
INTRODUCTION
Renal cell carcinoma (RCC) is one of the most common genitourinary neoplasm and accounts for approximately 3% of all malignancies worldwide with global incidence rates increasing 2–3% per year [1–2]. The clear cell renal cell carcinoma (ccRCC) is the main subtype of RCC and account for approximately 75% of all renal tumors [3]. Surgical management remains the most effective therapeutic alternative for ccRCC. However, there was still about one third patients who develop metastases subsequent to surgery [3]. The prognosis for patients with metastatic RCC is extremely poor largely because of its strong resistance to radiotherapy and chemotherapy and lack of effective therapeutic [4]. Although previous researches have revealed many genetic and epigenetic changes correlated with RCC genesis, there still lack of curative therapy for metastatic RCC and precise mechanism of RCC progression remains poorly understood [5–6]. Therefore, to identify a new reliable and sensible tumor marker for prognostic prediction is crucial for the patients with RCC.
Recent evidence increasingly points to the important role of long non-coding RNA (lncRNAs), the largest transcript class in human genome, which may play an important role in many cellular processes and multiple diseases including cancers [7–9]. Previous researches also reported the expression profile of lncRNA transcript in ccRCC tissues and indicated that there are many dysregulated lncRNAs which may allowed accurate identification of ccRCC tumor tissue [10, 11]. For example, CADM1-AS1 and NBAT-1, may correlated with the progression and worse survival in patients with ccRCC [4, 12]. However, lncRNAs remain poorly characterized in ccRCC.
In this study, through bioinformatics analysis, we performed an integrative analysis of two microarray datasets aimed to identify novel biomarkers of ccRCC. Selected DEGs had been examined in clinical samples by qRT-PCR. We found two high correlated DEGs, peejar and GFOD1 were associated with high ccRCC tumor stage, high ccRCC tumor grade and poor prognosis. Our findings may provide new prognostic biomarkers for patients with ccRCC.
RESULTS
Microarray datasets characteristics
ccRCC microarray datasets obtained from ArrayExpress database were included in this study. The E-GEOD-22541 dataset contained 24 primary ccRCC samples, including 8 samples with disease-free survival (DFS) less than or equal to 6 months represented synchronous metastases, 9 samples with DFS greater than or equal to 45 months represented metachronous metastases, and 7 samples without detectable distant metastases after at least 99 months follow up [13]. The E-MTAB-1050 dataset contained gene expression data from 13 primary tumors of ccRCC, and 25 corresponding mice xenograft ccRCC tumors [14]. The E-GEOD-53757 dataset contained 72 paired primary ccRCC samples and normal kidney tissue from the same patient [15]. Detailed description of sample characteristics and clinical variables are provided in the original report [15].
The gene expression of GFOD1 and peejar is a prognostic marker for ccRCC
Using the Significance Analysis of Microarrays (SAM) method, we identified 998 genes that were differentially expressed between 8 synchronous metastases patients and 16 metachronous metastases patients or patients without detectable metastases disease from E-GEOD-22541 dataset, with standard fold change superior to 1.6 (Supplementary Dataset S1). GO analysis results were shown in Supplementary File S5. Among those DEGs, 138 genes were corresponding to non-coding RNAs (ncRNAs), as known by align each probe sequence to NOCODE (www.noncode.org), LNCipedia (www.lncipedia.ord) [16] and AceView database (http://www.ncbi.nlm.nih.gov/ieb/research/acembly/) [17], using Biostrings bioconductor package (Supplementary Dataset S2). The gene expression correlation between 138 probeset corresponding to ncRNAs and remaining 850 probesets mainly corresponding to coding RNAs were calculated (Supplementary Dataset S3). There are 64 pairs of probesets, corresponding to non-coding or coding respectively, exhibited correlation coefficiency superior to 0.9 (Supplementary Dataset S4).
