
INTRODUCTION
Ewing sarcoma (ES) is a highly malignant tumor of bone and soft tissue that occurs in children, adolescents, and young adults. Tumors often grow in close proximity to bone but can occur as soft tissue masses [1-3]. ES cases show a balanced chromosomal translocation [4] that joins the EWS gene (EWing Sarcoma) located on chromosome 22 to an ETS family gene, which is most commonly either FLI1 (Friend Leukemia Insertion) located on chromosome 11, t(11;22) or ERG located on chromosome 21, t(21;22). The resulting fusion protein is termed EWS-FLI1 or EWS-ERG, respectively. Other infrequent variant fusion proteins are the products of ES translocations and are absent in non-tumor cells. FLI1 is an ETS family transcription factor with a conserved DNA binding domain. The carboxy terminal half of FLI1 contained in the EWS-FLI1 fusion protein retains its DNA binding domain. Therefore, EWS-FLI1 binds to DNA through the conserved ETS binding domain. However, the EWS-FLI1 fusion protein functions by a different mechanism than either EWS or FLI1 [5]. EWS-FLI1 is required to maintain the growth of ES cell lines, and when the expression level of EWS-FLI1 is reduced by alternative mechanisms, ES cell lines die in culture and xenografts in nude mice regress [6-13].
While the oncogenic activity of EWS-FLI1 is clear, the cell of origin for ES has been confounding due to the cytotoxic effects of expressing EWS-FLI1 in most primary cell types [14-16]. Previous studies have identified three primary cell types that are permissive for EWS-FLI1 expression and thus represent prime candidates for the elusive tumor cell of origin: (i) mesenchymal stem cells (MSCs) [17-19], (ii) neural crest stem cells [20], and (iii) embryonic osteochondrogenic progenitor cells [21].
Transgenic mouse models have been successfully developed for neoplasms with tumor-specific chromosomal translocations, including alveolar rhabdomyosarcoma, synovial sarcoma, myxoid liposarcomas, and clear cell sarcomas [22-27]. However, the same success has not been achieved in ES. When EWS-FLI1 was expressed ubiquitously under the native EWS promoter, either in utero or in adult mice, it resulted in lethality [16]. Because EWS-FLI1 induces apoptosis in mouse embryonic fibroblasts in vitro, the embryonic lethality resulting from broad transgenic expression was not surprising. When the expression of EWS-FLI1 was restricted to specific cell types, the animals survived but did not develop ES. EWS-FLI1 expression under the control of the Prx1 promoter resulted in developmental malformations in the limbs, but not tumor formation [28]. When these animals were crossed with p53 null mice, EWS-FLI1 expression accelerated the p53 null-induced formation of osteosarcoma and shifted the tumor histology from osteosarcoma to undifferentiated sarcoma. Moreover, EWS-FLI1 expression under the control of the Mx1 promoter resulted in the rapid development of myeloid/erythroid leukemia [29]. The Prx-1 promoter is active in the primitive mesenchyme of the early limb bud, while the Mx1 promoter is active in liver, spleen, bone marrow, and lymphoid tissues following induction with type I interferon (IFNα/β). A more recent attempt to create an ES transgenic mouse model utilized Cre-loxP-mediated somatic chromosomal translocation between the EWS and FLI1 locus to express the fusion protein in vivo [30]. However, this strategy did not lead to any malignant neoplasms; instead, the mice presented with cardiomyopathy followed by death [30].
Experimental ES models consist of murine xenografts from established human ES cell lines or as allografts of mouse bone marrow-derived mesenchymal progenitors transfected with EWS-FLI1 [17, 19, 21, 31, 32]. The expression of EWS-FLI1 in zebrafish also results in tumor formation, with higher incidences on the p53 null background [33]. However, these models lack the essential elements of tumor initiation, as they are derived from established tumors or cell lines transformed in vitro. Therefore, these models do not fulfill the need for a transgenic mouse that develops spontaneous ES driven by EWS-FLI1 expression.
RESULTS
To develop a clinically relevant ES mouse model for use in studying disease pathogenesis and testing novel therapies, we employed transgenic and non-transgenic approaches to express an EWS-FLI1 transgene in different tissues at different times. Overall, 16 alternative methods were tried in 6 independent laboratories (Table 1). For simplicity of discussion, these models will be referred to by the numbers provided in Table 1 in this manuscript.
Table 1: A summary of sixteen approaches employed by six independent laboratories to express an EWS-FLI1 transgene in mice.
Model # |
Target tissue |
Promoter |
Time of expression |
Inducible? |
Phenotype |
Lab |
#1 Runx2Cre-EF |
Osteoblast precursor |
Cre under Runx2 prom. EWS-FLI1 under Rosa26 prom |
Embryonal (E12.5) |
No |
No phenotype in two clones on WT or INK4a/ARF-/- background. Embryonic lethality (E13.5) in one clone. |
Moriggl |
# 2 OsxCre-EF |
Osteoblast precursor |
Cre under Osterix1 prom. EWS-FLI1 under Rosa26 prom. |
Embryonal (E14.5) or 3 weeks old |
Yes (Tet-off for Cre expression) |
Embryonal: Lethal 3 weeks : Facial deformities on WT p53 and Rb 3 weeks: Leukemia and reduced osteosarcoma in p53-/-Rb-/- background. |
Üren |
#3 Col1a2.3Cre-EF and Col1a3.6Cre-EF |
Osteoblasts |
Cre under Col1a2.3 or Col1a3.6 prom. EWS-FLI1 under EWS prom. |
Embryonal |
No |
Embryonic lethal |
Lee |
# 4 Cosco-EF |
Ubiquitous |
EWS-FLI1 under EWS prom. |
Embryonal |
No |
Embryonic lethal |
Delattre |
#5 Pgk-EF |
Ubiquitous |
EWS-FLI1 under Pgk prom. |
Embryonal |
No |
Embryonic lethal |
Delattre |
# 6 Nse-EF and Nse-EF-SV |
Neuronal tissue |
EWS-FLI1 under Nse prom. |
Embryonal |
No |
Embryonic lethal |
Delattre |
# 7 NEFL-EF |
Neuronal tissue |
EWS-FLI1 under dNEFL prom. |
Adult |
No |
EWS-FLI1 expressed in adult brain and cerebellum but no tumor |
Delattre |
# 8 MT-EF |
Ubiquitous |
EWS-FLI1 under Metallothionein prom. |
Variable |
Yes (ZnCl2 for EWS-FLI1 expression) |
No phenotype |
Delattre |
# 9 PLAPtTA-EF |
Ubiquitous |
tTA under PLAP prom. EWS-FLI1 under hCMV-TRE prom. |
Embryonal |
Yes (Tet-off for EWS-FLI1 expression) |
Embryonic lethal |
Delattre |
#10 COMET and COMETΔNeo |
Ubiquitous |
EWS-FLI1 under TREtight prom. |
Variable |
Yes (Tet-on for EWS-FLI1-luciferase expression) |
EWS-FLI1 toxicity during spermatogenesis in chimera mice, no F1 KI progeny. |
Delattre |
#11 Prx1Cre-EF |
Limb bud Mesenchyme |
Cre under Prx1 prom. EWS-FLI1 under EWS prom. |
Embryonal |
No |
Embryonic lethal |
Lee |
#12 Cre-TL-EF |
Mesenchymal and neural crest tissue |
Cre under Dermo1, Prx1, P0, Col1a2, or Sox9 prom. EWS-FLI1 under EWS prom. |
Embryonal |
No |
No phenotype on WT or INK4a/ARF-/- background |
Sweet-Cordero |
#13 RetroLTR-EF |
Mesenchymal stem cells |
EWS-FLI1 under Retroviral LTR |
Adult |
No |
Fibrosarcoma |
Nakamura |
#14 piggyBac-EF |
Mesenchymal stem cells |
EWS-FLI1 under CMV prom. |
Adult |
No |
Fibrosarcoma |
Nakamura |
#15 CreEP-TL-EF |
Not-tissue selective. Cre injected IM |
Cre under pMC1 prom. EWS-FLI1 under EWS prom. |
4 weeks old |
No |
Muscle degeneration |
Nakamura |
#16 Ad5Cre-EF |
Not-tissue selective. Virus injected IM, IP and IV |
Cre under Ad5-CMV prom. EWS-FLI1 under Rosa26 prom. |
1 day old |
No |
IV: No phenotype |
Üren |
Transgenic approaches: bone and bone precursors
EWS-FLI1 expression in osteoblast precursors via the Runx2 promoter (Model #1Runx2Cre-EF)
Runx2 is a master transcription factor for chondrocyte and osteoblast differentiation that regulates bone formation [34]. We established a conditional EWS-FLI1 mouse model in which the expression of the fusion protein was controlled by Cre recombinase driven by the Runx2 promoter in a 150 kB BAC transgene encompassing the Runx2 gene. Here, an improved Cre codon sequence was inserted into the coding exon adjacent to the START codon to drive expression from the bone-specific distal promoter [35] (Supplementary Figure S1). Cre-inducible ROSA26-loxP-STOP-loxP-EWS-FLI1 (E/F) mice [29], in which EWS-FLI1 is under control of the ROSA26 gene locus, were used. Therefore, EWS-FLI1 could be ubiquitously expressed following the removal of the STOP codon by Cre recombinase. To restrict and target EWS-FLI1 expression to the bone-forming lineage, E/F mice were crossed to Runx2-Cre mice. We used three different characterized Runx2-Cre transgenic mouse lines (#777, #784 and #1634) that gave different phenotypes. The highest Cre recombinase expression was observed in line #777 compared to lines #784 and #1634 [35].
An analysis of the tissues from E/F mice crossed with the #784 and #1634 Runx2-Cre transgenic lines (E/F+/-Runx2-Cre+ mice) showed Cre activity in the bone, bone marrow, calvaria, and testis (Supplementary Figure S2A). Offspring from these mice were viable and fertile (Supplementary Table S1). Careful and regular phenotype screening over 18 months did not reveal any obvious gross abnormalities or tumor formation. The excision of the STOP-cassette in the bone, testis, and isolated osteoblasts was confirmed by genomic PCR (Supplementary Figure S2B). However, EWS-FLI1 could not be detected at the mRNA level (Supplementary Figure S3A). We failed to detect EWS-FLI1 expression in compound animals of the #784 E/F+/- Runx2-Cre+ and #1634 E/F+/- Runx2-Cre+ lines. We also noted the loss of Cre expression in compound mesenchymal tissue, although Cre was well expressed in mice without the E/F transgene (Supplementary Figure S4). In order to confirm that the EWS-FLI1 locus was amenable to Cre-induced recombination, we isolated ear skin fibroblasts from these lines and expressed Cre recombinase using an adenoviral delivery system in cell culture (Supplementary Figure S5).
Crossing E/F mice with the #777 Runx2-Cre transgenic line resulted in early embryonic lethality around embryonic day 13.5 (E13.5), and no transgenic offspring could be generated (Supplementary Table S1). We hypothesized that EWS-FLI1 protein expression in the target tissues of the #777 Runx2-Cre line caused developmental problems that led to embryo resorption before E13.5 (two litters with 13 embryos were analyzed from E/F+/+ female versus the #777 Runx2-Cre male cross), which thereby excluded the usefulness of that model. We did not further trace the cause of death. Twelve litters of offspring from that cross were analyzed further. While genetic PCR showed that these offspring carried the E/F transgene, no #777 Runx2-Cre expression in parallel could be detected, suggesting negative selection and the loss of double transgenic embryos during early embryonic development.
EWS-FLI1 has a growth inhibitory effect in most primary cells, and the loss of p16INK4a enables the cells to tolerate EWS-FLI1 expression in vitro [14]. Therefore, we postulated that the lethal effect of EWS-FLI1 on Runx2-Cre-expressing cells could be relieved by deleting the p16ink4a locus. Moreover, the loss of p16INK4a is a relatively common event in ES [36]. Thus, we crossed E/F+/- Runx2-Cre+ mice (both #784 and #1634 lines) to an Ink4a-deficient background. The offspring of these mice were viable (Supplementary Table S1), and a careful analysis of these E/F+/- Runx2-Cre+ Ink4a-/- compound mice showed the same lack of EWS-FLI1-expressing offspring as seen in the E/F+/- Runx2-Cre+ mice. The deletion of the STOP-cassette was confirmed in the bone and testis, but no expression of EWS-FLI1 could be observed (Supplementary Figure S6). Ink4a-/- mice are prone to tumor formation of mainly fibrosarcoma, liposarcoma, angiosarcoma and lymphomas [37]. We observed 30 mice per genotype over 12 months and did not detect any changes in the frequency or histopathological type of tumors in E/F+/- Runx2-Cre+ Ink4a-/- compared to Ink4a-/- mice. Often, multiple different tumors occurred in single animals. However, we evaluated more than 10 tumors isolated from the different genotypes and could not detect Cre or EWS-FLI1 expression in any of them. Therefore, we concluded that the toxic effect of EWS-FLI1 expression could not be rescued by the loss of Ink4a protein.
