
Introduction
Inflammation is recognized as a major hallmark of cancer. As early as 1863, Rudolph Virchow speculated on a link between cancer and inflammation based on the observation of leukocyte infiltration in human breast cancer [1, 2]. It is generally accepted that up to 25% of malignancies are related to chronic inflammation, chronic infection, or both [3-5]. Numerous studies provide evidence that chronic inflammation facilitates resistance to growth inhibition, independent neoangiogenesis, apoptotic evasion, malignant transformation, and metastatic potential obtainment [6]. During tumor initiation, oxidative molecules including reactive oxygen species and reactive nitrogen species induced by tumor infiltrating immune cells induce epigenetic alterations in oncogenes or tumor suppressive genes, thereby promoting carcinogenesis[7-9]. On the other hand, during tumor progression and metastasis, cytokines or chemokines secreted by immune cells lead to an increase in cell survival, motility and invasiveness, such as epithelial-mesenchymal transition (EMT) [10-12]. Elucidating the molecular network between inflammation and cancer risk is of great significance for cancer prevention and treatment.
Once invaded by harmful microbes or foreign particles, germline-encoded pattern recognition receptors (PRRs) constitute the first line of defense. The PRR superfamily includes members of the Toll-like receptors (TLR), nucleotide-binding and oligomerization domain containing receptors (NOD-like receptors, NLRs), retinoic acid-inducible gene (RIG) I-like RNA helicases, C-type lectins, and AIMs like receptors (ALR) [13-15]. The molecular targets of PRRs usually include pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs). Binding of PAMPs or DAMPs to these receptors leads to an initiation of the host’s immune response by activation of inflammatory cells and a number of transcription factors such as NF-κB, STAT, and FOXO [16, 17]. The multimeric inflammasome complex senses all these processes.
Jurg Tschopp was the first to identify the inflammasome in 2002 [18]. Its structure consists of an assembly, either of the NLR proteins, NLRP1, NLRP3, NLRC4, NLRP6, and NAIP5 or the DNA-sensing complex of AIM2, a member of the interferon-inducible HIN-200 protein family [19]. Activation of inflammasome leads to NLR oligomerization and subsequent interaction with the adaptor protein ASC and the CARD domain of caspase-1. Caspase-1 in turn, regulates the maturation of proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 or the rapid inflammatory form of cell death called pyroptosis [20-22]. Notably, the level of IL-1β and IL-18 were found to be significantly elevated in various types of malignancies. These cytokines can facilitate pro-carcinogenic activity by triggering the secretion of VEGF, FGF2 and STAT3, and subsequently support cancer survival and distant metastasis [23-25]. Therefore, elucidating the molecular network of inflammasomes has become a novel strategy for cancer prevention research.
Inflammasome cascade signaling
Compared to TLRs that are usually located on the membrane, NLRs are intracellular molecules and classified into 22 and 34 isoforms in human and mouse genome, respectively. The NLRs are characterized by a tripartite structure, consisting of a carboxy-terminal leucine-rich repeat domain, a central nucleotide-binding oligomerization domain, and a variable N-terminal protein-protein interaction domain, which can be either a Pyrin domain (PYD), a caspase recruitment and activation domain (CARD), or a baculovirus inhibitor of apoptosis repeat domain (BIR) (Figure 1) [26, 27]. The common NLRs and their functions and ligands are summarized in Table 1. The leucine-rich repeat domain appears to act as a ligand sensing component of NLRs; however, the molecular basis of ligand-binding mechanisms of NLRs is poorly understood [28]. The nucleotide-binding oligomerization domain facilitates recruitment of pro-caspase-1 via interactions between pro-caspase-1 and adaptor protein ASC, which take place in the CARD domain [29]. The PYD domain of ASC interacts with NLRs and its CARD domain binds directly with pro-caspase-1. Once pro-caspase-1 is recruited to the inflammasome, it will be cleaved into a p35 and p10 fragments in a proximity-induced multimerization manner. The p35 fragment will subsequently be processed into the CARD and a p20 subunit. The p10 fragment together with 2 molecules of p20 will finally form an active caspase-1 enzyme, which converts pro- IL-1βand pro-IL-18 into their active forms (Figure 2) [30, 31]. Furthermore, pyroptosis could also be induced following caspase-1 activation and its activation is considered to be a critical mechanism fighting against Gram-negative and Gram-positive bacteria [32].
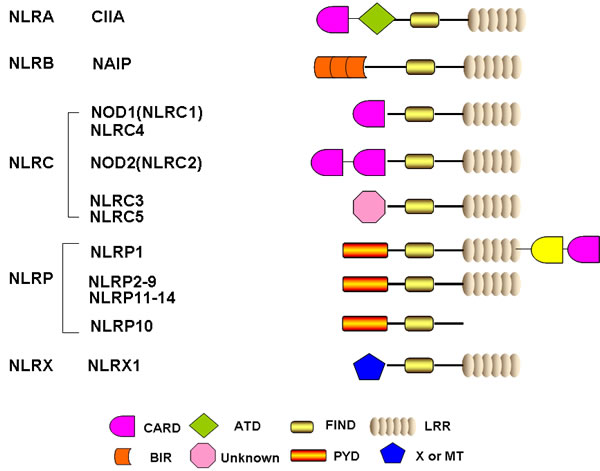
Figure 1: Schematic representation of the basic structure of individual NLR domain. Human NLRs were classified into five categories including NLRA, NLRB, NLRC, NLRP and NLRX. All 22 human NLRs contain a central NACHT domain and a C-terminal ligand sensing domain LRR, with the exception of NLRP10. The N-terminal domain of each NLR is specific and responsible for ascribing different biofunctions. CARD: caspase association and recruitment domain; ATD: acidic transactivation domain; FIND: function to find domain; PYD: pyrin domain; BIR: Baculoviral inhibition of apoptosis protein repeat domain; LRR: leucine-rich repeats; MT: targets NLRX1 to the mitochondria but no sequence homology with tranditional mitochondrial targeting sequence has been reported.
