou D, Silvius D, Fritzsch B, Xu PX. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development. 2004; 131:5561-5572.
19. Patrick AN, Cabrera JH, Smith AL, Chen XS, Ford HL, Zhao R. Structure-function analyses of the human SIX1-EYA2 complex reveal insights into metastasis and BOR syndrome. Nat Struct Mol Biol. 2013; 20:447-453.
20. Hu S, Mamedova A, Hegde RS. DNA-binding and regulation mechanisms of the SIX family of retinal determination proteins. Biochemistry. 2008; 47:3586-3594.
21. Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M, Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet. 1997; 15:157-164.
22. Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM, Jr., Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci USA. 2004; 101:8090-8095.
23. Lu K, Reddy R, Berika M, Warburton D, El-Hashash AH. Abrogation of Eya1/Six1 disrupts the saccular phase of lung morphogenesis and causes remodeling. Dev Biol. 2013; 382:110-123.
24. Lee SH, Kim J, Ryu JY, Lee S, Yang DK, Jeong D, Kim J, Lee SH, Kim JM, Hajjar RJ, Park WJ. Transcription coactivator Eya2 is a critical regulator of physiological hypertrophy. J Mol Cell Cardiol. 2012; 52:718-726.
25. Jin M, Mardon G. Distinct Biochemical Activities of Eyes absent During Drosophila Eye Development. Sci Rep. 2016; 6:23228.
26. Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009; 458:591-596.
27. Krishnan N, Jeong DG, Jung S-K, Ryu SE, Xiao A, Allis CD, Kim SJ, Tonks NK. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J Biol Chem. 2009; 284:16066-16070.
28. Okabe Y, Sano T, Nagata S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature. 2009; 460:520-524.
29. El-Hashash AH, Turcatel G, Varma S, Berika M, Al Alam D, Warburton D. Eya1 protein phosphatase regulates tight junction formation in lung distal epithelium. J Cell Sci. 2012; 125:4036-4048.
30. Yuan B, Cheng L, Chiang HC, Xu X, Han Y, Su H, Wang L, Zhang B, Lin J, Li X, Xie X, Wang T, Tekmal RR, Curiel TJ, Yuan ZM, Elledge R, et al. A phosphotyrosine switch determines the antitumor activity of ERβ. J Clin Invest. 2014; 124:3378-3390.
31. Wu K, Li Z, Cai S, Tian L, Chen K, Wang J, Hu J, Sun Y, Li X, Ertel A, Pestell RG. EYA1 phosphatase function is essential to drive breast cancer cell proliferation through cyclin D1. Cancer Res. 2013; 73:4488-4499.
32. Schulenburg A, Blatt K, Cerny-Reiterer S, Sadovnik I, Herrmann H, Marian B, Grunt TW, Zielinski CC, Valent P. Cancer stem cells in basic science and in translational oncology: can we translate into clinical application? J Hematol Oncol. 2015; 8:16.
33. Wu K, Li A, Rao M, Liu M, Dailey V, Yang Y, Vizio DD, Wang C, Lisanti MP, Sauter G, Russell RG, Cvekl A, Pestell RG. DACH1 Is a Cell Fate Determination Factor That Inhibits Cyclin D1 and Breast Tumor Growth. Mol Cell Biol. 2006; 26:7116-7129.
34. Chu Q, Han N, Yuan X, Nie X, Wu H, Chen Y, Guo M, Yu S, Wu K. DACH1 inhibits cyclin D1 expression, cellular proliferation and tumor growth of renal cancer cells. J Hematol Oncol. 2014; 7:73.
35. Liu Y, Zhou R, Yuan X, Han N, Zhou S, Xu H, Guo M, Yu S, Zhang C, Yin T, Wu K. DACH1 is a novel predictive and prognostic biomarker in hepatocellular carcinoma as a negative regulator of Wnt/β-catenin signaling. Oncotarget. 2015; 6:8621-8634. doi: 10.18632/oncotarget.3281.