EWS-FLI1 expression in osteoblast precursors via the Osterix-1 promoter (Model #2OsxCre-EF)
Considering that most ES cases develop in bone or in close proximity to the bone, we hypothesized that the targeted expression of EWS-FLI1 in osteoblast progenitors may induce ES. Osterix (Osx) is a zinc finger-containing transcription factor required for osteoblast differentiation [38], and the promoter restricts Cre expression to the osteoblast lineage [39]. We expressed EWS-FLI1 in osteoblast progenitors by crossing E/F mice with inducible Tet-Off-based Osx-Cre mice (Supplementary Figure S7). The expression of EWS-FLI1 in E/F+/- Osx-Cre+ mice with wild-type (WT) p53 and Rb was embryonically lethal. Thus, we delayed Cre expression, and hence EWS-FLI1 expression, until weaning, upon which 32.6% of these mice developed evident facial bone deformities at the age of 5-6 months (Figure 1A and 1B). Osx-Cre-mediated deletion of p53 and Rb in both Osx-Cre+ p53fl/+ pRbfl/+ and Osx-Cre+ p53fl/fl pRbfl/fl mice resulted in osteosarcoma, and the latency of the disease could be delayed using a doxycycline diet [40]. When we crossed E/F mice with Osx-Cre+ p53fl/fl pRbfl/fl mice, we observed embryonic lethality. The administration of doxycycline to pregnant mothers was required for viable births. When the EWS-FLI1 expression was delayed until weaning, E/F+/- Osx-Cre+ p53fl/+ pRbfl/+ mice did not develop sarcoma; instead, 45.2% of these mice presented with leukemia (Figure 1C and 1D). We confirmed EWS-FLI1 expression in the spleens and livers of these leukemic mice at the mRNA and protein levels (Supplementary Figures S8A and B). Immunophenotyping suggested that the leukemia in Osx-Cre+ p53fl/+ pRbfl/+ mice was similar to the previously reported EWS-FLI-induced erythroid/myeloid leukemia, in which EWS-FLI1 expression was driven by Mx1-Cre [29]. As in E/F+/- Mx1-Cre+ mice, spleen cells were enriched for CD43+/CD71+/CD117+/CD45- cell populations (Supplementary Figure S8C). Although Osx-Cre mainly targets osteoblast progenitors, it has also been shown to target non-osteoblast lineage cells in the bone marrow, such as stromal cells, adipocytes and perivascular cells [41]. Therefore, it is possible that EWS-FLI1 is expressed in the hematopoietic lineage and induces leukemia in E/F+/- Osx-Cre+ p53fl/+ pRbfl/+ mice as the result of leakiness of the Osx-Cre system. Alternatively, extracellular vesicle-mediated systemic Cre mRNA exchange may have occurred between osteoblast progenitor cells with the active Osx promoter and hematopoietic cells that would not otherwise express Cre [42].
EWS-FLI1 expression in osteoblasts via the Col1a1 promoter (Model #3Col1a2.3Cre-EF & Col1a3.6Cre-EF)
This mouse model was created by knocking-in (KI) the human FLI1 cDNA (spanning exons 5-9) with a C-terminal FLAG epitope into mouse Ews exon 7 (Supplementary Figure S9 and [16]). This approach closely mimics the translocation found in patients, as EWS-FLI1 is expressed under the control of native Ews promoter/enhancer elements. A loxP-flanked STOP-cassette was inserted in the antisense direction in Ews intron 6 to permit the conditional expression of EWS-FLI1 [see [16] for details]. The constitutive expression of EWS-FLI1 using an EIIa-Cre mouse failed to produce any viable pups [16]. To prevent cytotoxic effects and to direct EWS-FLI1 expression to osteogenic progenitors, we crossed an Ews-FLI1KI mouse to two transgenic Cre mice: Col 3.6-Cre and Col 2.3-Cre [43]. These transgenic lines direct Cre recombinase expression to the osteoblast lineage using 2.3-kb (Col 2.3-Cre) and 3.6-kb (Col 3.6-Cre) fragments of the rat Col1a1 promoter. Additional Cre expression in tendons was reported in the Col 3.6-Cre mouse only. Upon genotyping the pups at weaning and also at birth (more than 100 combined), no mice carrying both the Ews-FLI1KI and Cre genes could be obtained, suggesting embryonic lethality resulting from EWS-FLI1 expression in osteoblast progenitors. Alternatively, low but detectable expression of Cre in non-osseous tissues of the Col 2.3-Cre and Col 3.6-Cre mice (such as the kidney, liver and skin) [43] might have resulted in embryonic lethality.
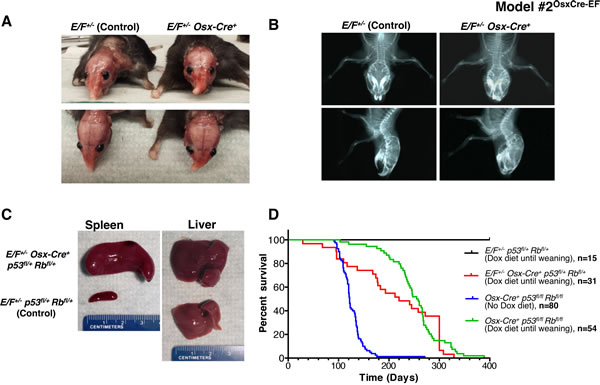
Figure 1: Postnatal expression of EWS-FLI1 in cells with the activated Osterix promoter in E/F+/- Osx-Cre+mice with or without p53 and pRb deletion in Model #2OsxCre-EF. A. Representative presentation of deformed nasal bone of an E/F+/- Osx-Cre+ mouse compared to an E/F+/- littermate control. B. X-ray imaging of an E/F+/- Osx-Cre+ mouse showing structural abnormalities in facial bones as compared to the clear facial bone structure of an E/F+/- littermate control. C. Representative photographs of spleens and livers collected from E/F+/- p53fl/+ pRbfl/+ (control) and E/F+/- Osx-Cre+ p53fl/+ pRbfl/+ littermates. Hepatomegaly and splenomegaly were consistently observed in E/F+/- Osx-Cre+ p53fl/+ pRbfl/+ mice displaying leukemia-like symptoms. D. Kaplan-Meier survival plots for the indicated genotypes: E/F+/- p53fl/+ pRbfl/+ (n = 15); E/F+/- Osx-Cre+ p53fl/+ pRbfl/+ (n = 31); and Osx-Cre+ p53fl/fl pRbfl/fl, (n = 54) mice with doxycline diet until weaning age. In addition, survival plot for Osx-Cre+ p53fl/fl pRbfl/fl (n = 80) mice with no doxycycline diet is presented.
Transgenic approaches: ubiquitous or neuronal tissue
Ubiquitous expression of EWS-FLI1 via the EWS or Pgk promoter (Model #4Cosco-EF and Model #5Pgk-EF)
We tried expressing EWS-FLI1 under broad and highly active promoters in transgenic mouse models to recapitulate the human ES phenotype. A cosmid containing the EWS promoter and the full EWS-FLI1 gene fusion were transfected into murine embryonic stem cells. We generated chimeric mice, but we were not able to obtain stable transgenic founder animals in the F0 generation (Supplementary Table S2, Supplementary Figure S10). We also tried expressing EWS-FLI1 under the control of a murine phosphoglucokinase-1 (Pgk-1) promoter (Supplementary Table S2, Supplementary Figure S11). We obtained two female F0 chimeric mice with the transgene, although one female was sterile and the progeny of the fertile mother were negative for the transgene.
Expression of EWS-FLI1 in neuronal tissues (Model #6Nse-EF and Nse-EF-SV and Model #7NEFL-EF)
As some ES cells express neuronal markers, including neuron-specific enolase (Nse), we prepared two transgenic constructs with the murine Nse promoter: one contained the EWS-FLI1 cDNA with the FLI1 polyadenylation site (Nse-EF), and the other contained the SV40 polyadenylation site (Nse-EF-SV). We transfected these two transgenes into murine embryonic stem cells to generate chimeric mice (Supplementary Table S2, Supplementary Figure S12). Among 32 chimeras we created for each model, only one Nse-EF chimera was transgenic, but no F1 was obtained from this founder. This result raised the hypothesis of embryonic toxicity in strains where EWS-FLI1 is expressed under constitutive (EWS, Pgk) or highly active (Nse) promoters. To overcome this problem, we expressed EWS-FLI1 under a minimally active neuron-specific promoter, neurofilament light chain (NEFL), as this gene was reported to be expressed in ES cells [44]. In Model #7NEFL-EF, we used the human NEFL promoter to drive the expression of EWS-FLI1 in mice (Supplementary Table S2, Supplementary Figure S13). Interestingly, four transgenic lines were obtained with Mendelian inheritance of the transgene. RT-PCR for the EWS-FLI1 transcript confirmed its specific expression in adult brain tissues (Supplementary Figure S13), but no tumor development was observed in these mice even after long-term surveillance (>1 year). This transgenic model may have expressed EWS-FLI1 too late in cellular differentiation to cause the growth of an embryonic tumor because NEFL is only expressed in post-mitotic neurons [45].
Inducible expression of EWS-FLI1 via ubiquitous promoters (Model #8 MT-EF, and Model #9PLAPtTA-EF)
In order to gain better control over the timing of EWS-FLI1 expression, we tried different inducible promoter systems. The metallothionein (Mt) promoter, which induces expression of a transgene upon administration of ZnCl2 to mice, was used to promote EWS-FLI1 expression in Model #8MT-EF (Supplementary Table S2, Supplementary Figure S14). Four stable transgenic lines were obtained. After ZnCl2 administration to the mice (24 h up to several weeks), various tissues were collected, but EWS-FLI1 expression was not detected in any of the tissues. We also used a tet-off system to control EWS-FLI1 expression (Supplementary Table S2, Supplementary Figure S15). In order to conditionally induce CMV-driven EWS-FLI1 expression upon doxycycline removal, two stable pUHD-10.3-EF strains were backcrossed with the LT1 strain that expresses the tetracycline-controlled transactivator (tTA) under the control of the human placental alkaline phosphatase (PLAP) promoter. Unfortunately, none of these backcrosses (Model #9PLAPtTA-EF) gave rise to double transgenic mice.
Spatiotemporal regulation of EWS-FLI1 expression (Model #10COMET and COMETΔNeo)
Learning from our past unsuccessful attempts (Supplementary Table S2) and from others [15, 16, 28, 29], we aimed to generate an inducible and conditional transgenic EWS-FLI1 mouse model. We created a knock-in (Gt(ROSA)26Sor locus) Cre/lox tetO inducible Mouse model for Ewing Tumor (COMET) (Supplementary Figure S16). For the conditional approach, an antisense EWS-FLI1 cassette was flanked with single mutated Lox sites (Lox66, Lox71). Upon Cre-mediated recombination, EWS-FLI1 is reversed and maintained in the sense orientation due to the generation of a double mutated lox72 site (unrecognized by the Cre) and a loxP site [46]. For the inducible strategy, a tet-On cassette containing a TREtight-inducible promoter oriented in the antisense direction was introduced in the Gt(ROSA)26Sor locus, as this was reported to improve its inducibility in this locus [47]. In order to track EWS-FLI1 expression using noninvasive bioluminescence imaging, an IRES luciferase (IRES Luc) cassette was added downstream of EWS-FLI1. A Frt-flanked neomycin selection cassette was also added. All of these cassettes were finally cloned into the pROSA26-1 vector to generate a pROSA26-1-COMET construct (Supplementary Figure S16). Western blotting against EWS-FLI1 (Figure 2A) confirmed the functionality of the COMET construct in vitro by co-transfecting a combination of COMET, Cre and rtTA plasmids in the presence or absence of doxycycline. Chemiluminescence only detected IRES-driven luciferase expression in the cells transfected with the COMET, Cre and rtTA constructs in a doxycycline dose-dependent manner (Figure 2B).
To generate Model #10COMET and COMETΔNeo, a knock-in strategy into the well-characterized Gt(ROSA)26Sor locus was favored over classical transgenesis (Supplementary Figure S16) due to its permanent accessibility for transcriptional activation in mice [48]. Homologous recombination was performed in murine embryonic stem cells with the pROSA26-1-COMET construct, and two positive clones (2C12, 2D8) were confirmed by Southern blotting (Supplementary Figure S17). We injected these two clones into mouse blastocysts. Among seven chimeric mice, only three generated agouti F1. However, none of the 31 F1 agoutis contained the COMET allele (Supplementary Table S3). This led us to speculate about the possible leaky expression of EWS-FLI1 leading to toxicity in Model #10COMET chimeric tissues. To overcome a described bidirectional activity of the PGK promoter that may have led to EWS-FLI1 expression [49], embryonic stem cell clones, 2C12 and 2D8, were transiently transfected with the pCAGGS-Flpe plasmid in order to delete the PGK-Neo selection cassette through FLPe/FRT-mediated excision (Supplementary Figure S16). Two clones deleted for the PGK-Neo cassette (1C3, 2A4) were injected into mouse blastocysts, giving rise to seven Model #10COMETΔNeo chimeric mice. Unfortunately, none of the 33 F1 agoutis contained the Model #10COMETΔNeo allele after backcrossing (Supplementary Table S3).