Table 1: Inflammasome and non-inflammasome forming NLRs, functions and their ligands
NLRs |
NLR family |
functions |
Ligands |
NLRP3 |
NLRP |
Interacts with caspase-1 and ASC; activates NF-κB signaling and IL-1β /IL-18 release |
Muramyl dipeptide, LPS, Bacterial and viral DNA/RNA, silica, amyloid-β fibrils, extracellular ATP |
NLRC4 |
LRC |
Interacts with caspase-1, ASC and NAIP; elevates NF-κB signaling and IL-1β /IL-18 release |
Flagellin from Salmonella, Legionella, Listeria, Pseudomonas |
NAIP |
NLRB |
Formation of NAIP/NLRC4 inflammasome complex |
Flagellin from Legionella |
NLRP6 |
NLRP |
Inflammasome complex formation with ASC and caspase-1; activates NF-κB signaling and IL-1β /IL-18 release |
Ligands unknown |
NLRP1 |
NLRP |
Inflammasome complex formation with ASC and caspase-1 |
Muramyl dipeptide, Toxoplasma gondii and Bacillus anthracis lethal toxin |
NLRP12 |
NLRP |
Inhibits IRAK1, TRAF3 and NIK, attenuates both canonical and non-canonical NF-κB signaling |
Ligands unknown |
NLRX1 |
NLRX |
Inhibits TRAF6 and attenuates canonical NF-κB signaling |
Viral RNA |
NLRC3 |
NLRC |
Inhibits TRAF6 and attenuates canonical NF-κB signaling |
Ligands unknown |
NOD1 |
NLRC |
Interacts with RIP2 and recruits RICK and CARD9 |
GM-tripeptide γ-d-Glu-DAP(iEDAP) d-lactyl-l-Ala-γ-Glu-meso-DAP-Gly (FK156) heptanolyl-γ-Glu-meso-DAP-Ala |
NOD2 |
NLRC |
Recruits RIP2 and activates NF-κB and MAPK pathways; negatively regulated by CARD8 |
Muramyl dipeptide MurNAc-l-Ala-g-d-Glu-l-Lys (M-TRlys) |
Alternatively, inflammasome could also be activated through a non-canonical pathway, which involves caspase-11 or caspase-8. Caspase-11 was found to be necessary for the maturation of IL-1β and IL-18 in enteric bacteria such as Escherichia coli, Citrobacter rodentium, and Vibrio cholera. After recruitment to the inflammasome, pro-caspase-11 is cleaved into the p26 subunit and subsequently interacts with caspase-1 [33]. Studies show that caspase-8 was necessary for the inflammasome activation in LPS-primed macrophages and dendritic cells [34]; however, the detailed interaction mode and mechanisms are poorly understood.
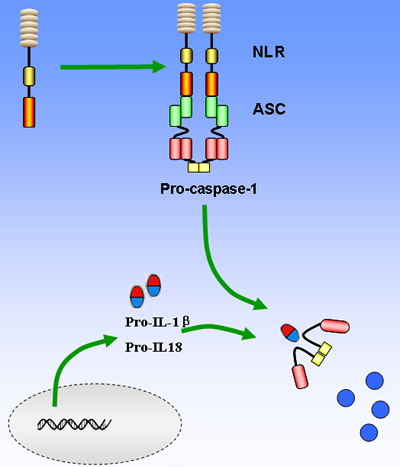
Figure 2: Basic mechanisms of activation of the main NLRs inflammasome. The recognition of PAMPs and/or DAMPs leads to NOD domain oligomerization, which in turn facilitates recruitment of pro-caspase-1 via the CARD domain interactions between pro-caspase-1 and adaptor protein ASC. Pro-caspase-1 will be then cleaved and converts pro- IL-1βand pro-IL-18 into their active forms to amplify the inflammatory response. On the other hand, caspase-1 can lead to cell pyropotosis with the consequence of membrane rupture and release of alarmins such as IL-1α and HMGB1.
Regulation of inflammasome activation
Given that IL-1β, IL-18 and pyroptotic death response have the potential to damage the host, tight control of inflammasome activation is of great significance for the prevention of disease progression. According to the 2-signal model of inflammasome induction, NF-κB is thought to serve as the first signal that primes NLR and pro-IL-1β expression [17, 22, 35]. Constitutive activation of NF-κB is shown in a wide variety of tumor types, such as lymphoma, liver cancer, lung cancer, breast cancer, etc [36, 37]. Besides, NF-κB is activated in response to carcinogenic processes such as tobacco, stress, obesity, alcohol, infectious agents, irradiation, etc [36, 37]. Furthermore, NF-κB controls the expression of the genes linked with proliferation, invasion, angiogenesis, and metastasis of cancer [38]. Besides, NF-κB activation further upregulates a series of inflammatory factors, such as TNFα, IL-6, IL-1 and IL-8, which constitute a positive feedback loop to induce cellular and DNA damage and to promote cell proliferation and transformation [36]. A previous study also demonstrated that NLRP3 promoter contains putative NF-κB binding site and NF-κB inhibition resulted in a significant reduction of NLRP3 expression [39]. Meanwhile, mounting evidence suggested that NLRP3 inflammasome formation is positively associated with NF-κB activity following drug treatment such as LPS, CPT-11, FGF-21, etc [40-42]. All these findings implied that the pro-tumorigenic ability of NF-κB might be attributed to inflammasome activation.
Similar to NF-κB, type I interferon is also important for inflammasome activation. AIM2 inflammasome activation following F.tularensis infection requires type I interferon stimuli, whereas macrophages deficient in the type I interferon secretion, result in reduced response of AIM2 inflammasome [43-45]. Although the precise mechanism of interferon signaling remains unclear, it has been proposed that type I interferon activates AIM2 inflammasome by generating cytosolic DNA from F.tularensis [44, 45]. However, type I interferon is also reported to inhibit inflammasome activation by two distinct mechanisms including the alteration of intracellular pro-IL-1β concentration and inhibiting caspase-1 activation [46]. The reduction of pro-IL-1β is determined by the capacity of type I interferon to induce the production of the anti-inflammatory cytokine, IL-10. IL-10 activation by STAT3 signaling pathway can inhibit the synthesis of pro-IL-1β and pro-IL-1α [47]. In addition, type I interferon is capable of suppressing caspase-1 activity by activation of the transcription factor, STAT1, subsequently inhibiting NLRP3 and NLRP1 inflammasome (Figure 3) [48]. Both in vitro and in vivo experiments further confirmed that IFN-β could suppress NLRP3 inflammasome, but the exact molecular mechanism that guides the preferential targeting of NLRP3 and NLRP1 inflammasome by type I interferon remains to be identified [49]. These data provide a duplex role of type I interferon in inflammasome modulation, which might be dependent on infected organisms or cell type and inflammation status. Besides the crosstalk between cytokines and inflammasomes, recent studies also suggest that the effector and memory T cells can block the activation of caspase-1 and IL-1β in macrophages and dendritic cells, mediated by CD40L, OX40L, and RANKL, which are all members of the TNF superfamily of ligands expressed on activated cells [50, 51]. Interestingly, although it is recognized that the T cells only target NLRP1 and NLRP3 inflammasomes, the underlying molecular mechanisms of how TNF ligands mediate the inhibition of caspase-1-dependent production of IL-1β is unknown and needs further investigation.