36. Yu Y, Davicioni E, Triche TJ, Merlino G. The homeoprotein six1 transcriptionally activates multiple protumorigenic genes but requires ezrin to promote metastasis. Cancer Res. 2006; 66:1982-1989.
37. Coletta RD, Christensen KL, Micalizzi DS, Jedlicka P, Varella-Garcia M, Ford HL. Six1 overexpression in mammary cells induces genomic instability and is sufficient for malignant transformation. Cancer Res. 2008; 68:2204-2213.
38. Chen K, Wu K, Gormley M, Ertel A, Wang J, Zhang W, Zhou J, Disante G, Li Z, Rui H, Quong AA, McMahon SB, Deng H, Lisanti MP, Wang C, Pestell RG. Acetylation of the cell-fate factor dachshund determines p53 binding and signaling modules in breast cancer. Oncotarget. 2013; 4:923-935. doi: 10.18632/oncotarget.1094.
39. Chen K, Wu K, Cai S, Zhang W, Zhou J, Wang J, Ertel A, Li Z, Rui H, Quong A, Lisanti MP, Tozeren A, Tanes C, Addya S, Gormley M, Wang C, et al. Dachshund binds p53 to block the growth of lung adenocarcinoma cells. Cancer Res. 2013; 73:3262-3274.
40. Towers CG, Guarnieri AL, Micalizzi DS, Harrell JC, Gillen AE, Kim J, Wang CA, Oliphant MU, Drasin DJ, Guney MA, Kabos P, Sartorius CA, Tan AC, Perou CM, Espinosa JM, Ford HL. The Six1 oncoprotein downregulates p53 via concomitant regulation of RPL26 and microRNA-27a-3p. Nat Commun. 2015; 6:10077.
41. Velasco-Velazquez MA, Homsi N, De La Fuente M, Pestell RG. Breast cancer stem cells. Int J Biochem Cell Biol. 2012; 44:573-577.
42. Wu K, Jiao X, Li Z, Katiyar S, Casimiro MC, Yang W, Zhang Q, Willmarth NE, Chepelev I, Crosariol M, Wei Z, Hu J, Zhao K, Pestell RG. Cell fate determination factor Dachshund reprograms breast cancer stem cell function. J Biol Chem. 2011; 286:2132-2142.
43. Iwanaga R, Wang CA, Micalizzi DS, Harrell JC, Jedlicka P, Sartorius CA, Kabos P, Farabaugh SM, Bradford AP, Ford HL. Expression of Six1 in luminal breast cancers predicts poor prognosis and promotes increases in tumor initiating cells by activation of extracellular signal-regulated kinase and transforming growth factor-beta signaling pathways. Breast Cancer Res. 2012; 14:R100. doi: 10.1186/bcr3219.
44. Xu H, Zhang Y, Altomare D, Pena MM, Wan F, Pirisi L, Creek KE. Six1 promotes epithelial-mesenchymal transition and malignant conversion in human papillomavirus type 16-immortalized human keratinocytes. Carcinogenesis. 2014; 35:1379-1388.
45. Micalizzi DS, Christensen KL, Jedlicka P, Coletta RD, Barón AE, Harrell JC, Horwitz KB, Billheimer D, Heichman KA, Welm AL, Schiemann WP, Ford HL. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-beta signaling. J Clin Invest. 2009; 119:2678-2690.
46. Guo F, Parker Kerrigan BC, Yang D, Hu L, Shmulevich I, Sood AK, Xue F, Zhang W. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J Hematol Oncol. 2014; 7:19
47. Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S, Chen Y, Wu K. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014; 7:87.
48. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012; 13:616-630.
49. Wu K, Yang Y, Wang C, Davoli MA, D’Amico M, Li A, Cveklova K, Kozmik Z, Lisanti MP, Russell RG, Cvekl A, Pestell RG. DACH1 inhibits transforming growth factor-beta signaling through binding Smad4. J Biol Chem. 2003; 278:51673-51684.