To determine whether the manipulated genetics of both the Model #10COMET and Model #10COMETΔNeo chimeric mice were functioning as designed, we measured bioluminescence. A clear signal was detected in the testes of both chimeric mouse models (Figure 2C). RNA from various organs of these chimeric mice was extracted, and RT-qPCR experiments for the expression levels of luciferase and EWS-FLI1 were performed (Figure 2D). The highest expression level of luciferase was found in the testis and correlated with EWS-FLI1 expression. In Model #10COMET, comparable mRNA levels were detected in the testis and spleen. However, bioluminescence was absent in the spleen (Figure 2C, white arrow), indicating that the protein may not be correctly processed in this organ.
To investigate the underlying cause of the leaky EWS-FLI1/luciferase expression, various organs were collected from Model #10COMET and COMETΔNeo chimeric mice. As untranslated transcripts of the Gt(ROSA)26Sor locus are expressed at high levels in the epididymis and the testis, we evaluated whether the EWS-FLI1/luciferase transcript might be expressed from the Gt(ROSA)26Sor promoter. In Model #10COMET chimeric mice, using RT-PCR, a fusion transcript between part of exon 1 of the RNA-gene trap ROSA 26 transcript (NR_027009.1) and the PGK promoter was detected only in the testis (Supplementary Figures S18A and B). However, this fusion transcript was expressed at very low levels and may have accounted for only a fraction of the high luciferase expression detected in the testes of Model #10COMET mice. In Model #10COMETΔNeo chimeric mice, a fusion transcript between exon 1 of the RNA-gene trap ROSA 26 transcript and the lox71 site (located just upstream of EWS-FLI1) could be detected in various organs (Supplementary Figures S18C and D). This fusion transcript was much more abundant than that was detected in Model #10COMETΔNeo mice and may have therefore accounted for a substantial part of the high luciferase expression detected in the testes of Model #10COMETΔNeo mice.
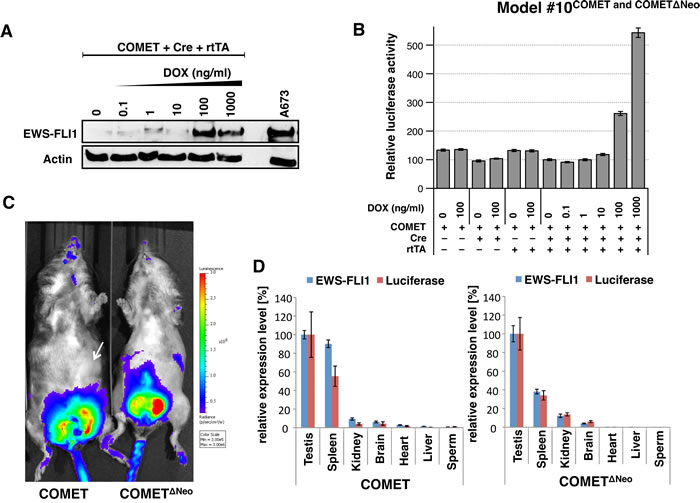
Figure 2: In vitro and in vivo expression of EWS-FLI1 and luciferase in Model #10COMET and COMETΔNeo. A. Western blotting against EWS-FLI1 and beta-actin was performed in triple-transfected (COMET + Cre + rtTA) 293T cells treated with increasing amounts of doxycycline (DOX). The A673 ES cell line is shown as the positive control for endogenous EWS-FLI1 expression (right lane). B. The COMET construct was transiently co-transfected in vitro with (+) or without (-) Cre recombinase and rtTA and in the presence or absence of DOX. Varying amounts of DOX (0 to 1,000 ng/ml) were added to the media. C. Models #10COMET (left) #10COMETΔNeo (right) chimera were imaged for in vivo luminescence measurements. D. Quantitative RT-PCR for EWS-FLI1 and luciferase in #10COMET (left panel) and #10COMETΔNeo (right panel) expression in various organs extracted from the imaged mice in panel C. Relative expression (to testis expression level) of duplicates with respective SD is shown.
Transgenic approaches: mesenchymal tissue
EWS-FLI1 expression in early limb bud mesenchyme via the Prx1 promoter (Model #11Prx1Cre-EF)
In parallel to using Cre driven by the Col1a1 promoter, we also targeted the expression of EWS-FLI1 to the early limb bud mesenchyme using Prx1-Cre transgenic mice (Supplementary Figure S19 and [50]). Again, no viable EWS-FLI1; Prx1-Cre positive pups were obtained at birth (more than 100 genotyped). Lin et al. reported using the same Prx1-Cre mouse to drive EWS-FLI1 expression in 3 transgenic lines [28]. These authors reported severe limb development defects in E14.5 embryos and embryonic lethality in one of their transgenic lines (EF-c), while two other transgenic lines showed milder limb deformities and a normal life span. Because the expression of EWS-FLI1 in our model was driven by the native EWS promoter, we speculated that the high levels of EWS-FLI1 expression in the early limb bud mesenchyme may have caused severe developmental defects in the limbs leading to embryonic lethality.
To circumvent the embryonic lethality of EWS-FLI1, in an earlier study, we crossed an EWS-FLI1KI mouse with a transgenic mouse expressing Cre fused to a mutated estrogen receptor driven by the CAG promoter (B6.Cg-Tg(CAG-Cre/Esr1)5Amc/J, Jackson Laboratory). Mice carrying both EWS-FLI1 and CreER genes were viable and healthy. However, the induction of EWS-FLI1 expression with tamoxifen led to the rapid death of the mice, demonstrating the cytotoxic effects of EWS-FLI1 even in somatic cells [16]. Collectively, these results highlight the difficulty in generating an ES mouse model and the importance of limiting EWS-FLI1 expression to a permissive cell type (presumed to be the EWS-FLI1 expression-tolerant tumor cell of origin).
Somatic chromosomal translocation between endogenous EWSR1 and FLI1 loci in mesenchymal and neural crest tissue (Model #12Cre-TL-EF)
We targeted mouse embryonic stem cells to insert a single loxP site into the Ewsr1 locus and a single loxP site into the Fli1 locus (Supplementary Figure S20). To avoid the potential loss of germline transmission, we chose to generate two separate strains instead of a single embryonic stem cell line in which both loci were targeted. This approach allowed us to delete the selectable markers prior to breeding the Ewsr1- and Fli1-targeted mice together without concern for extemporaneous recombination events. Ewsloxpurolox mice were made by inserting a lox-puromycinr-lox cassette between exons 8 and 9 of Ewsr1 on chromosome 9. Embryonic stem cell clones surviving puromycin selection were screened by Southern blot analysis for correct targeting (Figure 3A). In addition, Southern blot analysis allowed for a comparison between the intensities of the targeted Ewsloxpurolox and the non-targeted WT alleles in order to select clones that were likely to have a single targeted and single non-targeted Ewsr1 allele. We used a similar approach to generate the Fli1loxhygrolox mice. A lox-hygromycinr-lox cassette was inserted into the Fli1 locus on chromosome 11, between exons 5 and 6 (Supplementary Figure S20). As before, Southern blots were used to confirm correct targeting (Figure 3B). Targeted embryonic stem cells (from non-albino C57BL/6 cells) were injected into albino C57BL/6 blastocysts to generate chimeric mice. Once we obtained chimeras with suspected germline chimerism, we bred them to albino C57BL/6 mice and screened black pups for the presence of the lox-flanked cassettes. Mice carrying the cassettes were then bred to a CMV-Cre strain, in which Cre is expressed during implantation of the developing embryo [51]. Early Cre expression resulted in the deletion of the selection cassettes and the generation of mice with a single lox site in the middle of the Ewsr1 locus (Ewslox/wt) and in the middle of the Fli1 locus (Fli1lox/wt), which was confirmed by genomic DNA sequencing (Supplementary Figure S21A). Ewslox/wt and Fli1lox/wt mice were bred to generate homozygous Ewslox/lox and Fli1lox/lox mice (Supplementary Figure S20). These homozygous mice were used to generate all of the mice needed for the following experiments.
To test the feasibility of a reciprocal translocation event between chromosomes 9 and 11 in vivo, we first verified in vitro that this event could occur. Timed breedings between Ewslox/lox and Fli1lox/lox mice were set up to isolate mouse embryonic fibroblasts (MEFs). These Ewslox/wt; Fli1lox/wt MEFs were treated in vitro with adenovirus expressing Cre recombinase (Ad-Cre) (Figure 3C). PCR analysis using primers from the Ewsr1 and Fli1 introns from Ad-Cre-treated MEF genomic DNA confirmed that the recombination event could take place (Figure 3C). The presence of both the WT and lox alleles for Ewsr1 and Fli1 demonstrated that the recombination event was not 100%, which was as expected based on published models using this system in mice (Figure 3C). The Ews-Fli1 and Fli1-Ews junctions were sequenced to confirm that the single lox site was flanked by genomic DNA from each chromosome. To determine whether the Ews-Fli1 gene was transcribed into Ews-Fli1 mRNA, qRT-PCR was performed using primers that would span Ewsr1 exon 8 to FLI1 exon 6. Ews-Fli1 mRNA was detected in the MEF lines treated with Ad-Cre (Figure 3D). The Ews-Fli1 cDNA was then cloned using two rounds of nested PCR and sequenced to verify the correct splicing between Ewsr1 and Fli1. The CT values for the qPCR were relatively high, suggesting that Ews-Fli1 was expressed at a very low level. This low level of Ews-Fli1 expression may have resulted from the Ad-Cre infection; however, the batch of Ad-Cre had been used routinely in the lab to recombine lox sites found on a single chromosome with very high efficiency. Alternatively, the low level of detected Ews-Fli1 expression may have resulted from the loss of the cells expressing the translocation due to toxic effects. Attempts to quantify the levels of recombination using fluorescent in situ hybridization probes for chromosomes 9 and 11 were unsuccessful. However, given that the qPCR results confirmed that the translocation could be expressed, we hypothesized that even this low level of expression might be sufficient to initiate tumorigenesis in vivo.
To establish a physiologically relevant mouse model of ES, the Ewslox/lox; Cre+ and Fli1lox/lox colonies were crossed to generate cohorts of mice to observe for tumors over their life span. The mice in the experimental cohorts were Ewslox/+; Fli1lox/+; Cre+, and those in the control cohorts were Ewslox/+; Fli1lox/+; Cre-. We used 5 different Cre strains to generate tumor study cohorts: Col1a2-Cre (Cre+ n = 16, Cre- n = 6), Dermo1-Cre (Cre+ n = 25, Cre- n = 18), P0-Cre (Cre+ n = 20, Cre- n = 15), Prx1-Cre (Cre+ n = 25, Cre- n = 24), and Sox9-Cre (Cre+ n = 21, Cre- n = 15). Dermo1-Cre mice crossed with a LacZ reporter showed expression in mesenchymal tissues as early as E9.5; later, expression was detected in the condensing mesenchyme, chondrocytes and osteoblasts [52]. Col1a2-Cre showed a broader staining pattern than Dermo1-Cre, as LacZ expression was found starting around E8.5 in all dermal cells of the mouse as well as in the chondrocytes and osteoblasts of the developing bones [53]. The expression of Prx1-Cre began around E10 in the undifferentiated mesenchyme of the developing limb buds, increasing in expression with the development of the limb buds, in the mesenchyme between the limbs, and in the non-neural mesenchyme in the brain [50]. Some Prx1-expressing cells later expressed Sox9-Cre, giving us a subpopulation of Prx1-Cre cells to study [54]. The cells derived from the Sox9 lineage in the limb bud include chondrocytes and osteocytes, the cells that form tendons as well as the synovium (the soft tissue that lines the joints). A caveat of Sox9-Cre is that it has been shown to be expressed more broadly in various organ progenitor populations. To test whether a neural crest stem cell is the cell of origin for ES, we used the P0-Cre strain to drive Cre expression during the development of the neural crest lineage [55]. P0-Cre expression was shown to begin as early as E9.0 in tissues derived from the neural crest, and later in development, P0-Cre expression spread to the tissues derived from migrating neural crest cells. The notochord, which is not derived from the neural crest, also showed positive expression.
In addition to the above tumor study cohorts, a smaller cohort using CMV-Cre mice was observed to test whether the Cre expression that occurred very early in the developing embryo would show an embryonic lethal phenotype or a tumor phenotype [51]. Whereas Dermo1-Cre and Col1a2-Cre bred to E/F mice resulted in embryonic lethality, we obtained mice born at Mendelian ratios for all Cre strains without any lethality [29]. All of the CMV-Cre+ (n = 12) and CMV-Cre- (n = 9) mice were observed for >80 weeks without any differences. For the tumor cohorts with the mesenchymal and neural crest Cre strains, no signs of tumorigenesis were apparent after 70 weeks, so the cohorts were euthanized for analysis. Unfortunately, there were no differences between any of the Cre+ cohorts compared to the Cre- cohorts, and no obvious macroscopic tumors reminiscent of sarcomas were found (Supplementary Figures S22A-C). These results suggested either (i) the single hit of EWS-FLI1 expression in a subset of cells was not sufficient to induce tumor formation in the mouse (e.g. EWS-FLI1 might not have been expressed in enough cells to facilitate tumorigenesis) or (ii) the Ews-Fli1 translocation event never occurred in the first place.