Evolutionarily, autophagy is a cell-protective mechanism against harmful stress that facilitates catabolic processes and inhibits anabolic metabolism. Growing evidence indicates that autophagy is a critical process participating in cancer initiation and metastasis, growth, and drug resistance [52, 53]. Intriguingly, recent reports have also indicated that autophagy regulates various aspects of the immune response, such as antigen presentation, cell death, and cytokine secretion in immune cells [54]. In autophagy-deficient Atg16-/- mice, it was observed that the levels of IL-1β and IL-18 were significantly elevated following LPS treatment. However, the elevated IL-1β and IL-18 expression was not due to enhanced transcriptional activity, but instead was attributed to over-activation of caspase-1 [55]. Subsequent mechanistic studies demonstrated that the augmented caspase-1 activity might be due to the failure of autophagy-deficient cells to clear damaged mitochondrion [55]. When autophagy is inhibited, excessive reactive oxygen species (ROS) will accumulate in the damaged mitochondrion, resulting in the release of mitochondrion DNA into the cytoplasm that finally triggers activation of NLRP3 inflammasome (Figure 3) [56, 57]. However, detailed mechanisms accounting for how mitochondrial ROS or mitochondrial DNA activates inflammasome are still unclear. Meanwhile, an inflammasome-independent mechanism of autophagy-mediated regulation of IL-1β expression was recently identified. Autophagosome could degrade pro-IL-1β, thereby restraining the substrate for caspase-1 processing (Figure 3) [58, 59]. Alternatively, autophagy inhibition could also activate the transcription of pro-IL-1β in human peripheral blood mononuclear cells [59]. Recently, autophagy was reported to affect inflammasome activity by influencing IL-1β translocation from the endoplasmic reticulum and Golgi apparatus [60]. Lastly, autophagy machinery is believed to participate in clearing large inflammasome complexes from cells in order to prevent excessive cell damage by IL-1β and IL-18 [61]. Therefore, the autophagic process regulates inflammasome activation at several levels.
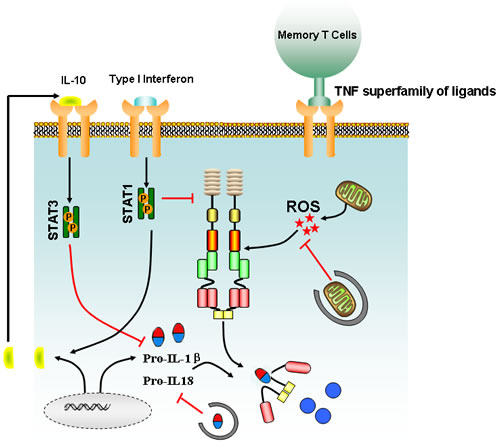
Figure 3: Main signaling involved in the regulation of inflammasome activation. Type I interferon signaling triggers the production of IL-10, which in turn acts on cells in an autocrine or paracrine manner to suppress the intracellular concentration of pro-IL-1β via the stat3 pathway. ROS burst from damaged mitochondrion could drive activation of inflammasome, but autophagy could block the accelerated IL-1β/IL-18 production via degrading the damaged mitochondrion and sequestering intracellular stores of pro-IL-1β and IL-18. Meanwhile, effector and memory T cells could also suppress inflammasome activation via a cognate mechanism mediated by TNF superfamily and their receptors.
Role of NOD-like receptors in cancer development
The abnormal activation of inflammasome is linked to various types of human disease, such as cryopyrinopathies, gout, asbestosis, silicosis, Alzheimer’s disease, and autoimmune diseases [62, 63]. To date, more than 70 inherited mutations have been identified associating with cryopyrinopathies occurrence, a large majority of which are situated within and around NLRP3 NACHT domain [64, 65]. These mutations are therefore believed to induce conformational changes that render NLRP3 constitutively active, resulting in continuous caspase-1 activation and release of IL-1β and IL-18 [65]. Besides, decreased NLRP3 expression and reduced IL-1β production have recently been linked with increased susceptibility to Crohn’s disease in humans [66, 67]. Moreover, inflammasome deregulation was also recorded to contribute to the pathogenesis of experimental autoimmune encephalomyelitis [68]. Significantly, accumulating evidence also suggested that NLRs are closely correlated to cancer occurrence, but conflicting evidence also exists, which might be due to the dual functions of inflammasomes in promoting carcinogenic inflammation or eliminating malignant cells via the pyrotosis death pathway.
NLRP3 signaling and its duplex role
NLRP3 is the most well-studied member of NLR family. It can be activated by a wide range of signals including infected pathogens, endogenous or environmental origins [69]. Based on current findings, 3 distinct mechanisms have been proposed to account for NLRP3 activation, including potassium efflux, phagolysosomal destabilization and mitochondrial ROS burst. Various bacterial pathogens can secret pore-forming toxins (e.g., nigericin from Streptomyces hygroscopicus, listeriolysin O from Listeria monocytogenes, pneumolysin from Streptococcus pneumoniiae, alpha-hemolysin from Escherichia coli) and subsequently activate the NLRP3 inflammasome by increasing potassium efflux [70-76]. In addition, bacterial and viral RNA is also reported to be an initial factor contributing to NLRP3 inflammasome assembly [77]. Moreover, extracellular ATP released from phagocytosed dying cells acts on purinergic receptor, P2X7 and induces pannexin-1 (PANX1) channels to promote potassium efflux and results in NLRP3 activation [78-80]. On the other hand, intracellular uptake of crystalline and particulate matters is also capable of causing lysosomal destabilization and release of cathepsin B, a sensor of NLRP3 [81-85]. Lysosome rupture-induced NLRP3 activation was also observed in cathepsin B-deficient cells, a phenomenon that may be attributed to potassium efflux (Figure 4) [86].