50. Yan W, Wu K, Herman JG, Brock MV, Zhou Y, Lu Y, Zhang Z, Yang Y, Guo M. Epigenetic silencing of DACH1 induces the invasion and metastasis of gastric cancer by activating TGF-β signalling. J Cell Mol Med. 2014; 18:2499-2511.
51. Yan W, Wu K, Herman JG, Brock MV, Fuks F, Yang L, Zhu H, Li Y, Yang Y, Guo M. Epigenetic regulation of DACH1, a novel Wnt signaling component in colorectal cancer. Epigenetics. 2013; 8:1373-1383.
52. Micalizzi DS, Wang C-A, Farabaugh SM, Schiemann WP, Ford HL. Homeoprotein Six1 increases TGF-beta type I receptor and converts TGF-beta signaling from suppressive to supportive for tumor growth. Cancer Res. 2010; 70:10371-10380.
53. McCoy EL1, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, Welm AL, Ford HL. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest. 2009; 119:2663-2670.
54. Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-β signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene. 2012; 31:552-562.
55. Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J, Wang J, Li Z, Addya S, Sorensen PH, Lisanti MP, Quong A, Ertel A, Pestell RG. Cell Fate Factor DACH1 Represses YB-1-Mediated Oncogenic Transcription and Translation. Cancer Res. 2014; 74:829-839.
56. Zhao F, Wang M, Li S, Bai X, Bi H, Liu Y, Ao X, Jia Z, Wu H. DACH1 inhibits SNAI1-mediated epithelial-mesenchymal transition and represses breast carcinoma metastasis. Oncogenesis. 2015; 4:e143.
57. Ono H, Imoto I, Kozaki K, Tsuda H, Matsui T, Kurasawa Y, Muramatsu T, Sugihara K, Inazawa J. SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. 2012; 31:4923-4934.
58. Pandey RN, Rani R, Yeo EJ, Spencer M, Hu S, Lang RA, Hegde RS. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene. 2010; 29:3715-3722.
59. Wu K, Katiyar S, Li A, Liu M, Ju X, Popov VM, Jiao X, Lisanti MP, Casola A, Pestell RG. Dachshund inhibits oncogene-induced breast cancer cellular migration and invasion through suppression of interleukin-8. Proc Natl Acad Sci U S A. 2008; 105:6924-6929.
60. Chen K, Wu K, Jiao X, Wang L, Ju X, Wang M, Di Sante G, Xu S, Wang Q, Li K, Sun X, Xu C, Li Z, Casimiro MC, Ertel A, Addya S, et al. The Endogenous Cell-Fate Factor Dachshund Restrains Prostate Epithelial Cell Migration via Repression of Cytokine Secretion via a CXCL Signaling Module. Cancer Res. 2015; 75:1992-2004.
61. Han N, Yuan X, Wu H, Xu H, Chu Q, Guo M, Yu S, Chen Y, Wu K. DACH1 inhibits lung adenocarcinoma invasion and tumor growth by repressing CXCL5 signaling. Oncotarget. 2015; 6:5877-5888. doi: 10.18632/oncotarget.3463.
62. Watanabe A, Ogiwara H, Ehata S, Mukasa A, Ishikawa S, Maeda D, Ueki K, Ino Y, Todo T, Yamada Y, Fukayama M, Saito N, Miyazono K, Aburatani H. Homozygously deleted gene DACH1 regulates tumor-initiating activity of glioma cells. Proc Natl Acad Sci U S A. 2011; 108:12384-12389.
63. Tadjuidje E, Wang TS, Pandey RN, Sumanas S, Lang RA, Hegde RS. The EYA tyrosine phosphatase activity is pro-angiogenic and is inhibited by benzbromarone. PloS One. 2012; 7:e34806.
64. El-Hashash AH, Al Alam D, Turcatel G, Bellusci S, Warburton D. Eyes absent 1 (Eya1) is a critical coordinator of epithelial, mesenchymal and vascular morphogenesis in the mammalian lung. Dev Biol. 2011; 350:112-126.