We have shown that the Ews-Fli1 reciprocal translocation event did not induce tumor formation when expressed in mouse tissues using specific Cre strains. To test whether the loss of the tumor suppressors Ink4a and Arf would enable tumor formation or whether EWS-FLI1 was able to alter the tumors formed with Ink4a/Arf loss, cohorts of mice were generated such that Cre expression would delete the Ink4a/Arf locus as well as induce Ews-Fli1 translocation. The main groups of this study consisted of mice with the genotype Ews lox/+; Fli1lox/+; Ink4a/Arfflox/flox; Cre+, which were tested using the following Cre strains: Col1a2-Cre, Dermo1-Cre, P0-Cre, Prx1-Cre, or Sox9-Cre. The control mice lacked Cre (Ewslox/+; Fli1lox/+; Ink4a/Arfflox/flox; Cre-) or were hemizygous for the floxed Ink4a/Arf allele (Ewslox/+; Fli1lox/+; Ink4a/Arfflox/+; Cre- and Ewslox/+; Fli1lox/+; Ink4a/Arfflox/+; Cre+). In addition, cohorts of mice with the genotypes Ewslox/lox; Ink4a/Arfflox/flox; Cre+ and Ewslox/lox; Ink4a/Arfflox/flox; Cre- using Dermo1-Cre, P0-Cre and Prx1-Cre were generated to test the effects of Ink4a/Arf loss itself on tumorigenesis. Mice were observed until they appeared moribund or they reached the age of 70 weeks. The survival curves for these mice demonstrated that EWS-FLI1 did not enhance or change the tumor phenotype of these mice (Supplementary Figures S22D-F). A summary of the types of tumors and the number of mice with each type of tumor are presented (Supplementary Table S4). The loss of Ink4a/Arf alone either with or without EWS-FLI1 expression led to decreased survival of the mice such that the majority of the Ink4a/Arfflox/flox; Cre+ mice died earlier than the control mice. To confirm that EWS-FLI1 was not expressed in the tumors that developed in these mice, total RNA from tumor tissues was isolated and qRT-PCR for Ews-Fli1 was performed. By normalizing to a line that did not express mouse Ews-Fli1 and using a positive control for mouse Ews-Fli1 expression (mouse EWS-FLI1-overexpressing MEFs), we demonstrated that none of the tumors from the Ink4a/Arf tumor study expressed Ews-Fli1 at levels above background (Supplementary Figure S21B).
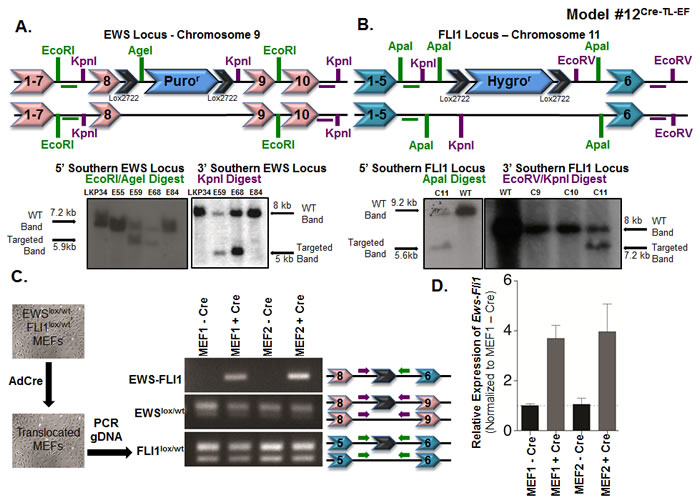
Figure 3: Somatic chromosomal translocation between endogenous Ewsr1 and Fli1 loci in Model #12Cre-TL-EF. A. Targeting mouse embryonic stem cells to insert a lox-puromycinr-lox cassette between exons 8 and 9 of the Ewsr1 locus. Genomic DNAs from the embryonic stem cell clones were EcoRI/AgeI (left) or KpnI (right) digested and were analyzed for the 5’ and 3’ integrations using Southern blot. Green and purple horizontal bars represent the probes used in the Southern blots. B. Targeting mouse embryonic stem cells to insert a lox-hygromycinr-lox cassette between exons 5 and 6 of the Fli1 locus. Genomic DNAs from the embryonic stem cell clones were ApaI (left) or EcoRV/KpnI (right) digested and were analyzed for the 5’ and 3’ integrations using Southern blot. Green and purple bars represent the probes used in the Southern blots. C. Schematic illustration for adenoviral Cre infection of Ewslox/wt; Fli1lox/wt MEFs in vitro. Genomic PCR was used to detect the translocated and untranslocated Ews and Fli1 chromosomes. The locations of the primers used are presented in the schematic. D. qPCR for Ews-Fli1 on total RNA from adenoviral Cre-treated MEFs. Hprt was used as the control gene, and samples were normalized to uninfected MEFs.
Expression of EWS-FLI1 in murine mesenchymal stem cells (MSCs) (Model #13RetroLTR-EF and Model #14piggyBac-EF)
MSCs are characterized by self-renewal activities and multi-directional differentiation [56]. It was expected that the cellular plasticity of MSCs might help EWS-FLI1 to function as a proper ES oncogene. We expressed EWS-FLI1 in bone marrow-derived MSCs (BM-MSCs; [57]) using retrovirus- or transposon-mediated gene transfer. FLAG-tagged EWS-FLI1 was inserted into the pMYs-IRES-Neo retroviral vector (Supplementary Figure S23). BM-MSCs of a BALB/c background were transduced with the retrovirus and then injected into sublethally irradiated (8 Gy) BALB/c recipients via the tail vein. All recipients (n = 5) developed multiple tumors in the lungs three weeks after injection. Histologically, the tumors exhibited storiform growth of spindle cells with frequent mitotic figures, which was consistent with the characteristics of fibrosarcoma (Figure 4A). The EWS-FLI1 transposon construct was generated using the piggyBac (PB) transposon/transposase system [58] (Supplementary Figure S24). BM-MSCs were transfected with PB-EF along with mRNA of the piggyBac transposase to increase the transfection efficiencies. The transfectants were transplanted into irradiated recipients as above (n = 5) or transplanted subcutaneously (n = 9). Again, 100% of the recipients developed fibrosarcoma-like tumors within 8 weeks (Figure 4B). Thus, although EWS-FLI1 expression could induce neoplastic transformation of BM-MSCs, the lesions did not recapitulate the morphologic characteristics of the small, round cells observed in human ES.
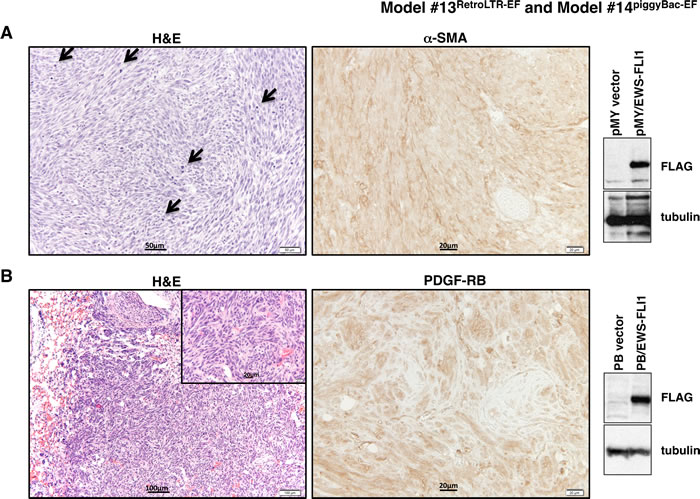
Figure 4: Histopathological analysis of tumors from Model #13RetroLTR-EF and Model #14piggyBac-EF. A. Fibrosarcoma developed in Model #13RetroLTR-EF. The storiform pattern of spindle-shaped, pleomorphic tumor cells is remarkable. Frequent mitotic figures (arrows) indicate aggressive tumor growth (left). α-SMA is a marker of smooth muscle and myofibroblastic cells. Human fibrosarcoma stains positive for α-SMA, whereas ES stains negative (middle). Expression of EWS-FLI1 in tumor tissue was confirmed using an anti-FLAG M2 antibody (right). B. Fibrosarcoma with a similar histology as (A) was also induced in Model #14piggyBac-EF. Invasive growth of the tumor in lung tissue is noted (left). PDGF-RB is a mesenchymal marker that is frequently positive in human fibrosarcoma and negative in ES (middle). The expression of EWS-FLI1 was confirmed by western blotting (right).
Non-transgenic approaches: localized Cre delivery
Localized Cre delivery via in vivo electroporation in EWS-FLI1 translocation mice (Model #15CreEP-TL-EF)
Localized expression of EWS-FLI1 was also tested in the Cre-loxP-mediated somatic chromosomal translocation model mice [30]. A Cre expression plasmid, pMC1-Cre, was delivered into the muscles of the lower legs of Ewsr1fl/+:Fli1fl/+ mice via electroporation (Supplementary Figure S25). The delivery of Cre via electroporation in vivo was successfully achieved to induce pleomorphic rhabdomyosarcoma in adult mice [59]. The mice received in vivo electroporation of the Cre plasmid four times and were observed for 1 year. No CreEP-TL-EF mice (n = 12) developed neoplastic lesions in the muscle, though the mice did show localized degeneration of the muscle fibers (Supplementary Figure S26), which was comparable to the damage in the myocardium in Cag-Cre:Ewsr1fl/+:Fli1fl/+ mice [30].
Localized adenovirus-mediated Cre delivery in E/F mice (Model #16Ad5Cre-EF)
Expressing EWS-FLI1 in most tissues resulted in embryonic lethality, as described in earlier models. To circumvent the reliance on tissue-specific promoters and to overcome embryonic lethality, we utilized adenovirus-delivered Cre to allow for the localized expression of EWS-FLI1 at different stages of postnatal development in E/F mice (Supplementary Figure S27). Adenovirus-Cre-mediated recombination has been successfully used to generate sporadic lung cancer [60], pancreatic adenocarcinoma [61], and colon cancer [62] mouse models. We hypothesized that the target cell may be present or susceptible to EWS-FLI1 tumorigenicity at certain stages of postnatal development. Therefore, adenovirus-Cre (Ad5-Cre) was delivered via intramuscular (IM), intraperitoneal (IP), or intravenous (IV) injections at different ages.
For IM delivery, E/F mice were injected with 109 plaque-forming units (pfu) Ad5-Cre in the left leg and 109 pfu Ad5-eGFP in the right leg at 1 day, 1 week, or 2 weeks of age. Cre mediated removal of the STOP-cassette 48 h after IM delivery of Ad5-Cre was confirmed using genomic DNA PCR (Supplementary Figure S28A). We observed muscle loss, limping, and prolonged clasping of the hind leg in the Ad5-Cre injected leg, but not in the Ad5-eGFP injected leg (Figure 5A and Supplementary Figure S28B). Significant muscle loss was observed in the Ad5-Cre-injected left leg compared to the Ad5-eGFP-injected right leg of the same mouse, which was quantitated as the width of the quadriceps femoris muscle (Figure 5B and 5C). EWS-FLI1 expression has been previously reported to induce caspase 3 transcription, which subsequently triggers apoptosis in vivo [16]. EWS-FLI1 expression in cultured cardiac muscle cells also induced apoptosis in vitro and cardiomyopathy in vivo [30]. Hence, the observed myopathy in the legs injected with Ad5-Cre may have been due to the apoptotic effect of EWS-FLI1 in the myocytes in which Cre-mediated recombination may have occurred. This phenotype was more evident in mice injected at day 1 postnatal compared to the mice injected at 1 or 2 weeks, in which the penetrance of the phenotype observed was 90.9%, 9.1%, and 11.1%, respectively (Figure 5A and Supplementary Figure S28C). This observation may have indicated a differential sensitivity of murine muscle cells to EWS-FLI1 expression during early postnatal development. Alternatively, the variability in the penetrance of the phenotype may have been because the dosage per body weight was much higher in the 1-day-old mice compared to the older mice injected with the same number of Ad5-Cre viral particles. None of the IM-injected mice developed sarcoma when they were observed up to 9 months of age.
For IP delivery, E/F mice were injected with 109 pfu of Ad5-Cre at 1 day or 1 week of age. As the negative control, we used 109 pfu Ad5-eGFP injected littermates of the same age and genotype. Cre expression was confirmed 48 h after IP delivery of Ad5-Cre-eGFP using Maestro fluorescence imaging (Supplementary Figure S29A). Three weeks following injection in 1-day-old mice, most of the Ad5-Cre mice presented with abdominal distention (Figure 5D and Supplementary Figure S29B). The Ad5-Cre-injected E/F mice also showed significantly shortened small intestines (Figures 5E and 5F). Additionally, we observed malformed livers in the Ad5-Cre-injected mice; the liver had a more globular shape compared to the normal disc shape (Supplementary Figure S29C). The same phenotypes were present in the mice injected with Ad5-Cre at 1 week of age (Supplementary Figure S29D). The E/F mice intraperitoneally injected with Ad5-Cre at 1 week of age survived up to 6 months, but none of them developed any detectable ES. Histopathological analysis of the intestines did not reveal any defects in the villi or crypt structures in the majority of the intestine, but there were small areas of necrosis and perforation in the wall in multiple locations in each mouse, which may have led to leakage into the peritoneal cavity that caused distention of the abdomen (Supplementary Figure S30). The growth arrest and apoptotic effects of EWS-FLI1 expression may explain the defects in small intestine development [14-16, 63]. The ability of EWS-FLI1 to antagonize β-catenin/TCF-mediated transcription may have also contributed to the shortened small intestine phenotype [64, 65]. Furthermore, as an effort to deliver Cre recombinase systematically, we injected 109 pfu Ad5-Cre intravenously in 3-week-old E/F mice; their littermates were injected with 109 pfu Ad5-eGFP as a control. Over 7.5 months of follow-up, neither the Ad5-Cre- nor the Ad5-eGFP-injected mice presented with any evident phenotype.