Notably, mitochondrial ROS generation is considered to be one of the most important mechanisms of NLRP3 activation. Pharmacological inhibition of mitochondrial ROS burst has been shown to prevent NLRP3-inflammasome formation [81, 82, 87]. Although the detailed molecular mechanisms of ROS-mediated NLRP3 activation remain largely unclear, calcium influx mediated by the transient receptor potential melastatin 2 (TRPM2) has been suggested to be a possible reason [87]. Extracellular calcium has been shown to activate the calcium sensing receptor (CASR) and thus lead to the release of calcium stores from the endoplasmic reticulum (ER), eventually triggering the formation of NLRP3 inflammasome (Figure 4) [88-90]. On the other hand, mitochondrial ROS burst is an upstream event leading to the loss of mitochondrial membrane potential, a pivotal event in inducing intrinsic apoptosis. Interestingly, overexpression of the anti-apoptotic protein, BCL2, was shown to limit the activation of NLRP3 inflammasome, indicating that apoptosis-regulated proteins might be closely correlated with NLRP3 activation [91, 92]. More recently, cIAP1, cIAP2, and XIAP have also been linked with inflammasome activation. cIAP1 and cIAP2 were found to enhance inflammasome activation by ubiquitinating and stabilizing caspase-1 and consequently, promoting Il-1β release, whereas concurrent inhibition of cIAP1, cIAP2, and XIAP was shown to limit caspase-1 activation [93-95]. Overall, these studies place the mitochondria as a potential player for inflammasome activation. However, the precise role of mitochondria in mediating NLRP3 inflammasome formation and subsequent promotion of carcinogenesis awaits clarification.
With regards to pro-tumorigenic ability, NLRP3 polymorphism is shown to be associated with melanoma susceptibility, colorectal cancer prognosis, and overall survival of myeloma [96]. In a Swedish case-control study, NLRP3 variant (rs35829419) was significantly more common in male patients than in controls (OR, 2.22; CI, 1.27-3.86) and showed strong association with nodular melanoma (OR, 2.89; CI: 1.33-6.30) [97]. It has been suggested that NLRP3 activation could suppress NK and T cell-mediated anti-tumor actions in a sarcoma mouse model and metastatic melanoma, whereas the population of myeloid-derived suppressor cells and Tregs were increased [98]. Consistently, NLRP3 silencing resulted in a 5-fold reduction in the number of tumor-associated myeloid-derived suppressor cells found in host mice, and NLRP3-/- MDSCs were less efficient to reach the tumor site, demonstrating the critical role of NLRP3 in preventing cancer occurrence by modulating host immunity [99]. Furthermore, it was found that NLRP3-deficient mice generated less pulmonary metastasis in an orthotopic transplant mouse model of mammary adenocarcinoma [100]. In addition, chemotherapeutic agents, gemcitabine and 5-fluorouracil were shown to activate NLRP3-medaited inflammasome formation in myeloid-derived suppressor cells, leading to IL-1β production that is capable of inducing IL-17 secretion from CD4+ T cells and blunting the anticancer efficacy of chemotherapeutic drugs [101]. Accordingly, gemcitabine and 5-fluorouracil exert increased antitumor effects when tumors were established in NLRP3-/- or Caspase-1-/- mice, and NLRP3 activation by chemotherapeutic drugs is considered to be a positive regulator to promote cancer growth [101]. All these findings suggest the pro-tumorigenic role of NLRP3 in cancer development.
Although several lines of evidence have indicated the pro-carcinogenic activities of NLRP3, its role in cancer development remains controversial. NLRP3-/- mice were shown to be more susceptible to cancer and the number of colon polyps in the AOM-DSS mouse model and the accelerated tumor growth in the carcinogenesis model was accompanied with drastically low levels of colonic IL-18, suggesting that NLRP3 may play a protective role against neoplasia formation and IL-18 might be closely associated with colon cancer initiation [102, 103]. Of note, IL-18 knockout mice generated more tumors than controls after administering AOM-DSS (azoxymethane- dextran sodium sulfate), whereas injection of recombinant IL-18 successfully restrained disease progression, which might be associated with MyD88-related pathway [104]. Similar anti-carcinogenic role of NLRP3 was also observed in hepatocellular carcinoma. Both mRNA and protein levels of NLRP3 were significantly down-regulated in the hepatic parenchymal cells derived from liver cancer biopsies compared to non-cancerous samples [105]. In this context, it is logical to deduce that NLRP3 may play a duplex role in controlling cancer growth. Thus, on one hand, NLRP3 could promote tumor cell survival through activation of NF-κB-stat1/3 pathway or by limiting cytotoxic immune cells infiltration, but on the other, it could suppress malignant progression by triggering mitochondrial apoptotic pathway or by enhancing immune-cytokine levels in the tumor microenvironment. In addition, the role of NLRP3 in cancer development might be tissue or cell dependent. For example, NLRP3 exhibits a protective role for colon cancer, but pro-carcinogenic effects for gastric and prostate malignancies [106]. Therefore, a much more comprehensive analysis of NLRP3 using conditional knockout models and pharmacological activators or inhibitors is needed to decode the precise effects of NLRP3 on cancer development in the future.
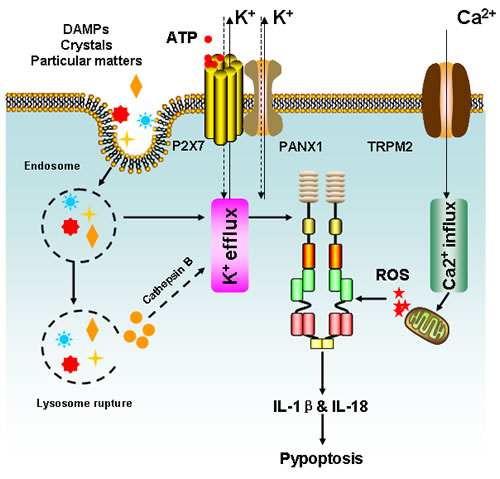
Figure 4: Simplified mechanisms for NLRP3 inflammasome activation. Three distinct machineries have been proposed to account for NLRP3 activation, including K+ efflux, phagolysosomal destabilization and mitochondrial ROS burst. Extracellular ATP released from dying cells acts on purinergic receptor P2X7 and prompts pannexin-1 (PANX1) channels to enhance K+ efflux and result in NLRP3 activation. Meanwhile, PAMPs such as pore-forming toxins are also capable to facilitate K+ efflux and activate NLRP3 inflammasome. Besides, K+ efflux could be activated by crystals or particular maters, which enter the cells via endocytosis and trigger NLRP3 inflammasome via cathepsin B following lysosome rupture. Finally, intracellular Ca2+ accumulation could result in mitochondrion damage and lead to ROS burst, which may activate the NLRP3 inflammasome either directly or by inducing K+ efflux. Following NLRP3 activation, IL-1β and IL-18 will be greatly produced and results in inflammation or pyroptotic cell death.