DEGs were identified from dataset E-MTAB-1050, using SAM method at false discovery rate = 0.05, and standard fold change cut off = 1.6. GO analysis and KEGG pathway analysis results were shown in Supplementary Files S6 and S7. After overlapping the 64 pair of probesets and the 2910 DEGs (Supplementary Dataset S5), finally 4 pair of genes were selected for next step. There are two DEGs, 219821_s_at and 230179_at, with Pearson correlation coefficiency reaching 0.939 between their signal intensity. They are corresponding to GFOD1 and peejar respectively, after align probe sequences of those two probesets to Aceview database [17] (Supplementary File S1). Gene GFOD1 maps on chromosome 6, from 13487893 to 13363496, on the reverse strand, while gene peejar maps to chromosome 6, from 13363310 to 13358058, on the reverse strand too (NCBI 37, August 2010). There were only 186 bp distance between those two genes on their chromosome, which indicated that GFOD1 gene and peejar gene maybe transcriptional coupling neighboring genes [18]. Their probesets signal intensity had been presented in Figure 1.
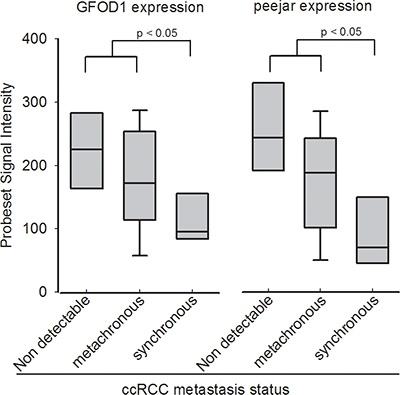
Figure 1: Probeset signal intensity of 219821_s_at (GFOD1) and 230179_at (peejar) from Dataset E-GEOD-22541. Patients with non-detectable metastasis after at least 99 months follow-up shown highest GFOD1 and peejar expression in tumor samples. Patients with synchronous metastasis disease shown lowest GFOD1 and peejar expression in tumor samples, and significant difference compared to patients with non-detectable or metachronous metastasis disease.
The gene expression of GFOD1 and peejar decreased with ccRCC tumor stage progression
The signal intensity of probesets 219821_s_at and 230179_at were extracted from microarray dataset E-GEOD-53757. Both genes exhibited lower expression in normal kidney tissue samples, while significantly elevated expression in stage I ccRCC tumor samples. During disease progression, the gene expression was gradually decreased. There were significant difference between gene expression in early stage (stage I and II) tumor tissue samples and late stage (stage III and IV) tumor tissue samples, which indirectly confirmed the observation that decrease of their gene expression linked to patient’s disease progression and poor prognosis (Figure 2).
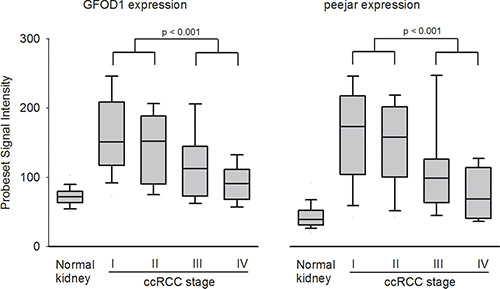
Figure 2: Probeset signal intensity of 219821_s_at (GFOD1) and 230179_at (peejar) from Dataset E-GEOD-53757. Both probesets shown lowest gene expression from 72 normal kidney samples, with highest average gene expression in 24 ccRCC Stage I tumor samples, and gradually decreasing in 19 stage II tumor samples, 14 stage II tumor samples and 15 stage IV samples. There were significant difference between gene expression from patients’ samples with early stage and late stage diseases.
The gene expression of GFOD1 and peejar decreased with ccRCC tumor grade progression
We further evaluated the gene expression of GFOD1 and peejar by qRT-PCR in an independent sample of 50 CCRCC tumor samples and 36 non-tumor kidney tissues. After reference genes normalization, the relative expression intensity (Ct) was highest in normal kidney tissue samples, and lowest in tumor samples with low grade G1. There were significant difference between gene expression in low or intermediate grade (G1 and G2) tumor tissue samples and high grade (G3 and G4) tumor tissue samples, which indirectly confirmed the observation that decrease of their gene expression linked to patient’s disease progression and poor prognosis (Figure 3).