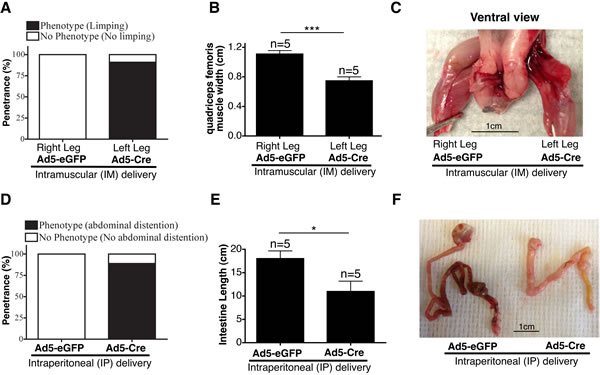
Figure 5: Intramuscular and intraperitoneal delivery of adenovirus-Cre in 1-day-old E/F+/+ mice in Model #16Ad5Cre-EF. A. Penetrance of limping phenotype observed in Ad5-Cre-injected left leg vs. Ad5-eGFP-injected right leg (n = 11) at an age of 1 day. B. Comparison of quadriceps femoris muscle width between an Ad5-eGFP-injected right leg and Ad5-Cre-injected left leg. C. Representative image showing muscle atrophy observed in Ad5-Cre-injected leg. D. Penetrance of abdominal distention phenotype observed in 1-day-old mice IP-injected with Ad5-Cre (n = 9) vs. Ad5-eGFP (n = 7). E. Comparison of intestine length (from stomach to rectum) in 1-day-old mice IP injected with Ad5-Cre vs. Ad5-eGFP. F. Representative image showing shortened intestines observed in Ad5-Cre-injected mice compared to littermate mice of the same genotype injected with Ad5-eGFP.
DISCUSSION
Several pediatric sarcomas contain tumor-specific chromosomal translocations that produce chimeric proteins with novel functions. These neoplasms include alveolar rhabdomyosarcoma, myxoid liposarcoma, synovial sarcoma, and clear cell sarcoma harboring PAX3:FKHR t(2;13), TLS-CHOP t(12;16), SYT-SSX t(X;18), and EWS-ATF1 t(12;22) chromosomal translocations, respectively [22, 23, 66, 67]. Similar to the role of EWS-FLI1 in ES, both Pax3:Fkhr and SYT-SSX have been shown to play critical roles in the malignant phenotypes of rhabdomyosarcoma and synovial sarcoma cell lines, respectively. These observations led to development of useful transgenic mouse models for both tumors. When PAX3:FKHR expression was targeted by the Myf6 promoter in terminally differentiating skeletal muscle, mice developed alveaolar rhabdomyosarcoma with a very low penetrance (< 1%). However, when the transgene was expressed on the Ink4a/Arf or p53 null background, the penetrance increased to 30-40% [24]. Similarly, the expression of the TLS-CHOP chromosomal translocation product under the control of the Prx1 promoter (in early mesodermal tissue) induced myxoid liposarcomas only on the p53 null background [25]. Moreover, the expression of the EWS-ATF1 chromosomal translocation product in neural crest-derived cells resulted in clear cell sarcomas [26], and when SYT-SSX expression was induced in Myf5-expressing myoblasts, 100% of mice developed synovial sarcoma-like tumors [27]. Interestingly, the induction of SYT-SSX expression through Hprt-Cre, Pax3-Cre, or Pax7-Cre resulted in embryonic lethality, and SYT-SSX expression in Myf6-expressing myocytes or Myf6-expressing myofibers resulted in myopathy but no tumors. Therefore, tumor-specific chromosomal translocation products can produce specific types of sarcomas in mice when they are expressed at the right time, in the right cell population and, in some cases, with the help of deleting tumor suppressors.
The successes in developing clinically relevant mouse models for rhabdomyosarcoma, myxoid liposarcoma, clear cell sarcoma, and synovial sarcoma have not been translated to ES, regardless of an exhaustive series of modeling attempts. Several factors have contributed to the failure of developing an ES transgenic mouse model by expressing EWS-FLI1. In most cell types, the expression of EWS-FLI1 induces growth arrest or apoptosis [14-16, 63]. Our findings are consistent with the detrimental effect of EWS-FLI1 expression in sensitive tissues, as we repeatedly observed embryonic lethality or developmental defects. Cooperation from the loss of additional tumor suppressor genes was not observed in our experiments, demonstrating that the correct dosage and timing of EWS-FLI1 expression was not met in these models despite multiple efforts and protocols used. The loss of p53 and Rb or p16 and p19 did not result in ES formation in the presence of EWS-FLI1.
The exact cell of origin for ES remains uncertain, which further impairs our ability to target the correct ontologic cell at the correct developmental stage. Even though many publications support the hypothesis that MSCs may be the cell type of origin [17, 18, 68-71], the lack of lineage-specific promoters as well as a range of MSC phenotypes prevents targeting EWS-FLI1 expression in these cells. In our studies, we used several promoters (Model #11Prx1Cre-EF, Model #12Cre-TL-EF, Models #13RetroLTR-EF and #14piggyBac-EF) that can target EWS-FLI1 expression in different mesenchymal tissues without any success in generating ES in mice.
For Model #1, we concluded that the cells expressing high levels of Cre, which resulted in high EWS-FLI1 expression, did not survive. Following Cre-induced recombination, cells either died by apoptosis or were cleared by the immune system. Bone development is a dynamic process, and normal cells with lost Cre and EWS-FLI1 expression overtook the proper development of bone structures. One possible explanation for our negative results with the E/F+/- Runx2-Cre+ lines could be that the binding of EWS-FLI1 onto the Runx2 promoter negatively regulates Runx2 activity, which could inhibit differentiation of mesenchymal cells [72]. One could assume that EWS-FLI1 down-regulates Runx2 at the transcriptional level, and this is consistent with the lack of Cre expression, which itself retains a certain degree of toxicity.
EWS-FLI1 expression driven by Col 2.3-Cre or Col 3.6-Cre proved fatal to developing embryos. The original study reported robust Cre activity in osteoblast progenitors in E18.5 to postnatal day 5 animals [43]. However, it is unknown whether Cre activity is detected earlier in development. Furthermore, low but detectable levels of Cre activity were reported in the non-osseous tissues of these mice, suggesting the possibility that non-osseous EWS-FLI1 expression caused embryonic lethality. Targeted expression of EWS-FLI1 in the early limb bud mesenchyme (E9.5) with Prx1-Cre was also fatal. Based on the abundant EWS expression levels in most tissues, it can be surmised that the native EWS promoter drives the high level of EWS-FLI1 expression upon Prx1-Cre activation, leading to embryonic lethality. Embryonic lethality was also observed in the Prx1-Cre-driven EWS-FLI1 transgenic line [28].
Accumulating evidence suggests that EWS-FLI1 may support oncogenic phenotypes beyond its role as a DNA-binding transcription factor. Recent epigenomic studies have identified EWS-FLI1 as an epigenetic driver. Similarly to MYC, EWS-FLI1 may function as a transcriptional amplifier of gene expression by binding to open promoters of widely expressed genes; EWS-FLI1 may also activate novel enhancers and superenhancers [73, 74]. It is therefore possible that the tissue- or stage-specific chromatin accessibility to key genes of ES tumorigenesis differs between mice and men, thus vexing our attempts at modeling [75].
Alternative splicing is emerging as an important mechanism in carcinogenesis [76]. Alternative splicing of the same gene may result in two proteins with opposite functional roles. BCL2L1 and FAS Receptor (TNR6) are two examples that can create both pro-apoptotic and anti-apoptotic protein products as a result of excluding or including a specific exon. EWS-FLI1 interacts with key proteins in the splicing complex and regulates alternative splicing of a specific set of genes that do not always overlap with transcriptional target genes (FLI1 target genes) [77-81]. Therefore, EWS-FLI1 may function through regulating transcription of certain genes and regulating translation of a different set of genes. The homology between human and mouse genes does not always cover the alternative splicing sites. Due to differences in alternative splicing sites between mice and humans, the expression of EWS-FLI1 in human cells may potentially result in a different set of alternatively spliced products than the products in murine cells. If these sets of genes are critical for ES development, then creating an ES model in mice by expressing EWS-FLI1 may not be possible.
Another difference between the human and mouse genomes involves microsatellite sequences. EWS-FLI1 regulates target gene expression through GGAA microsatellite response elements [82-84]. The number of GGAA microsatellite motifs and their distance to promoters significantly alter target gene expression [85-87]. Furthermore, recent findings indicate that ES patients preferentially carry the A risk allele (compared to T) of the rs79965208 single-nucleotide polymorphism (SNP). An ES genome-wide association study identified microsatellite susceptibility variants near the EGR2 gene. The increase in the length of GGAA microsatellites may therefore contribute to EWS-FLI1 oncogenesis [88], but this microsatellite and SNP are not conserved in mice. Therefore, even though the human and mouse genomes may have similar EWS-FLI1 target genes, their expression patterns may be completely different due to differences in GGAA microsatellite motifs between the human and mouse genomes.
Human ES cells express high levels of cell surface protein CD99, which is routinely used for confirming diagnosis [89-92]. In addition to its diagnostic value, inhibition of CD99 expression or engagement by CD99 antibodies stop growth of ES cell lines both in culture and in xenograft models [93-97]. Mouse homolog of CD99 has less than 50% amino acid identity to its human counterpart [98, 99]. Therefore, biological pathways regulated by human CD99 and mouse CD99 may show significant differences. Because human CD99 is a critical component of ES pathogenesis, it is possible that the lack of a comparable protein in mouse cells may be responsible for the lack of ES development from EWS-FLI1 expression.
ES patients harbor a balanced chromosomal translocation, which generates two chimeric fusion genes, EWS-FLI1 and FLI1-EWS. Many researchers in the field could not detect expression of FLI1-EWS mRNA or protein in ES cell lines or human tumor samples. However, a recent publication provided data suggesting that FLI1-EWS is expressed in ES cells and more importantly FLI1-EWS expression is required for EWS-FLI1 mediated transformation [100]. If this hypothesis is true, the lack of FLI1-EWS component in our attempts might have been responsible for the failed mouse models .
In summary, the ectopic expression of EWS-FLI1 in murine MSCs, neural crest-derived stem cells, or osteochondrogenic progenitor cells could transform primary cells into malignant tumor cells. However, achieving tumor growth in animals with intact immune systems is a higher-level challenge than anchorage-independent growth or tumor development in immunocompromised mice with cells transformed in vitro. Hence, it remains possible that an EWS-FLI1-driven sarcoma model can be generated, although the number of failed attempts at creating a transgenic model suggest that this challenge has yet to be accomplished. This comprehensive analysis of those models that have been attempted, without success, should allow future investigations to advance without repeating unsuccessful models.
MATERIALS AND METHODS
All procedures involving mice were approved by the respective institutions’ animal care and use committees.
Model #1Runx2Cre-EF
Mice were kept under standardized conditions at the Decentralized Biomedical Facility of the Medical University of Vienna. E/F mice harboring a Cre-inducible EWS-FLI1 knocked into the ubiquitous ROSA26 locus (ROSA26loxP-STOPloxP-HA-EWS-FLI1 allele) [29] were crossed to three different Runx2-Cre isoforms [35]. Runx2 was expressed from promoters p1 and p2 [101]. These strains were generated with a BAC (bacterial artificial chromosome) approach with different transgenic integrations and copy numbers (#777, #784, #1634). E/F+/-Runx2-Cre+ mice were also bred to mice deficient in p16INK4A and p19ARF proteins [37]. All mice were on mixed background (Sv129 and C57BL/6).
Model #2OsxCre-EF
Mice
E/F mice were generously provided by Dr. S. Baker (St. Jude Children’s Research Hospital) and were on a C57BL/6 background. Osx-Cre mice on a C57BL/6J background were obtained from the Jackson Laboratories (Bar Harbor, ME). Osx-Cre+p53fl/fl pRbfl/fl animals were generously provided by Dr. Stuart Orkin (Harvard University) and were on a C57BL/6J 129 FVB/n hybrid background. E/F mice were maintained as homozygotes (E/F+/+) and bred with either Osx-Cre+ or Osx-Cre+p53fl/fl pRbfl/fl animals. Mice received doxycycline diet (2000mg/kg diet, Harlan Laboratories) throughout the pregnancy (average 21 days) and withdrawn at the weaning age of pups (postnatal day 21). Mice were euthanized if the animals showed signs of pain and distress or when the animals reached the age of 300 days.