65. Wang CA, Jedlicka P, Patrick AN, Micalizzi DS, Lemmer KC, Deitsch E, Casás-Selves M, Harrell JC, Ford HL. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012; 122:1895-1906.
66. Liu D, Li L, Zhang XX, Wan DY, Xi BX, Hu Z, Ding WC, Zhu D, Wang XL, Wang W, Feng ZH, Wang H, Ma D and Gao QL. SIX1 promotes tumor lymphangiogenesis by coordinating TGFβ signals that increase expression of VEGF-C. Cancer Res. 2014; 74:5597-5607.
67. Velasco-Velazquez M, Xolalpa W, Pestell RG. The potential to target CCL5/CCR5 in breast cancer. Expert Opin Ther Targets. 2014; 18:1265-1275.
68. Casimiro MC, Velasco-Velazquez M, Aguirre-Alvarado C, Pestell RG. Overview of cyclins D1 function in cancer and the CDK inhibitor landscape: past and present. Expert Opin Investig Drugs. 2014; 23:295-304.
69. Powe DG, Dhondalay GK, Lemetre C, Allen T, Habashy HO, Ellis IO, Rees R, Ball GR. DACH1: its role as a classifier of long term good prognosis in luminal breast cancer. PloS One. 2014; 9:e84428.
70. Wu K, Katiyar S, Witkiewicz A, Li A, McCue P, Song LN, Tian L, Jin M, Pestell RG. The cell fate determination factor dachshund inhibits androgen receptor signaling and prostate cancer cellular growth. Cancer Res. 2009; 69:3347-3355.
71. Nan F, Lü Q, Zhou J, Cheng L, Popov VM, Wei S, Kong B, Pestell RG, Lisanti MP, Jiang J, Wang C. Altered expression of DACH1 and cyclin D1 in endometrial cancer. Cancer Biol Ther. 2009; 8:1534-1539.
72. Zhu H, Wu K, Yan W, Hu L, Yuan J, Dong Y, Li Y, Jing K, Yang Y, Guo M. Epigenetic silencing of DACH1 induces loss of transforming growth factor-beta1 antiproliferative response in human hepatocellular carcinoma. Hepatology. 2013; 58:2012-2022.
73. Zhang Q, Yuan Y, Cui J, Xiao T, Jiang D. MiR-217 Promotes Tumor Proliferation in Breast Cancer via Targeting DACH1. J Cancer. 2015; 6:184-191.
74. Kahlert C, Lerbs T, Pecqueux M, Herpel E, Hoffmeister M, Jansen L, Brenner H, Chang-Claude J, Blaker H, Kloor M, Roth W, Pilarsky C, Rahbari NN, Scholch S, Bork U, Reissfelder C, et al. Overexpression of SIX1 is an independent prognostic marker in stage I-III colorectal cancer. Int J Cancer. 2015; 137:2104-2113.
75. Li Z, Tian T, Hu X, Zhang X, Nan F, Chang Y, Lv F, Zhang M. Six1 mediates resistance to paclitaxel in breast cancer cells. Biochem Biophys Res Commun. 2013; 441:538-543.
76. Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, Ford HL. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007; 67:3036-3042.
77. Imam JS, Buddavarapu K, Lee-Chang JS, Ganapathy S, Camosy C, Chen Y, Rao MK. MicroRNA-185 suppresses tumor growth and progression by targeting the Six1 oncogene in human cancers. Oncogene. 2010; 29:4971-4979.
78. Zhao H, Xu Z, Qin H, Gao Z, Gao L. MiR-30b regulates migration and invasion of human colorectal cancer via SIX1. Biochem J. 2014; 460:117-125.
79. Wu W, Ren Z, Liu H, Wang L, Huang R, Chen J, Zhang L, Li P, Xiong Y. Core promoter analysis of porcine Six1 gene and its regulation of the promoter activity by CpG methylation. Gene. 2013; 529:238-244.
80. Feng GW, Dong LD, Shang WJ, Pang XL, Li JF, Liu L, Wang Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. Eur Rev Med Pharmacol Sci. 2014; 18:811-816.