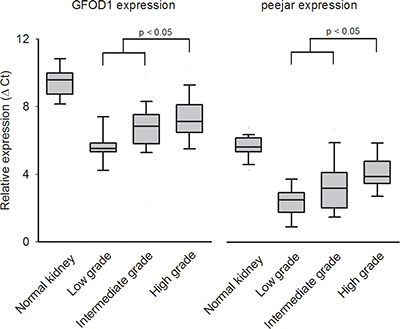
Figure 3: The relative expression (Ct value) from qRT-PCR experiment. Higher Ct value indicated lower gene expression. Both GFOD1 and peejar exhibited highest Ct value in normal kidney tissue samples, and lowest Ct value in ccRCC patients’ tumor samples with low grade disease. There were significant difference between expression levels from patients’ samples with low to intermediate grade and high grade diseases.
DISCUSSION
lncRNAs are a class of RNA molecules arbitrarily defined as being longer than 200 nucleotides and it was initially thought to be a transcriptional noise without protein coding potential [7]. However, lncRNAs have gained massive attention for their crucial roles in gene regulation in recent years [4]. More and more evidence showed lncRNAs may regulate protein coding genes expression at both transcriptional and post-transcriptional level and they were also proved to contribute in tumor development and can be used as biomarkers and prognosis factors [19–20].
Recently, roles of lncRNAs in genitourinary carcinomas have attracted much interest from urological researchers and are becoming a hot spot in renal cancer research [4, 12, 21]. Yao et al. reported lncRNA CADM1-AS1, which is located in the antisense direction of a coding exon of tumor suppressor gene CADM1, was decreased in tumor tissues of ccRCC patients and it may regulate CADM1 expression on proliferation, apoptosis and migration via the expression pattern of “CADM1-AS1/CADM1 mRNA gene pairs” in vitro [12]. However, the few examples described above are just a tip of the iceberg as the field of lncRNAs is currently evolving and the work of annotation and characterization of lncRNAs in ccRCC is ongoing [22].
In this study, we retrieve public available ArrayExpress database to identify DEGs which may link to different disease-free survivals. The bioinformatics analysis showed that peejar and GFOD1 expression were significantly elevated in ccRCC tissues, but gradually decreased during cancer progression. We further checked these findings in 50 ccRCC and 36 normal kidney tissues and found the expression of peejar and GFOD1 was significantly higher in low to intermediate grade tumors compared to high-grade tumors, which indirectly confirmed the observation that decrease of their gene expression linked to patient’s tumor progression.
However, our study has several limitations. The study population was small and the number of patients with T4 or M1 disease was limited, and it is difficult to compare the qRT-PCR results from patients’ samples with early stage disease and late stage disease. The lncRNAs candidates identified here may not represent the complete lncRNA populations underlying ccRCC biological behavior. Although our study revealed the expression of patterns and dysregulation of peejar and GFOD1, their function still remains unknown. peejar and GFOD1 was identified after overlapping two lists of differentially expressed genes. Despite some cancer cells might also expressing GFOD1 and peejar, we are expecting that those GFOD1-expressing tumor infiltrated immune cells, like NK cells, Eosinophil cells and Macrophage cells might contributed to the prognosis (Supplementary Files S8 and S9). Future immunohistochemistry or flow cytometry studies on those immune cells will be focused on checking whether their the quantity difference or GFOD1-expressing level difference could predict the clinical outcome.
In conclusion, our study demonstrated for the first time that lncRNA peejar and mRNA GFOD1 expression was significantly increased in ccRCC and decreased during tumor progression. Our findings indicate the potential roles of lncRNA in ccRCC, and provide useful information for discovery of new biomarkers and therapeutic targets.
MATERIALS AND METHODS
Public microarray datasets
ccRCC microarray datasets and corresponding clinical data were retrieved from public available ArrayExpress database [23]. Gene expression profiling was performed using GeneChip human genome U133 Plus 2.0 array (HG-U133Plus2) from Affymetrix (Santa Clara, CA). This array offers 54.000 probe sets for screening 38,500 human genes. Microarray data preprocessing was conducted using the R software and packages from the Bioconductor project [24]. Raw data were collected from CEL files and preprocessed with the Robust Multi-chip Average (RMA) algorithm for background correction, quantile normalization, and median polish summarization [25]. After RMA preprocessing, and nonparametric batch adjustments using ComBat [26], a set of probe ID-centric gene expression values were available for downstream analysis. Differentially expressed genes (DEGs) between any two groups were determined using Significance Analysis of Microarrays (SAM) method with a false discovery rate (FDR) below 0.05, and permutation of 1000. MAplot and histogram were used for analysis processing quality control and PCA analysis to evaluate the quality of processed data (Supplementary File S2).