Flow cytometry
For cell surface analysis of splenocytes, harvested spleens were minced into small pieces in 1x PBS to release the blood cells, which were subsequently strained using 70 μm cell strainer (Fisher, Cat No. 352350) to prepare a single cell suspension. The strained mix was then subjected to centrifugation at 350 g for 5 min. Pelleted cells were resuspended in 1x PBS and spleen cell count was determined using hemocytometer. 0.25 μl of CD43-PE (Clone S11, BioLegend, Cat No. 143205), 0.5 μl of CD45-Alexa Fluor-488 (Clone 30-F111, BioLegend, Cat No. 103122), 1 μl of CD71-Brilliant Violet 421 (Clone RI7217, BioLegend, Cat No. 113813), and 2.5 μl of CD117-APC (Clone 2B8, BioLegend, Cat No. 105812) fluorescent conjugated primary antibodies were used to stain one million splenocytes in 100 μl 1x PBS for 20 min on ice in the dark. The stained cells were washed twice with 1x PBS. The cells were then resuspended in 500 μl of 1x PBS and analyzed using flow cytometer.
Western blot
Total protein extracts were prepared from spleens and livers of E/F+/- Osx-Cre+ p53fl/+ pRbfl/+ mice using the following protocol. Small fragments (~2mm3) of flash frozen spleens were homogenized using hand held mortar mixer (VWR, Cat. No: 47747-370) in 200 μl phospholysis buffer (50 mM HEPES pH 7.9, 100 mM sodium chloride, 4.0 mM sodium pyrophosphate, 10 mM EDTA, 10 mM sodium fluoride, and 1% Triton X-100 v:v) containing 2.0 mM sodium vanadate, 1.0 mM PMSF, 4.0 µg/ml aprotinin, and 4.0 µg/ml leupeptin. Once the spleens were fully homogenized, the lysates were incubated on ice for 30 min. Next, the lysates were subjected to centrifugation for 10 min at 16,000 g at 4oC. Proteins were denatured in 5x Laemmli sample buffer and subjected to SDS-PAGE (10% polyacrylamide). Resolved proteins were transferred to 0.45 μm Immobilon-P PVDF membrane (Millipore, Cat No. IPVH00010). The membranes were blocked in 5% nonfat dry milk in 1x TTBS (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Tween 20 v:v) for 2 hrs. Dilutions for primary antibodies were anti-FLI1 (Santa Cruz Biotechnology, Cat No. sc-356) at 1:1000, anti-HA (Roche, Cat No. 1867423) at 1:500, and anti-actin-horseradish peroxidase (C-11, Santa Cruz Biotechnology, Cat No. sc-1615 ) at 1:5000. Primary antibodies were added to the membranes in 5% nonfat dry milk in 1x TTBS for 2hrs at room temperature. The membranes were then washed three times in 1x TTBS and incubated for 1h with 1:5000 dilution of horseradish peroxidase-linked anti-rabbit (GE Healthcare, Cat No. LNA934V/AG) or anti-rat secondary antibody (R&D, Cat No. HAF005) prepared in 5% nonfat dry milk. Blots were washed three times in 1x TTBS and then developed using Immobilon Western Chemiluminescent HRP Substrate per the manufacturer’s instructions (Millipore Corporation, Cat No. WBKLS0100). Chemiluminescence was detected using a Fujifilm LAS-3000 imaging system.
RT-qPCR
RNA was extracted from ~50 mg flash frozen spleen fragments using TRIzol according to the manufacturer’s protocol (Invitrogen, Cat No. 15596-018). Extracted RNA was reverse transcribed to cDNA using QuantiTect reverse transcription kit (Qiagen, Cat No. 205311) as described by the manufacturer using Applied Biosytems Veriti Thermal Cycler. Real-time quantitative PCR was performed in an Eppendorf Mastercycler realplex using KiCqStart SYBR Green qPCR ReadyMix (Sigma-Aldrich, Cat No. KCQS00) per manufacturer’s protocol. Data were analyzed for expression relative to 18S rRNA using the comparative Ct method. Forward and reverse qPCR primers for EWS-FLI1 were CAGCCTCCCACTAGTTACCC and GTTGAGGCCAGAATTCATG, respectively.
Model #3Col1a2.3Cre-EF and Col1a3.6Cre-EF and Model #11Prx1Cre-EF
Ews-FLI1KI mice (C57BL/6 background) had been described earlier [16]. Col1a2.3-Cre and Col1a3.6-Cre mice were generated previously [43] and Prx1-Cre mouse was purchased from Jackson Laboratory. Ews-FLI1KI mice were crossed with Col1a2.3-Cre, Col1a3.6-Cre or Prx1-Cre and pups were genotyped at weaning and at birth (n>100 for each cross). No viable pups carrying both the EWS-FLIKI and Cre alleles were obtained in any of the crosses.
Models #4Cosco-EF, #5Pgk-EF, #6Nse-EF and Nse-EF-SV, #7NEFL-EF, #8MT-EF, #9PLAPtTA-EF, and #10COMET and COMETΔNeo
Generation of the targeting vector and COMET mouse
The 6158 bp COMET cassette (Supplementary Figure S16) was gene synthetized (EpochBiolabs, INC. Missouri city, TX) and cloned into Sma1 digested pUC19. The resulting pUC19-COMET plasmid was digested with Nhe1, the resulting COMET cassette was subcloned into Xba1 digested pROSA26-1 vector (gift from P. Soriano). The resulting pROSA26-1-COMET plasmid was linearized using SacII and purified. This targeting construct was electroporated into CK35 (129 Stevens Pasteur background) mouse embryonic stem cells for homologous recombination. G418-resistant clones were isolated, screened by PCR for homologous recombination, and confirmed by southern blot (150 bp EcoR1/HindIII fragment of pROSA-5’, gift from P. Soriano). Two positive embryonic stem cell clones (2C12, 2D8) were injected into C57BL/6J blastocysts to generate the first generation of chimeras (Model #10COMET). FRT flanked Neomycin selection cassette was deleted in vitro by transient transfection of pCAGGS-Flpe (Gene Bridges GmbH, Heidelberg, Germany) plasmid into 2C12, 2D8 ES clone according to manufacturer protocol. The deleted subclones 2A3 (derived from 2C12) and 1C3 (derived from 2D8) were injected into C57BL/6J blastocysts to generate the second generation of chimeras (Model #10COMETΔNeo).
Quantitative RT-PCR
cDNAs were synthesized from 1µg of RNA using the GeneAmp RNA PCR core Kit (Applied Biosystem, Courtaboeuf, France). Quantitative PCR analyses were performed using SYBR green (Applied Biosystem). The following primers were used: EWS-FLI1 (5’-GCCAAGCTCCAAGTCAATATAGC-3’, 5’-GAGGCCAGAATTCATGTTATTGC-3’); luciferase (5’-TTACACGGCGATCTTTCCGCCC-3’, 5’-AGTTGCGCGGAGGAGTTGTGTT-3’). Reactions were run on an ABI/PRISM 7500 (Applied Biosystem) and analyzed using the 7500 system SDS software. For detection of splice variant between Gt(ROSA)26Sor transcripts and COMET transcript, the following primers were used (EWSR1-kozac-Rev 5’-TCCGTGGACGCCATGGTGAATT-3’,ROSA26-EX1-Fw 5’-CTGCCGGGGCCGCCTAAAGAA-3’) with Phusion High-Fidelity DNA polymerase (Finnzymes, Finland); 35 cycles of 98°C for 7 seconds, 60°C for 20 seconds, and 72°C for 30 seconds.
Luciferase assay
293T cells were plated into six well plates and grown in DMEM (Invitrogen, Cergy Pontoise, France) supplemented with 10% fetal calf serum. The day after, cells were CaCl2 transfected for six hours using 1µg of pUC19-COMET alone and/or pIRES-Cre (Cre recombinase cloned into Xma1-Not1 digested pIRES) and/or pTet-On Advanced (Takara Bio Europe/Clontech, France) and 20 ng of pREP7-Rluc, (kindly provided by Keji Zhao). Twenty-four hours post-transfection, doxycycline (0 to 1000 ng/ml) was added to the media. Forty-eight hours post-transfection, cells were lysed and assayed for luciferase activity using the dual luciferase reporter assay system (Promega, Charbonnieres-les-bain, France) according to the manufacturer’s instructions. Firefly activity was normalized to Renilla luciferase activity to adjust differences in transfection efficiency.
Antibodies
β-actin antibody (A-5316) was purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France). Anti-FLI1 antibody (7.3) was described previously [102].
Noninvasive bioluminescence imaging
Approximately twenty minutes prior imaging, mice were injected intraperitoneally with 10 µl/g of body weight of sterile D-luciferin PBS solution (15 mg/ml). Mice were imaged using an IVIS Spectrum system (Caliper Life Sciences S.A., Villepinte, France) for 1 minute. Anesthesia was administered in an induction chamber with 2.5% isoflurane and reduced to 1.5% while the animal was in the IVIS imaging device. The data are reported as radiance (photons/sec/cm2/steradian).
Model #12Cre-TL-EF
Mice
Generation of Ewsloxpurolox/wt, Fli1loxhygrolox/wt and EWS-FLI1-V5 mice was carried out using standard gene targeting protocols. Correctly targeted embryonic stem cells were injected into albino C57BL/6 blastocysts by the Transgenic Facility at Stanford University. Southern blot analyses on embryonic stem cell clones were performed following standard protocols. Sox9-Cre, Dermo1-Cre, P0-Cre and Col1α2-Cre mice were obtained from researchers that made the mice. Prx1-Cre mice were obtained from the Jackson Laboratory (B6.Cg-Tg(Prrx1-cre)1Cjt/J Stock Number: 005584). CMV-Cre mice were obtained from the Jackson Laboratory (B6.C-Tg(CMV-cre)1Cgn/J Stock Number: 006054). Ink4a/Arf floxed mice were a kind gift from Dr. Ron DePinho [103]. At the end of the tumor study, mice were euthanized and a necropsy was carried out. Gross abnormalities by eye were noted and chunks of tumors were collected and snap frozen in liquid nitrogen for future study. Tumors chunks were crushed cold using a mortar and pestle chilled by liquid nitrogen and the ground tissue was resuspended in Trizol.
Cell culture
MEFs were isolated as following standard procedures from E13.5 embryos and cultured in DMEM with 10% BGS and 100U/100ug/2 mM Pen/Strep/Glutamine (Invitrogen). Mouse bone progenitor cells were isolated as previously described and cultured in α-MEM supplemented with 20% fetal bovine serum (Omega Scientific) and 100U/100μg/2 mM Pen/Strep/Glutamine (Invitrogen) [104]. Adenoviral GFP or Cre infections were performed overnight in culture. Hematopoietic cells were isolated following standard protocols and immediately resuspended in TRIzol [105].
RNA analysis
RNA was isolated using TRIzol reagent (Invitrogen) following the manufacturer’s specifications. cDNA was synthesized using either the DyNAmo cDNA synthesis kit (New England Biolabs, F470) or the Maxima First Strand cDNA synthesis kit (Thermo, K1642). qPCR was performed using SYBR Green (Applied Biosystems).
Primers
EWS lox genomic DNA TGGCCAGGCTATAAAACTACTTCCA TGCTGGGATGACTAGTTACAATTCC
FLI1 lox genomic DNA GTGACGGAGATCCCGAATTCTTTCC AACTGGGCCAGCCAACGCTTTC
EWS-FLI1 CAATATAGCCAACAGAGCAGCA GCTCCTCTCCTGACAGAGTCAT
GGGCAGCAGCCTCCTACTA TTCCATGCTCCTCTCCTGAC
HPRT TGACACTGGTAAAACAATGCA GGTCCTTTTCACCAGCAAGCT
Ink4a TGAGGCCGGATTTAGCTCTGCTC TCCGCTGCAGACAGACTTGCCAG
Arf TGAGGCCGGATTTAGCTCTGCTC CTTGGTCACTGTGAGGATTC
Models #13RetroLTR-EF and #14piggyBac-EF
BM-MSCs were obtained from 6-week-old BALB/c mice (Crea Japan), according to the previously described method [57]. Self-renewal activities of BM-MSCs were confirmed by in vitro clonogenic assays, and differentiation into multiple lineages were confirmed by induction toward osteogenic, chondrogenic, neuronal, adipogenic and myogenic differentiation [56]. A FLAG-tagged EWS-FLI1 was subcloned into pMYs-ires-neo retroviral vector, and retroviral packaging was carried out using PLAT-E cells (a gift from T. Kitamura). BM-MSCs were then transduced with the EWS-FLI1 retrovirus and injected from tail vein into irradiated BALB/c mice (model #13). EWS-FLI1 was also subcloned into piggyBac transposon plasmid (a gift from P. Liu), and BM-MSCs were transfected with PB-EF and transposase mRNA generated by using an mMESSAGE mMACHINE T7 Ultra kit (Ambion). The cells were transplanted subcutaneously or injected via tail vein into irradiated recipients. Expression of EWS-FLI1 was confirmed by western blotting using anti-FLAG M2 and anti-beta-tubulin antibodies (Sigma).