81. Campbell RM, Tummino PJ. Cancer epigenetics drug discovery and development: the challenge of hitting the mark. J Clin Invest. 2014; 124:64-69.
82. Saha MN, Qiu L, Chang H. Targeting p53 by small molecules in hematological malignancies. J Hematol Oncol. 2013; 6:23.
83. Zhang L, Yang N, Huang J, Buckanovich RJ, Liang S, Barchetti A, Vezzani C, O’Brien-Jenkins A, Wang J, Ward MR, Courreges MC, Fracchioli S, Medina A, Katsaros D, Weber BL, Coukos G. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005; 65:925-932.
84. Vincent A, Hong SM, Hu C, Omura N, Young A, Kim H, Yu J, Knight S, Ayars M, Griffith M, Van Seuningen I, Maitra A, Goggins M. Epigenetic silencing of EYA2 in pancreatic adenocarcinomas promotes tumor growth. Oncotarget. 2014; 5:2575-2587. doi: 10.18632/oncotarget.1842.
85. Sun Y, Li X. The canonical wnt signal restricts the glycogen synthase kinase 3/fbw7-dependent ubiquitination and degradation of eya1 phosphatase. Mol Cell Biol. 2014; 34:2409-2417.
86. Sun Y, Kaneko S, Li XK, Li X. The PI3K/Akt signal hyperactivates Eya1 via the SUMOylation pathway. Oncogene. 2015; 34:2527-2537.
87. Liu X, Xu Y, Pang Z, Guo F, Qin Q, Yin T, Sang Y, Feng C, Li X, Jiang L, Shu P, Wang Y. Knockdown of SUMO-activating enzyme subunit 2 (SAE2) suppresses cancer malignancy and enhances chemotherapy sensitivity in small cell lung cancer. J Hematol Oncol. 2015; 8:67.
88. Rayapureddi JP, Kattamuri C, Steinmetz BD, Frankfort BJ, Ostrin EJ, Mardon G, Hegde RS. Eyes absent represents a class of protein tyrosine phosphatases. Nature. 2003; 426:295-298.
89. Jung SK, Jeong DG, Chung SJ, Kim JH, Park BC, Tonks NK, Ryu SE, Kim SJ. Crystal structure of ED-Eya2: insight into dual roles as a protein tyrosine phosphatase and a transcription factor. FASEB J. 2010; 24:560-569.
90. Park H, Jung SK, Yu KR, Kim JH, Kim YS, Ko JH, Park BC, Kim SJ. Structure-based virtual screening approach to the discovery of novel inhibitors of eyes absent 2 phosphatase with various metal chelating moieties. Chem Biol Drug Des. 2011; 78:642-650.
91. Park H, Ryu SE, Kim SJ. Structure-based de novo design of Eya2 phosphatase inhibitors. J Mol Graph Model. 2012; 38:382-388.
92. Pandey RN, Wang TS, Tadjuidje E, McDonald MG, Rettie AE, Hegde RS. Structure-activity relationships of benzbromarone metabolites and derivatives as EYA inhibitory anti-angiogenic agents. PloS One. 2013; 8:e84582.
93. Krueger AB, Dehdashti SJ, Southall N, Marugan JJ, Ferrer M, Li X, Ford HL, Zheng W, Zhao R. Identification of a selective small-molecule inhibitor series targeting the eyes absent 2 (Eya2) phosphatase activity. J Biomol Screen. 2013; 18:85-96.
94. Krueger AB, Drasin DJ, Lea WA, Patrick AN, Patnaik S, Backos DS, Matheson CJ, Hu X, Barnaeva E, Holliday MJ, Blevins MA, Robin TP, Eisenmesser EZ, Ferrer M, Simeonov A, Southall N, et al. Allosteric inhibitors of the Eya2 phosphatase are selective and inhibit Eya2-mediated cell migration. J Biol Chem. 2014; 289:16349-16361.
95. Smith A, Roda D, Yap T. Strategies for modern biomarker and drug development in oncology. J Hematol Oncol. 2014; 7:70.