Patients and samples
We included 50 ccRCC tumor samples and 36 non-tumor kidney tissues for qRT-PCR analyses. The tumors were staged according to the TNM system developed by the American Joint Committee on Cancer and the International Union against Cancer [27] and graded according to Fuhrman’s nuclear grading system [28]. Out of the 50 tumors, 10 in low grade G1, 23 in intermediate grade G2, 15 in high grade G3, and 2 in high grade G4, while 35 in stage I, 9 in stage II, and 6 in stage III. The written informed consent was obtained from all participants. The study was approved by the Ethics Committee of Ren Ji hospital, China. The detailed clinical information is provided in Supplementary File S3.
RNA extraction and qRT-PCR experiments
Frozen tissues were grossly dissected into TRIZOL for RNA extraction following standard protocols using dry ice pre cold pestle and motor. Using an Anakytik Jena scandrop spectrophotometer, the ratio of the absorbance of each RNA at 260 and 280 nm (A260:A280) was measured as an indicator of RNA purity. The mRNA integrity was assessed by the absorption curve, which showed two clear rRNA bands of 28S and 18S. One microgram of total RNA was used for cDNA synthesis with QuantiTect reverse transcription kit (Qiagen). qRT-PCR was performed on an ABI 7900HT instrument using QuantiFast SYBR green PCR kit (Qiagen). A combination of 4 genes, HMBS, PPIA, ATP5J and TBP, being stably expressed in tissues from ccRCC, has been used as reference genes for gene expression internal control [29]. Primer sequences of targets are provided in Supplementary File S4. Each amplification was run in duplicate. The cycle number of each target was normalized against the geometric mean of the cycle numbers from 4 reference genes. The calculation of delta Ct value was performed as follows: Ct(target) = Ct (target) – GM{Ct(HMBS), Ct(PPIA), Ct(ATP5J), Ct(TBP)}.
Statistical analysis
The DEGs expression differences between the tumor and normal kidney samples were analyzed using Student’s t-test with SPSS software (version 17.0, SPSS). A two-sided p value less than 0.05 was considered to be statistically significant.
CONFLICTS OF INTEREST
The authors declare no competing interests.
GRANT SUPPORT
This study was funded by grants from National Natural Science Foundation of China (81402084, 81472378 and 81272841) and Advanced Technology Popularization Program of Shanghai Municipal Commission of Health and Family Planning (2013SY027). All these study sponsors have no roles in the study design, in the collection, analysis, and interpretation of data.
REFERENCES
1. Siegel RL, Miller KD, Jamal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5–29.
2. Bhatt JR, Finelli A. Landmarks in the diagnosis and treatment of renal cell carcinoma. Nat Rev Urol. 2014; 11:517–525.
3. Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009; 373:1119–1132.
4. Xue S, Li QW, Chen JP, Guo Y, Yang FQ, Zheng JH. Decreased expression of long non-coding RNA NBAT-1 is associated with poor prognosis in patients with clear cell renal cell carcinoma. Int J Clin Exp Pathol. 2015; 8:3765–3774.
5. Ha YS, Lee GT, Modi P, Kwon YS, Ahn H, Kim WJ, Kim IY. Increased Expression of Androgen Receptor mRNA in Human Renal Cell Carcinoma Cells is Associated with Poor Prognosis in Patients with Localized Renal Cell Carcinoma. J Urol. 2015; 194:1441–1448.
6. Motzer RJ, Hutson TE, McCann L, Deen K, Choueiri TK. Overall survival in renal-cell carcinoma with pazopanib versus sunitinib. N Engl J Med. 2014; 370:1769–1770.