Other NLRs in carcinogenesis
Besides NLRP3, a number of NLRs have also been shown to be associated with cancer progression. NLRC4 was identified as a downstream transcriptional target of p53, indicating the tumor suppressive role of NLRC4 [107]. Mice lacking NLRC4 had significantly increased tumor numbers and burden compared to the wild-type controls in the AOM-DSS colon cancer model, but no differences in inflammation severity were noted, implying that tumor regulation by NLRC4 might be mostly cell intrinsic and not through down-regulation of inflammation [108, 109]. Similar to NLRC4, both NLRP6 and NLRP12 were also found to play a critical role in AOM-DSS tumorigenesis. A significant increase in tumor number and burden was observed in NLRP6-deficient mice compared to wild-type controls after chemical induction. But unlike NLRC4, NLRP6-mediated protection against tumor formation is attributed to hematopoietic cells rather than intestinal epithelial cells, because similar numbers of tumor were observed between NLRP6 deficient mice and irradiated wild-type mice that were transplanted with NLRP6 deficient bone marrow [110-112]. By contrast, NLRP6 deficient mice that received wild-type bone marrow transplant were shown to have reduced tumorigenic ability, similar to that of wild-type animals [111]. In addition, genetic profiling of tumors from wild-type and NLRP6 deficient mice exhibited a significant increase in the number of genes in the Wnt and Notch signaling cascade from a set of 1,884 genes, supporting a novel role of NLRP6 in controlling intestinal proliferation [112]. Of note, proinflammatory cytokines such as TNFα and IL-6 were elevated in the tumor microenvironment, whereas the level of IL-18 was significantly reduced. Meanwhile, IL-18 silencing in NLRP6 deficient mice has been associated with increased colon cancer development, indicating the pivotal role of cytokines in mediating the anti-carcinogenic activities of NLRP6 [110]. Similar to NLRP6, NLRP12 was also considered to be a tumor suppressive molecule as shown in ex vivo and in vivo carcinogenic animal models. Mice lacking NLRP12 were found to be more susceptible to DSS-injury, accompanied by increased body weight loss, enhanced pathology scores coupled with severe inflammatory cell infiltration and high levels of cytokine production [113-115]. The AOM-DSS mouse model also revealed that NLRP12 deficient mice had accelerated colon tumor development and progression, which was demonstrated with over-activation of NF-κB signaling pathway and enhanced gene expression such as CXCL12 and CXCl13 [116, 117]. Taken together, the NLRP6/12-mediated protective mechanisms against tumorigenesis provide a complex network involving interactions between hematopoietic cells, cytokines, and epithelial cells and further show that experimental validation is needed to pinpoint the precise signaling transduction mode underlying their anti-carcinogenic effects.
Double-edged swords of pyroptosis
Pyroptosis is a critical self-protection mechanism responding to pathogen invasion by inducing pro-inflammatory cell death. Unlike apoptosis, pyroptosis is characterized by cytoplasmic swelling and early cellular membrane rupture, which happens following caspase activation, nuclear condensation and DNA fragmentation [118]. Although the precise mechanisms underlying pyroptosis induction still remains elusive, the products released from dead cells may limit malignant cell survival and proliferation by activating the innate immune response. Increasing evidence validates that dying tumor cells following chemotherapy might activate the NLRP3 inflammasome of dendritic cells via P2X7 purinergic receptors, thus priming tumor-specific interferon-γ-producing T lymphocytes to limit cancer growth [119]. Moreover, mice lacking P2X7 or NLRP3 failed to prime interferon-γ-producing CD8+ T cells after chemotherapy, and anthracycline-treated breast cancer patients with P2X7 mutation developed metastatic lesions more rapidly than normal individuals [100]. Notably, a novel therapeutic strategy is in development to foster dendritic cells-mediated anti-tumor immunity via acceleration of pyroptosis of cancer cells by oncolytic viruses [120]. However, several studies have also indicated that pyroptosis might contribute to tumorigenic ability after inflammasome activation [121, 122]. These conflicting findings may be attributed to differences in the redox status of model cells and specific molecules involved in the process. For example, the reduced form of HMGB1 released from dying cells could trigger dendritic cells to induce anti-tumor immune response, while the oxidized form of HMGB1 would be unable to activate the immune response [123, 124]. In addition, the role of pyroptosis in cancer development might critically depend on the cell type. Pyroptosis of immune cells might bring harmful consequences to tumor immunoediting, while cancer cell pyroptosis would improve anti-cancer immunity. Overall, impaired pyroptosis has been considered to be a potential mechanism linking chronic inflammation to cancer initiation, and pyroptosis targeting is becoming a novel strategy to prevent cancer and improve cancer therapeutic efficacy.
Conclusions
Remarkable advancements in recent years have greatly increased our understanding of NLRs function and the associated inflammasome in host defense and disease pathogenesis. NLR containing inflammasomes are not only important for fighting against bacterial, fungi and viruses, but also appear to be a critical step in mediating cancer initiation and progression. Inflammasome activation would create a pro-inflammatory microenvironment for inducing malignant transformation, and suppress local immunity caused by NK or T cells. In addition, chemotherapeutic agents were found to activate inflammasome defense, which had positive feedback to support cancer growth. Notably, inflammasome-related autophagy is also believed to significantly contribute to cancer drug resistance and metastasis. All these findings greatly highlight the role of inflammasome as a novel target to prevent and treat cancer.