Model #15 CreEP-TL-EF
Generation of Ewsr1fl/+:Fli1fl/+ mice (C57BL/6) was described elsewhere [30]. Genotypes of mice were confirmed by genomic PCR. The Cre expression vector pMC1-Cre (a gift by T. Yagi) was delivered by in vivo electroporation. The gastrocnemius muscles of 8-week-old mice were injected with 50 µg of plasmid DNA, and the site of inoculation was immediately given electric pulses with a CUY21EDIT Square Wave Electroporator (NEPAGENE) using a CUY560-3 electrode. The pulse was 50 ms in duration at a voltage of 50V. Cre expression was confirmed 2 weeks after electroporation by RT-PCR.
Model #16Ad5Cre-EF
Mice
Both adenovirus-Cre (Ad5-CMV-Cre-eGFP) and adenovirus-eGFP (Ad5-CMV-eGFP) were purchased from University of Iowa Viral Vector Core Facility (Iowa city, IA). For intramuscular (IM) delivery E/F+/+ mice were injected with 20μl of 109pfu Ad5-CMV-Cre-eGFP in the left leg and 20μl of 109pfu Ad5-CMV-eGFP in the right leg at 1 day, 1 week, or 2 weeks of age using 31guage 3/8 inch Thinpro insulin syringe (Terumo). For IP delivery, E/F+/+ mice were injected with 20μl of 109pfu Ad5-Cre at 1 day or 1 week of age. Littermate mouse of the same age and genotype were injected with 20μl of 109pfu Ad5-eGFP as a control. For IV delivery, 20μl of 109pfu Ad5-Cre was intravenously injected to 3 weeks old E/F+/+ mice. Their littermates were injected with 20μl of 109pfu Ad5-eGFP as a control.
PCR
DNA extracted from E/F+/- and E/F+/+ mice livers were used as negative controls while DNA from leukemic E/F+/- Mx1-Cre+ mouse liver was used as a positive control. DNAs extracted from muscles of E/F+/+ mice injected with Ad5-eGFP in the right legs were also used as negative controls. DNA was isolated using wizard genomic DNA purification kit per manufacturer’s protocol (Promega, A1120) Extracted DNAs were subjected to PCR to confirm that Cre-mediated recombination has occurred. A forward primer located in the endogenous Rosa26 locus (GATCCACTAGTTCTAGAGCGGC) and a reverse primer that lies shortly after the first loxP site (GAGTTGTTATCAGTAAGGGAGC) were used to detect the knock-in allele. On the hand, the same forward primer in combination with a reverse primer that lies within the EWS-FLI1 sequence (GGTATCATAAGCACCAGTG) were used to detect the removal of the STOP-cassette.
ACKNOWLEDGMENTS AND FUNDING
Model #1Runx2Cre-EF
The generation of this model was supported by charity funds collected by St. Anna Kinderkrebsforschung e.V. and from support from the Deutsche Forschungsgemeinschaft Collaborative Research Centre 1149 ‘Trauma’ (INST 40/492-1), “Immunobone” (SPP 1468 Tu220/6-2). We thank Dr. S. Baker for providing E/F mice.
Models #2OsxCre-EF and #16Ad5Cre-EF
Support for this work came from the Children’s Cancer Foundation (Baltimore MD), St. Baldrick’s Foundation, Go4theGoal, a Burroughs Wellcome Clinical Scientist Award in Translational Research (J.T.), and the NIH RC4CA156509 (J.T.), R01CA133662 (J.T.), R01CA138212 (J.T.). We wish to thank the Flow Cytometry/Cell sorting and Histopathology & Tissue Shared Resource (HTSR) Shared Resources at the Lombardi Comprehensive Cancer Center (Georgetown University), which are supported by a grant P30 CA51008 (PI Louis Weiner) from the National Cancer Institute. In addition, we would like to thank Kelli Schanze for her excellent technical support.
Models #4Cosco-EF, #5Pgk-EF, #6Nse-EF and Nse-EF-SV, #7NEFL-EF, #8MT-EF, #9PLAPtTA-EF, and #10COMET and COMETΔNeo
This work was supported by grants from the Institut National de la Santé et de la Recherche Médicale, the Institut Curie, the Institut National du Cancer, the Ligue Nationale contre le Cancer (Equipe labellisée and CIT program), the Réseau National des Génopoles, Agence National de la Recherche, the société Française des Cancers de l’Enfant, and the following associations: Courir pour Mathieu, Dans les pas du Géant, Olivier Chape, Les Bagouzamanon and les Amis de Claire.
Model #12Cre-TL-EF
We thank Dr. Charles Chan and Dr. Irving Weissman for helpful discussions. We especially thank Dr. Charles Chan for teaching us to isolate embryonic mouse bone progenitors, for reagents and for FACS training. We thank Dr. Hong Zeng and the rest of the members of the Transgenic Facility at Stanford University for help in generating the chimeras and discussions about ES targeting. We thank Dr. Ronald DePinho for the conditional Ink4a/Arf mice. We thank Dr. David Ornitz for the Dermo1-Cre mice. We thank Dr. Mohan Subburaman and Dr. Peter Angel for the Col1a2-Cre mice. We thank Dr. Benoit de Crombrugghe for the Sox9-Cre mice. We thank Dr. Joe Kissil for the P0-Cre mice.
CONFLICTs OF INTEREST
The authors declare no conflict of interest.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Arndt CA and Crist WM. Common musculoskeletal tumors of childhood and adolescence. The New England journal of medicine. 1999; 341:342-352.
2. Potratz J, Dirksen U, Jurgens H and Craft A. Ewing sarcoma: clinical state-of-the-art. Pediatric hematology and oncology. 2012; 29:1-11.
3. Tsokos M, Alaggio RD, Dehner LP and Dickman PS. Ewing sarcoma/peripheral primitive neuroectodermal tumor and related tumors. Pediatric and developmental pathology. 2012; 15:108-126.
4. Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G and et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992; 359:162-165.
5. May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R and Denny CT. The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Molecular and Cellular Biology. 1993; 13:7393-7398.
6. Tanaka K, Iwakuma T, Harimaya K, Sato H and Iwamoto Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. The Journal of clinical investigation. 1997; 99:239-247.
7. Toretsky JA, Connell Y, Neckers L and Bhat NK. Inhibition of EWS-FLI-1 fusion protein with antisense oligodeoxynucleotides. J Neurooncol. 1997; 31:9-16.
8. Kovar H, Aryee DN, Jug G, Henockl C, Schemper M, Delattre O, Thomas G and Gadner H. EWS/FLI-1 antagonists induce growth inhibition of Ewing tumor cells in vitro. Cell Growth Differ. 1996; 7:429-437.
9. Maksimenko A, Lambert G, Bertrand JR, Fattal E, Couvreur P and Malvy C. Therapeutic potentialities of EWS-Fli-1 mRNA-targeted vectorized antisense oligonucleotides. Ann N Y Acad Sci. 2003; 1002:72-77.
10. Ouchida M, Ohno T, Fujimura Y, Rao VN and Reddy ES. Loss of tumorigenicity of Ewing’s sarcoma cells expressing antisense RNA to EWS-fusion transcripts. Oncogene. 1995; 11:1049-1054.
11. Chansky HA, Barahmand-Pour F, Mei Q, Kahn-Farooqi W, Zielinska-Kwiatkowska A, Blackburn M, Chansky K, Conrad EU 3rd, Bruckner JD, Greenlee TK and Yang L. Targeting of EWS/FLI-1 by RNA interference attenuates the tumor phenotype of Ewing’s sarcoma cells in vitro. J Orthop Res. 2004; 22:910-917.
12. Dohjima T, Lee NS, Li H, Ohno T and Rossi JJ. Small interfering RNAs expressed from a Pol III promoter suppress the EWS/Fli-1 transcript in an Ewing sarcoma cell line. Mol Ther. 2003; 7:811-816.
13. Kovar H, Ban J and Pospisilova S. Potentials for RNAi in sarcoma research and therapy: Ewing’s sarcoma as a model. Semin Cancer Biol. 2003; 13:275-281.
14. Deneen B and Denny CT. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 2001; 20:6731-6741.
15. Lessnick SL, Dacwag CS and Golub TR. The Ewing’s sarcoma oncoprotein EWS/FLI induces a p53-dependent growth arrest in primary human fibroblasts. Cancer cell. 2002; 1:393-401.
16. Sohn EJ, Li H, Reidy K, Beers LF, Christensen BL and Lee SB. EWS/FLI1 oncogene activates caspase 3 transcription and triggers apoptosis in vivo. Cancer research. 2010; 70:1154-1163.
17. Riggi N, Cironi L, Provero P, Suva ML, Kaloulis K, Garcia-Echeverria C, Hoffmann F, Trumpp A and Stamenkovic I. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer research. 2005; 65:11459-11468.
18. Riggi N, Suva ML, Suva D, Cironi L, Provero P, Tercier S, Joseph JM, Stehle JC, Baumer K, Kindler V and Stamenkovic I. EWS-FLI-1 expression triggers a Ewing’s sarcoma initiation program in primary human mesenchymal stem cells. Cancer research. 2008; 68:2176-2185.
19. Castillero-Trejo Y, Eliazer S, Xiang L, Richardson JA and Ilaria RL Jr. Expression of the EWS/FLI-1 oncogene in murine primary bone-derived cells Results in EWS/FLI-1-dependent, ewing sarcoma-like tumors. Cancer research. 2005; 65:8698-8705.
20. von Levetzow C, Jiang X, Gwye Y, von Levetzow G, Hung L, Cooper A, Hsu JH and Lawlor ER. Modeling initiation of Ewing sarcoma in human neural crest cells. Plos One. 2011; 6:e19305.
21. Tanaka M, Yamazaki Y, Kanno Y, Igarashi K, Aisaki K, Kanno J and Nakamura T. Ewing’s sarcoma precursors are highly enriched in embryonic osteochondrogenic progenitors. The Journal of clinical investigation. 2014; 124:3061-3074.
22. De Giovanni C, Landuzzi L, Nicoletti G, Lollini PL and Nanni P. Molecular and cellular biology of rhabdomyosarcoma. Future Oncol. 2009; 5:1449-1475.
23. Haldar M, Randall RL and Capecchi MR. Synovial sarcoma: from genetics to genetic-based animal modeling. Clinical orthopaedics and related research. 2008; 466:2156-2167.
24. Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA and Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004; 18:2614-2626.
25. Charytonowicz E, Terry M, Coakley K, Telis L, Remotti F, Cordon-Cardo C, Taub RN and Matushansky I. PPARgamma agonists enhance ET-743-induced adipogenic differentiation in a transgenic mouse model of myxoid round cell liposarcoma. The Journal of clinical investigation. 2012; 122:886-898.
26. Yamada K, Ohno T, Aoki H, Semi K, Watanabe A, Moritake H, Shiozawa S, Kunisada T, Kobayashi Y, Toguchida J, Shimizu K, Hara A and Yamada Y. EWS/ATF1 expression induces sarcomas from neural crest-derived cells in mice. The Journal of clinical investigation. 2013; 123:600-610.
27. Haldar M, Hancock JD, Coffin CM, Lessnick SL and Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer cell. 2007; 11:375-388.
28. Lin PP, Pandey MK, Jin F, Xiong S, Deavers M, Parant JM and Lozano G. EWS-FLI1 induces developmental abnormalities and accelerates sarcoma formation in a transgenic mouse model. Cancer research. 2008; 68:8968-8975.
29. Torchia EC, Boyd K, Rehg JE, Qu C and Baker SJ. EWS/FLI-1 induces rapid onset of myeloid/erythroid leukemia in mice. Molecular and cellular biology. 2007; 27:7918-7934.
30. Tanaka M, Yamaguchi S, Yamazaki Y, Kinoshita H, Kuwahara K, Nakao K, Jay PY, Noda T and Nakamura T. Somatic chromosomal translocation between Ewsr1 and Fli1 loci leads to dilated cardiomyopathy in a mouse model. Sci Rep. 2015; 5:7826.
31. Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, Abaan OD, Chou TH, Dakshanamurthy S, Brown ML, Uren A and Toretsky JA. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nature medicine. 2009; 15:750-756.
32. Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, Albanese C, Toretsky JA and Uren A. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. The Journal of clinical investigation. 2011; 121:148-160.
33. Leacock SW, Basse AN, Chandler GL, Kirk AM, Rakheja D and Amatruda JF. A zebrafish transgenic model of Ewing’s sarcoma reveals conserved mediators of EWS-FLI1 tumorigenesis. Disease models & mechanisms. 2012; 5:95-106.
34. Jeong JH, Jin JS, Kim HN, Kang SM, Liu JC, Lengner CJ, Otto F, Mundlos S, Stein JL, van Wijnen AJ, Lian JB, Stein GS and Choi JY. Expression of Runx2 transcription factor in non-skeletal tissues, sperm and brain. J Cell Physiol. 2008; 217:511-517.
35. Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, Kirilov M, Mandic V, Takacz A, Schmidt-Ullrich R, Ostermay S, Schinke T, Spanbroek R, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010; 11:517-531.
36. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, Reynaud S, Rio-Frio T, Hedlund E, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer discovery. 2014; 4:1342-1353.
37. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D and DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996; 85:27-37.
38. Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR and de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002; 108:17-29.
39. Rodda SJ and McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006; 133:3231-3244.
40. Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH, Alt FW, McMahon AP and Orkin SH. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev. 2008; 22:1662-1676.
41. Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM and Long F. Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. Plos One. 2014; 9:e85161.
42. Zomer A, Maynard C, Verweij FJ, Kamermans A, Schafer R, Beerling E, Schiffelers RM, de Wit E, Berenguer J, Ellenbroek SIJ, Wurdinger T, Pegtel DM and van Rheenen J. In Vivo Imaging Reveals Extracellular Vesicle-Mediated Phenocopying of Metastatic Behavior. Cell. 2015; 161:1046-1057.
43. Liu F, Woitge HW, Braut A, Kronenberg MS, Lichtler AC, Mina M and Kream BE. Expression and activity of osteoblast-targeted Cre recombinase transgenes in murine skeletal tissues. Int J Dev Biol. 2004; 48:645-653.
44. Horowitz M, DeLaney, T, Malawer, M, Tsokos M. Ewing’s sarcoma family of tumors: Ewing’s sarcoma of bone and soft tissue and the peripheral primitive neuroectodermal tumors. (Philadelphia: Lippincott). 1993.
45. Beaudet L, Charron G, Houle D, Tretjakoff I, Peterson A and Julien JP. Intragenic regulatory elements contribute to transcriptional control of the neurofilament light gene. Gene. 1992; 116:205-214.
46. Albert H, Dale EC, Lee E and Ow DW. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995; 7:649-659.
47. Strathdee D, Ibbotson H and Grant SG. Expression of transgenes targeted to the Gt(ROSA)26Sor locus is orientation dependent. PLoS One. 2006; 1:e4.
48. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999; 21:70-71.
49. Scacheri PC, Crabtree JS, Novotny EA, Garrett-Beal L, Chen A, Edgemon KA, Marx SJ, Spiegel AM, Chandrasekharappa SC and Collins FS. Bidirectional transcriptional activity of PGK-neomycin and unexpected embryonic lethality in heterozygote chimeric knockout mice. Genesis. 2001; 30:259-263.
50. Logan M, Martin JF, Nagy A, Lobe C, Olson EN and Tabin CJ. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002; 33:77-80.
51. Schwenk F, Baron U and Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995; 23:5080-5081.
52. Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA and Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003; 130:3063-3074.
53. Florin L, Alter H, Grone HJ, Szabowski A, Schutz G and Angel P. Cre recombinase-mediated gene targeting of mesenchymal cells. Genesis. 2004; 38:139-144.
54. Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T and de Crombrugghe B. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. P Natl Acad Sci USA. 2005; 102:14665-14670.
55. Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S and Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999; 212:191-203.
56. Shiota M, Heike T, Haruyama M, Baba S, Tsuchiya A, Fujino H, Kobayashi H, Kato T, Umeda K, Yoshimoto M and Nakahata T. Isolation and characterization of bone marrow-derived mesenchymal progenitor cells with myogenic and neuronal properties. Exp Cell Res. 2007; 313:1008-1023.
57. Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF and Prockop DJ. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood. 2004; 103:1662-1668.
58. Cadinanos J and Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007; 35:e87.
59. Tsumura H, Yoshida T, Saito H, Imanaka-Yoshida K and Suzuki N. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene. 2006; 25:7673-7679.
60. Meuwissen R, Linn SC, van der Valk M, Mooi WJ and Berns A. Mouse model for lung tumorigenesis through Cre/lox controlled sporadic activation of the K-Ras oncogene. Oncogene. 2001; 20:6551-6558.
61. Ueda S, Fukamachi K, Matsuoka Y, Takasuka N, Takeshita F, Naito A, Iigo M, Alexander DB, Moore MA, Saito I, Ochiya T and Tsuda H. Ductal origin of pancreatic adenocarcinomas induced by conditional activation of a human Ha-ras oncogene in rat pancreas. Carcinogenesis. 2006; 27:2497-2510.
62. Hung KE, Maricevich MA, Richard LG, Chen WY, Richardson MP, Kunin A, Bronson RT, Mahmood U and Kucherlapati R. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. P Natl Acad Sci USA. 2010; 107:1565-1570.
63. Zwerner JP, Guimbellot J and May WA. EWS/FLI function varies in different cellular backgrounds. Experimental cell research. 2003; 290:414-419.
64. Navarro D, Agra N, Pestana A, Alonso J and Gonzalez-Sancho JM. The EWS/FLI1 oncogenic protein inhibits expression of the Wnt inhibitor DICKKOPF-1 gene and antagonizes beta-catenin/TCF-mediated transcription. Carcinogenesis. 2010; 31:394-401.
65. Fevr T, Robine S, Louvard D and Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol. 2007; 27:7551-7559.
66. Crozat A, Aman P, Mandahl N and Ron D. Fusion of Chop to a Novel Rna-Binding Protein in Human Myxoid Liposarcoma. Nature. 1993; 363:640-644.
67. Sonobe H, Takeuchi T, Taguchi T, Shimizu K, Iwata J, Furihata M and Ohtsuki Y. Further characterization of the human clear cell sarcoma cell line HS-MM demonstrating a specific t(12;22)(q13;q12) translocation and hybrid EWS/ATF-1 transcript. J Pathol. 1999; 187:594-597.
68. Burns JS, Abdallah BM, Schroder HD and Kassem M. The histopathology of a human mesenchymal stem cell experimental tumor model: support for an hMSC origin for Ewing’s sarcoma? Histology and histopathology. 2008; 23:1229-1240.
69. Lin PP, Wang Y and Lozano G. Mesenchymal Stem Cells and the Origin of Ewing’s Sarcoma. Sarcoma. 2011; 2011.
70. Potikyan G, France KA, Carlson MR, Dong J, Nelson SF and Denny CT. Genetically defined EWS/FLI1 model system suggests mesenchymal origin of Ewing’s family tumors. Laboratory investigation. 2008; 88:1291-1302.
71. Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P and Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer cell. 2007; 11:421-429.
72. Li X, McGee-Lawrence ME, Decker M and Westendorf JJ. The Ewing’s sarcoma fusion protein, EWS-FLI, binds Runx2 and blocks osteoblast differentiation. J Cell Biochem. 2010; 111:933-943.
73. Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, Formey A, Patel A, Gymrek M, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer cell. 2014; 26:668-681.
74. Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schonegger A, Datlinger P, Kubicek S, Bock C and Kovar H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015; 10:1082-1095.
75. Kovar H. Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin Ther Targets. 2014; 18:1315-1328.
76. Chen J and Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene. 2015; 34:1-14.
77. Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, Yu S, Liu X, Paulsen MT, Ljungman ME, Wu CH, Lawlor ER, Uren A, et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. P Natl Acad Sci USA. 2015; 112:E1307-1316.
78. Knoop LL and Baker SJ. EWS/FLI alters 5 ‘-splice site selection. Journal of Biological Chemistry. 2001; 276:22317-22322.
79. Yang L, Chansky HA and Hickstein DD. EWS center dot Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. Journal of Biological Chemistry. 2000; 275:37612-37618.
80. Chansky HA, Hu M, Hickstein DD and Yang L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res. 2001; 61:3586-3590.
81. Kovar H, Jug G, Hattinger C, Spahn L, Aryee DN, Ambros PF, Zoubek A and Gadner H. The EWS protein is dispensable for Ewing tumor growth. Cancer Res. 2001; 61:5992-5997.
82. Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, Boucher KM, Watkins WS, Jorde LB, Graves BJ and Lessnick SL. Microsatellites as EWIS/FLI response elements in Ewing’s sarcoma. P Natl Acad Sci USA. 2008; 105:10149-10154.
83. Luo W, Gangwal K, Sankar S, Boucher KM, Thomas D and Lessnick SL. GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene. 2009; 28:4126-4132.
84. Gangwal K, Close D, Enriquez CA, Hill CP and Lessnick SL. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing’s Sarcoma. Genes Cancer. 2010; 1:177-187.
85. Monument MJ, Johnson KM, Grossmann AH, Schiffman JD, Randall RL and Lessnick SL. Microsatellites with Macro-Influence in Ewing Sarcoma. Genes-Basel. 2012; 3:444-460.
86. Beck R, Monument MJ, Watkins WS, Smith R, Boucher KM, Schiffman JD, Jorde LB, Randall RL and Lessnick SL. EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet-Ny. 2012; 205:304-312.
87. Monument MJ, Johnson KM, McIlvaine E, Abegglen L, Watkins WS, Jorde LB, Womer RB, Beeler N, Monovich L, Lawlor ER, Bridge JA, Schiffman JD, Krailo MD, et al. Clinical and Biochemical Function of Polymorphic NR0B1 GGAA-Microsatellites in Ewing Sarcoma: A Report from the Children’s Oncology Group. Plos One. 2014; 9:e104378. doi: 10.1371/journal.pone.0104378.
88. Grunewald TG, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, Aynaud MM, Mirabeau O, Cidre-Aranaz F, Tirode F, Zaidi S, Perot G, Jonker AH, Lucchesi C, et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet. 2015; 47:1073-1078.
89. Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H and Salzer-Kuntschik M. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991; 67:1886-1893.
90. Kovar H, Dworzak M, Strehl S, Schnell E, Ambros IM, Ambros PF and Gadner H. Overexpression of the pseudoautosomal gene MIC2 in Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. Oncogene. 1990; 5:1067-1070.
91. Fellinger EJ, Garin-Chesa P, Triche TJ, Huvos AG and Rettig WJ. Immunohistochemical analysis of Ewing’s sarcoma cell surface antigen p30/32MIC2. The American journal of pathology. 1991; 139:317-325.
92. Perlman EJ, Dickman PS, Askin FB, Grier HE, Miser JS and Link MP. Ewing’s sarcoma—routine diagnostic utilization of MIC2 analysis: a Pediatric Oncology Group/Children’s Cancer Group Intergroup Study. Human pathology. 1994; 25:304-307.
93. Kreppel M, Aryee DN, Schaefer KL, Amann G, Kofler R, Poremba C and Kovar H. Suppression of KCMF1 by constitutive high CD99 expression is involved in the migratory ability of Ewing’s sarcoma cells. Oncogene. 2006; 25:2795-2800.
94. Scotlandi K, Baldini N, Cerisano V, Manara MC, Benini S, Serra M, Lollini PL, Nanni P, Nicoletti G, Bernard G, Bernard A and Picci P. CD99 engagement: an effective therapeutic strategy for Ewing tumors. Cancer research. 2000; 60:5134-5142.
95. Sohn HW, Choi EY, Kim SH, Lee IS, Chung DH, Sung UA, Hwang DH, Cho SS, Jun BH, Jang JJ, Chi JG and Park SH. Engagement of CD99 induces apoptosis through a calcineurin-independent pathway in Ewing’s sarcoma cells. The American journal of pathology. 1998; 153:1937-1945.
96. Rocchi A, Manara MC, Sciandra M, Zambelli D, Nardi F, Nicoletti G, Garofalo C, Meschini S, Astolfi A, Colombo MP, Lessnick SL, Picci P and Scotlandi K. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. The Journal of clinical investigation. 2010; 120:668-680.
97. Scotlandi K, Perdichizzi S, Bernard G, Nicoletti G, Nanni P, Lollini PL, Curti A, Manara MC, Benini S, Bernard A and Picci P. Targeting CD99 in association with doxorubicin: an effective combined treatment for Ewing’s sarcoma. European journal of cancer. 2006; 42:91-96.
98. Bixel G, Kloep S, Butz S, Petri B, Engelhardt B and Vestweber D. Mouse CD99 participates in T-cell recruitment into inflamed skin. Blood. 2004; 104:3205-3213.
99. Suh YH, Shin YK, Kook MC, Oh KI, Park WS, Kim SH, Lee IS, Park HJ, Huh TL and Park SH. Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs. Gene. 2003; 307:63-76.
100. Elzi DJ, Song M, Houghton PJ, Chen Y and Shiio Y. The role of FLI-1-EWS, a fusion gene reciprocal to EWS-FLI-1, in Ewing sarcoma. Genes & cancer. 2015; 6:452-461.
101. Stock M and Otto F. Control of RUNX2 isoform expression: the role of promoters and enhancers. J Cell Biochem. 2005; 95:506-517.
102. Melot T, Gruel N, Doubeikovski A, Sevenet N, Teillaud JL and Delattre O. Production and characterization of mouse monoclonal antibodies to wild-type and oncogenic FLI-1 proteins. Hybridoma. 1997; 16:457-464.
103. Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS and DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes & development. 2003; 17:3112-3126.
104. Chan CK, Chen CC, Luppen CA, Kim JB, DeBoer AT, Wei K, Helms JA, Kuo CJ, Kraft DL and Weissman IL. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature. 2009; 457:490-494.
105. Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S, McLaughlin ME, Weissman IL, Butte AJ, Passegue E and Sage J. Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell. 2008; 3:416-428.
106. Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M and Sutcliffe JG. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990; 5:187-197.