7. Ponting CP, Oliver PL, Reik W. Evolution and functions of long non-coding RNA. Cell. 2009; 136:629–641.
8. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol cell. 2011; 43:904–914.
9. Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ, Lam WL. Human cancer long non-coding RNA transcriptomes. PloS One. 2011; 6:e25915.
10. Deng M, Blondeau JJ, Schmidt D, Perner S, Müller SC, Ellinger J. Identification of novel differentially expressed lncRNA and mRNA transcripts in clear cell renal cell carcinoma by expression profiling. Genom Data. 2015; 5:173–175.
11. Qin C, Han Z, Qian J, Bao M, Li P, Ju X, Zhang S, Zhang L, Li S, Cao Q, Lu Q, Li J, Shao P, et al. Expression pattern of long non-coding RNAs in renal cell carcinoma revealed by microarray. PLoS One. 2014; 9:e99372.
12. Yao J, Chen Y, Wang Y, Liu S, Yuan X, Pan F, Geng P. Decreased expression of a novel lncRNA CADM1-AS1 is associated with poor prognosis in patients with clear cell renal cell carcinomas. Int J Clin Exp Pathol. 2014; 7:2758–2767.
13. Wuttig D, Zastrow S, Füssel S, Toma MI, Meinhardt M, Kalman K, Junker K, Sanjmyatav J, Boll K, Hackermüller J, Rolle A, Grimm MO, Wirth MP. CD31, EDNRB and TSPAN7 are promising prognostic markers in clear-cell renal cell carcinoma revealed by genome-wide expression analyses of primary tumors and metastases. Int J Cancer. 2012; 131:E693–704.
14. Sivanand S, Peña-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, Yamasaki T, McBride DJ, Gillen J, Wolff NC, Morlock L, Lotan Y, Raj GV, et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med. 2012; 4:137ra75.
15. von Roemeling CA, Radisky DC, Marlow LA, Cooper SJ, Grebe SK, Anastasiadis PZ, Tun HW, Copland JA. Neuronal pentraxin 2 supports clear cell renal cell carcinoma by activating the AMPA-selective glutamate receptor-4. Cancer Res. 2014; 74:4796–4810.
16. Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K, Vandesompele J, Mestdagh P. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013; 41:D246–251.
17. Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006; 7:S12.1–14.
18. Wang GZ, Lercher MJ, Hurst LD. Transcriptional coupling of neighboring genes and gene expression noise: evidence that gene orientation and noncoding transcripts are modulators of noise. Genome Biol Evol. 2011; 3:320–331.
19. Zhang HM, Yang FQ, Yan Y, Che JP, Zheng JH. High expression of long non-coding RNA SPRY4-IT1 predicts poor prognosis of clear cell renal cell carcinoma. Int J Clin Exp Pathol. 2014; 7:5801–5809.
20. Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011; 10:38.
21. Martens-Uzunova ES, Böttcher R, Croce CM, Jenster G, Visakorpi T, Calin GA. Long noncoding RNA in prostate, bladder, and kidney cancer. Eur Urol. 2013; 65:1140–1151.
22. Yarmishyn AA, Kurochkin IV. Long noncoding RNAs: a potential novel class of cancer biomarkers. Front Genet. 2015; 6:145.
23. Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG, Oezcimen A, Rocca-Serra P, Sansone SA. ArrayExpress—a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 2003; 31:68–71.
24. Reimers M, Carey VJ. Bioconductor: an open source framework for bioinformatics and computational biology. Methods Enzymol. 2006; 411:119–134.
25. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–264.
26. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using Empirical Bayes methods. Biostatistics. 2007; 8:118–127.
27. Kim SP, Alt AL, Weight CJ, Costello BA, Cheville JC, Lohse C, Allmer C, Leibovich BC. Independent validation of the 2010 American Joint Committee on Cancer TNM classification for renal cell carcinoma: results from a large, single institution cohort. J Urol. 2011; 185:2035–2039.
28. Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982; 6:655–663.
29. Bjerregaard H, Pedersen S, Kristensen SR, Marcussen N. Reference genes for gene expression analysis by real-time reverse transcription polymerase chain reaction of renal cell carcinoma. Diagn Mol Pathol. 2011; 20:212–217.