Despite mounting evidence listed above suggesting the potential of the inflammasome as a promising marker for cancer prevention, contrary data also exists to imply that inflammasome signaling could behave as a kind of anti-cancer mechanism. Mice lacking NLRP3 or NLRC4 show higher susceptibility to colon cancer following AOM-DSS treatment, and aberrant inflammasome formation leads to inhibition of tumor suppressor genes such as p53 and over-activation of oncogenes such as Wnt. What’s more, inflammasome-mediated pyroptosis is also considered to play a critical role in recruiting dendritic cells to limit cancer growth. Based on the conflicting evidence, a number of questions remain unanswered. Whether or not inflammasome-related carcinogenesis is cell dependent is an important question. A second question is whether a specific NLR would exhibit different bioactivity correlating with a cancer stage. Meanwhile, there are 22 NLR members in humans and it is unknown how these NLR molecules are activated or how they interact with each other. The complex network is awaits elucidation. There is also a lot of interest to identify novel ligand-receptor binding molecules, novel signaling pathway and novel targets for cancer prevention or therapy. The inflammasome is becoming a significant research topic in tumor microenvironment field and there is every likelihood that it could be developed as important biomarker for cancer diagnosis or prognosis prediction. Meanwhile, drug discovery targeting inflammasome modulation is also expected to improve cancer therapeutic efficacy to successfully reduce cancer risk. Taken together and given the emerging role of inflammasome in cancer development, understanding its signaling network and pathological significance might bring novel strategies for malignancy therapy and prevention.
Acknowledgments
This work was supported by the National Natural science Foundation of China (81402173, 81573651), Pearl River S&T Nova Program of Guangzhou (201506010098) and Combined Scientific Project Funded by Guangdong Provincial Science and Technology Agency and Guangdong Provincial Academy of Traditional Chinese Medicine (2014A020221047).
Conflict of Interests
No potential conflicts of interest were disclosed.
References
1. Balkwill F and Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001; 357(9255):539-545.
2. R V. Diekrankhaften Geschwülste. Berlin: August Hirschwald. 1863.
3. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006; 441(7092):431-436.
4. Hussain SP and Harris CC. Inflammation and cancer: an ancient link with novel potentials. International journal of cancer. 2007; 121(11):2373-2380.
5. Mantovani A, Allavena P, Sica A and Balkwill F. Cancer-related inflammation. Nature. 2008; 454(7203):436-444.
6. Shalapour S and Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. The Journal of clinical investigation. 2015; 125(9):3347-3355.
7. Reuter S, Gupta SC, Chaturvedi MM and Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free radical biology & medicine. 2010; 49(11):1603-1616.
8. Khansari N, Shakiba Y and Mahmoudi M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent patents on inflammation & allergy drug discovery. 2009; 3(1):73-80.
9. Bartsch H and Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbeck’s archives of surgery / Deutsche Gesellschaft fur Chirurgie. 2006; 391(5):499-510.
10. Coffelt SB and de Visser KE. Cancer: Inflammation lights the way to metastasis. Nature. 2014; 507(7490):48-49.
11. Yang L. TGFbeta and cancer metastasis: an inflammation link. Cancer metastasis reviews. 2010; 29(2):263-271.
12. Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B, Giordano A, Tin S, Wu Q, Garza RJ, Cristofanilli M, Mani SA, Croix DA, Ueno NT, Woodward WA, Luthra R, et al. Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PloS one. 2015; 10(7):e0132710.
13. Takeda K and Akira S. Toll-like receptors in innate immunity. International immunology. 2005; 17(1):1-14.
14. Huysamen C and Brown GD. The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS microbiology letters. 2009; 290(2):121-128.
15. Yoneyama M and Fujita T. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine & growth factor reviews. 2007; 18(5-6):545-551.
16. Kono H and Rock KL. How dying cells alert the immune system to danger. Nature reviews Immunology. 2008; 8(4):279-289.
17. Gross O, Thomas CJ, Guarda G and Tschopp J. The inflammasome: an integrated view. Immunological reviews. 2011; 243(1):136-151.
18. Martinon F, Burns K and Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell. 2002; 10(2):417-426.
19. Lechtenberg BC, Mace PD and Riedl SJ. Structural mechanisms in NLR inflammasome signaling. Current opinion in structural biology. 2014; 29:17-25.
20. Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nature reviews Immunology. 2010; 10(10):688-698.
21. Shin S and Brodsky IE. The inflammasome: Learning from bacterial evasion strategies. Seminars in immunology. 2015; 27(2):102-110.
22. Rathinam VA, Vanaja SK and Fitzgerald KA. Regulation of inflammasome signaling. Nature immunology. 2012; 13(4):333-342.
23. Tas F, Tilgen Yasasever C, Karabulut S, Tastekin D and Duranyildiz D. Clinical significance of serum interleukin-18 (IL-18) levels in patients with gastric cancer. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2015; 70:19-23.
24. Fabbi M, Carbotti G and Ferrini S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. Journal of leukocyte biology. 2015; 97(4):665-675.
25. Kim JW, Koh Y, Kim DW, Ahn YO, Kim TM, Han SW, Oh DY, Lee SH, Im SA, Kim TY, Heo DS and Bang YJ. Clinical Implications of VEGF, TGF-beta1, and IL-1beta in Patients with Advanced Non-small Cell Lung Cancer. Cancer research and treatment : official journal of Korean Cancer Association. 2013; 45(4):325-333.
26. Franchi L, Warner N, Viani K and Nunez G. Function of NOD-like receptors in microbial recognition and host defense. Immunological reviews. 2009; 227(1):106-128.
27. Siegel RM. Caspases at the crossroads of immune-cell life and death. Nature reviews Immunology. 2006; 6(4):308-317.
28. Suarez R and Buelvas N. [Inflammasome: activation mechanisms]. Investigacion clinica. 2015; 56(1):74-99.
29. de Alba E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). The Journal of biological chemistry. 2009; 284(47):32932-32941.
30. Bryan NB, Dorfleutner A, Rojanasakul Y and Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol. 2009; 182(5):3173-3182.
31. Ippagunta SK, Malireddi RK, Shaw PJ, Neale GA, Vande Walle L, Green DR, Fukui Y, Lamkanfi M and Kanneganti TD. The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization. Nature immunology. 2011; 12(10):1010-1016.
32. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD and Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nature immunology. 2010; 11(12):1136-1142.
33. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M and Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature. 2011; 479(7371):117-121.
34. Gurung P and Kanneganti TD. Novel roles for caspase-8 in IL-1beta and inflammasome regulation. The American journal of pathology. 2015; 185(1):17-25.
35. Hoesel B and Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Molecular cancer. 2013; 12:86.
36. Fan Y, Mao R and Yang J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein & cell. 2013; 4(3):176-185.
37. DiDonato JA, Mercurio F and Karin M. NF-kappaB and the link between inflammation and cancer. Immunological reviews. 2012; 246(1):379-400.
38. Prasad S, Ravindran J and Aggarwal BB. NF-kappaB and cancer: how intimate is this relationship. Molecular and cellular biochemistry. 2010; 336(1-2):25-37.
39. Qiao Y, Wang P, Qi J, Zhang L and Gao C. TLR-induced NF-kappaB activation regulates NLRP3 expression in murine macrophages. FEBS letters. 2012; 586(7):1022-1026.
40. Li Q, Zhang X, Wang W, Li L, Xu Q, Wu X and Gu Y. CPT-11 activates NLRP3 inflammasome through JNK and NF-kappaB signalings. Toxicology and applied pharmacology. 2015; 289(2):133-141.
41. Liu MH. FGF-21 alleviates diabetes-associated vascular complications: Inhibiting NF-kappaB/NLRP3 inflammasome-mediated inflammation? International journal of cardiology. 2015; 185:320-321.
42. Xiang P, Chen T, Mou Y, Wu H, Xie P, Lu G, Gong X, Hu Q, Zhang Y and Ji H. NZ suppresses TLR4/NF-kappaB signalings and NLRP3 inflammasome activation in LPS-induced RAW264.7 macrophages. Inflammation research : official journal of the European Histamine Research Society [et al]. 2015; 64(10):799-808.
43. Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP and Alnemri ES. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nature immunology. 2010; 11(5):385-393.
44. Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM and Monack DM. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(21):9771-9776.
45. Henry T, Brotcke A, Weiss DS, Thompson LJ and Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. The Journal of experimental medicine. 2007; 204(5):987-994.
46. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P and Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011; 34(2):213-223.
47. Dickensheets HL and Donnelly RP. IFN-gamma and IL-10 inhibit induction of IL-1 receptor type I and type II gene expression by IL-4 and IL-13 in human monocytes. J Immunol. 1997; 159(12):6226-6233.
48. Detjen KM, Farwig K, Welzel M, Wiedenmann B and Rosewicz S. Interferon gamma inhibits growth of human pancreatic carcinoma cells via caspase-1 dependent induction of apoptosis. Gut. 2001; 49(2):251-262.
49. Malhotra S, Rio J, Urcelay E, Nurtdinov R, Bustamante MF, Fernandez O, Oliver B, Zettl U, Brassat D, Killestein J, Lechner-Scott J, Drulovic J, Chan A, Martinelli-Boneschi F, Garcia-Merino A, Montalban X, et al. NLRP3 inflammasome is associated with the response to IFN-beta in patients with multiple sclerosis. Brain : a journal of neurology. 2015; 138(Pt 3):644-652.
50. Masters SL, Mielke LA, Cornish AL, Sutton CE, O’Donnell J, Cengia LH, Roberts AW, Wicks IP, Mills KH and Croker BA. Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO reports. 2010; 11(8):640-646.
51. Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, Schneider P and Tschopp J. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009; 460(7252):269-273.
52. Rebecca VW and Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016; 35(1):1-11.
53. Jiang X, Overholtzer M and Thompson CB. Autophagy in cellular metabolism and cancer. The Journal of clinical investigation. 2015; 125(1):47-54.
54. Pan H, Chen L, Xu Y, Han W, Lou F, Fei W, Liu S, Jing Z and Sui X. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget. 2016: doi: 10.18632/oncotarget.6908.
55. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T and Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008; 456(7219):264-268.
56. Zhou R, Yazdi AS, Menu P and Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469(7329):221-225.
57. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW and Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011; 12(3):222-230.
58. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA and Lavelle EC. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. The Journal of biological chemistry. 2011; 286(11):9587-9597.
59. Crisan TO, Plantinga TS, van de Veerdonk FL, Farcas MF, Stoffels M, Kullberg BJ, van der Meer JW, Joosten LA and Netea MG. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PloS one. 2011; 6(4):e18666.
60. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D and Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. The EMBO journal. 2011; 30(23):4701-4711.
61. Martins JD, Liberal J, Silva A, Ferreira I, Neves BM and Cruz MT. Autophagy and inflammasome interplay. DNA and cell biology. 2015; 34(4):274-281.
62. Lamkanfi M, Vande Walle L and Kanneganti TD. Deregulated inflammasome signaling in disease. Immunological reviews. 2011; 243(1):163-173.
63. Hoffman HM and Brydges SD. Genetic and molecular basis of inflammasome-mediated disease. The Journal of biological chemistry. 2011; 286(13):10889-10896.
64. Masters SL, Simon A, Aksentijevich I and Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annual review of immunology. 2009; 27:621-668.
65. Dowds TA, Masumoto J, Zhu L, Inohara N and Nunez G. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. The Journal of biological chemistry. 2004; 279(21):21924-21928.
66. Li J, Moran T, Swanson E, Julian C, Harris J, Bonen DK, Hedl M, Nicolae DL, Abraham C and Cho JH. Regulation of IL-8 and IL-1beta expression in Crohn’s disease associated NOD2/CARD15 mutations. Human molecular genetics. 2004; 13(16):1715-1725.
67. Villani AC, Lemire M, Fortin G, Louis E, Silverberg MS, Collette C, Baba N, Libioulle C, Belaiche J, Bitton A, Gaudet D, Cohen A, Langelier D, Fortin PR, Wither JE, Sarfati M, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nature genetics. 2009; 41(1):71-76.
68. Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA and Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010; 184(9):4610-4614.
69. Sutterwala FS, Haasken S and Cassel SL. Mechanism of NLRP3 inflammasome activation. Annals of the New York Academy of Sciences. 2014; 1319:82-95.
70. Munoz-Planillo R, Franchi L, Miller LS and Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009; 183(6):3942-3948.
71. Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N’Guessan P, Witzenrath M, Netea MG, Chakraborty T, Suttorp N and Opitz B. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol. 2010; 184(2):922-930.
72. Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E and Hornung V. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. European journal of immunology. 2010; 40(6):1545-1551.
73. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA and Ting JP. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009; 183(3):2008-2015.
74. Duncan JA, Gao X, Huang MT, O’Connor BP, Thomas CE, Willingham SB, Bergstralh DT, Jarvis GA, Sparling PF and Ting JP. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009; 182(10):6460-6469.
75. Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ and Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009; 30(4):556-565.
76. Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD and Fitzgerald KA. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell host & microbe. 2009; 5(5):487-497.
77. Li J, Hu L, Liu Y, Huang L, Mu Y, Cai X and Weng C. DDX19A Senses Viral RNA and Mediates NLRP3-Dependent Inflammasome Activation. J Immunol. 2015; 195(12):5732-5749.
78. Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM and Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006; 440(7081):228-232.
79. Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P and Nunez G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007; 26(4):433-443.
80. Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G and Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(23):8067-8072.
81. Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA and Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(26):9035-9040.
82. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT and Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008; 320(5876):674-677.
83. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS and Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008; 453(7198):1122-1126.
84. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ and Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nature immunology. 2008; 9(8):857-865.
85. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA and Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature immunology. 2008; 9(8):847-856.
86. Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I, Corradin G and Tschopp J. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PloS one. 2009; 4(8):e6510.
87. Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS and Qiao L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nature communications. 2013; 4:1611.
88. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL and Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012; 492(7427):123-127.
89. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM and Horng T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(28):11282-11287.
90. Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schoneberg T, Schaefer M, Krugel U, Smajilovic S, Brauner-Osborne H, Baerwald C and Wagner U. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nature communications. 2012; 3:1329.
91. Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B and Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007; 129(1):45-56.
92. Faustin B, Chen Y, Zhai D, Le Negrate G, Lartigue L, Satterthwait A and Reed JC. Mechanism of Bcl-2 and Bcl-X(L) inhibition of NLRP1 inflammasome: loop domain-dependent suppression of ATP binding and oligomerization. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(10):3935-3940.
93. Hawkins CJ, Uren AG, Hacker G, Medcalf RL and Vaux DL. Inhibition of interleukin 1 beta-converting enzyme-mediated apoptosis of mammalian cells by baculovirus IAP. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93(24):13786-13790.
94. Labbe K, McIntire CR, Doiron K, Leblanc PM and Saleh M. Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasome. Immunity. 2011; 35(6):897-907.
95. Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O’Reilly L, Mason K, Gross O, Ma S, Guarda G, Anderton H, Castillo R, Hacker G, Silke J and Tschopp J. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012; 36(2):215-227.
96. Cook GP, Savic S, Wittmann M and McDermott MF. The NLRP3 inflammasome, a target for therapy in diverse disease states. European journal of immunology. 2010; 40(3):631-634.
97. Verma D, Bivik C, Farahani E, Synnerstad I, Fredrikson M, Enerback C, Rosdahl I and Soderkvist P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment cell & melanoma research. 2012; 25(4):506-513.
98. Chow MT, Sceneay J, Paget C, Wong CS, Duret H, Tschopp J, Moller A and Smyth MJ. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer research. 2012; 72(22):5721-5732.
99. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, McElvania-Tekippe E, Serody JS and Ting JP. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer research. 2010; 70(24):10161-10169.
100. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nature medicine. 2009; 15(10):1170-1178.
101. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, Boireau W, Simon B, Ryffel B, Connat JL, Kanellopoulos J, Martin F, Rebe C, Apetoh L and Ghiringhelli F. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nature medicine. 2013; 19(1):57-64.
102. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M and Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010; 32(3):379-391.
103. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros P, Beauchemin N, Vallance BA and Saleh M. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010; 32(3):367-378.
104. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M and Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010; 185(8):4912-4920.
105. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, Zhao W, Huai W, Guo P and Han L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Laboratory investigation; a journal of technical methods and pathology. 2014; 94(1):52-62.
106. Xu Y, Li H, Chen W, Yao X, Xing Y, Wang X, Zhong J and Meng G. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PloS one. 2013; 8(11):e77955.
107. Sadasivam S, Gupta S, Radha V, Batta K, Kundu TK and Swarup G. Caspase-1 activator Ipaf is a p53-inducible gene involved in apoptosis. Oncogene. 2005; 24(4):627-636.
108. Hu B, Elinav E and Flavell RA. Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle. 2011; 10(12):1936-1939.
109. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC and Flavell RA. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(50):21635-21640.
110. Chen GY, Liu M, Wang F, Bertin J and Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011; 186(12):7187-7194.
111. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI and Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011; 145(5):745-757.
112. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D and Chamaillard M. NOD-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(23):9601-9606.
113. Jeru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, Grimprel E, Landman-Parker J, Hentgen V, Marlin S, McElreavey K, Sarkisian T, Grateau G, Alnemri ES and Amselem S. Mutations in NALP12 cause hereditary periodic fever syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(5):1614-1619.
114. Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, Schneider M, Roney KE, O’Connor BP, Moore CB, Morrison A, Sutterwala FS, Bertin J, Koller BH, Liu Z and Ting JP. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol. 2010; 185(8):4515-4519.
115. Borghini S, Tassi S, Chiesa S, Caroli F, Carta S, Caorsi R, Fiore M, Delfino L, Lasiglie D, Ferraris C, Traggiai E, Di Duca M, Santamaria G, D’Osualdo A, Tosca M, Martini A, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis and rheumatism. 2011; 63(3):830-839.
116. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, Green DR, Lamkanfi M and Kanneganti TD. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer cell. 2011; 20(5):649-660.
117. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RM, Davis BK, Uronis JM, Herfarth HH, Jobin C, Rogers AB and Ting JP. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity. 2012; 36(5):742-754.
118. Bergsbaken T, Fink SL and Cookson BT. Pyroptosis: host cell death and inflammation. Nature reviews Microbiology. 2009; 7(2):99-109.
119. Zitvogel L, Kepp O, Galluzzi L and Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nature immunology. 2012; 13(4):343-351.
120. Li QX, Liu G and Wong-Staal F. Oncolytic virotherapy as a personalized cancer vaccine. International journal of cancer. 2008; 123(3):493-499.
121. Masters SL, Gerlic M, Metcalf D, Preston S, Pellegrini M, O’Donnell JA, McArthur K, Baldwin TM, Chevrier S, Nowell CJ, Cengia LH, Henley KJ, Collinge JE, Kastner DL, Feigenbaum L, Hilton DJ, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity. 2012; 37(6):1009-1023.
122. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM and Feldstein AE. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014; 59(3):898-910.
123. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature medicine. 2007; 13(9):1050-1059.
124. Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR and Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008; 29(1):21-32.