
INTRODUCTION
New Zealand (NZ) has the highest incidence rate of melanoma in the world [1], with new registrations occurring at a rate of 36.9 per 100,000 in the NZ population in 2012, when age-standardized to the WHO standard population (Wellington: Ministry of Health. www.health.govt.nz/publication/cancer-new-registrations-and-deaths-2012).
The cause of the high melanoma incidence in NZ is thought to be mainly due to the high levels of solar UV-radiation exposure, which the NZ population is exposed to, especially during the summer months. Solar UV radiation exposure is widely accepted as a key risk factor for cutaneous melanomas, although the latter may arise on both sun-exposed as well as non-sun-exposed skin sites [2]. Many melanomas develop in association with nevi on the skin, the number of which is proportional to childhood sun exposure, as well as to genetic factors [3]. In contrast, melanomas may also develop with no prior association with nevi. The sequencing of melanoma genomes has shown that melanomas frequently contain high mutation loads; on average melanomas contain the highest mutation load of all cancer types, including high levels of UV signature mutations, such as C>T nucleotide transitions [4, 5].
Based on a number of Next Generation Sequencing studies involving hundreds of melanomas, a molecular disease model of melanoma has been proposed, whereby ~40-50% of melanomas from patients carry mutations in the BRAF gene, with ~90% of these BRAF mutations being a BRAFV600E mutation, and a further 20% carry mutations in the NRAS gene [6]. Almost half a decade of Next Generation Sequencing studies of cutaneous melanoma has recently been reviewed [7], including one of the most recent and most extensive exome sequencing studies carried out to date. The Cancer Genome Atlas (TCGA) melanoma skin cancer study investigated exome sequences of 333 melanomas [8], and revealed recurrent aberrant sequence variants in a number of genes, including BRAF, NRAS, TP53, PPP6C, NF1, CDKN2A, PTEN, ARID2, DDX3X, RAC1, IDH1, RB1, MAP2K1, HRAS, KRAS, KIT, and CDK4.
BRAF and NRAS mutations result in activation of the MEK-ERK signalling cascade [9]. This observation has led to extensive efforts to develop drugs targeting this pathway. Two such drugs, Vemurafenib and Dabrafenib are BRAF inhibitors that inhibit mutant BRAF proteins containing V600E [10, 11], and prevent the activation of the MAP kinase pathway, resulting in antitumor effects such as inhibition of cell proliferation and induction of apoptosis [12].
In recent years BRAF inhibitor drugs have led to significantly improved outcomes for melanoma patients [9]. In addition, immune checkpoint inhibitors have been shown to significantly improve melanoma patient survival, and it is notable that better response rates to immune checkpoint inhibitors have been reported in melanomas with high mutation loads, particularly those containing NRAS mutations [13]. As progressively more therapies tailored towards oncogenic mutations are under development, it is important to understand the incidence of commonly mutated genes within a given population, especially in regard to targeted therapy options that may be available. Although the incidence rates of recurrent driver mutations in melanoma have been widely reported in different populations in the world, it remains unknown how frequent these mutations are in NZ melanomas.
Here we have investigated the mutation frequencies in 20 genes represented on the Sequenom MelaCarta MassARRAY in NZ melanomas. We included melanomas from both North and South Islands, constituting the first comprehensive mutation analysis of clinical melanoma samples in NZ.
RESULTS
Patients and samples
Genomic DNAs were isolated from 529 metastatic melanoma samples, from a total of 529 patients. Samples were analyzed using the Sequenom MassARRAY MelaCarta panel of recurrently mutated melanoma genes, and also by direct Sanger sequencing of BRAF exon 15, which served to identify the full extent of BRAF exon 15 mutations, and to provide validation of the BRAF MelaCarta mutation data.
Overall, 453 melanoma samples were successfully analyzed by Sanger sequencing, and 466 melanoma samples were successfully analyzed by Sequenom MelaCarta. Not all melanomas analyzed by sequencing were analyzed by MelaCarta and vice versa. The patients were diagnosed at four different geographically dispersed localities in NZ (Figure 1A), and were predominantly Caucasian (243 males, 162 females, 61 of unknown gender, Figure 1B). The most common location for metastasis in this cohort was lymph nodes (34.1% of the patients) followed by skin (11.4%) and brain (11.2%, Figure 1C). The age of the melanoma patients ranged from 18 to 95 years with the median being 66 years of age (Figure 1D).
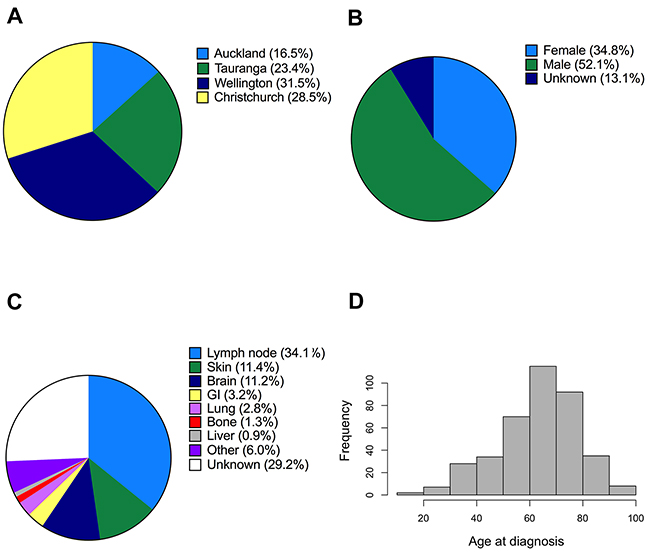
Figure 1: Description of metastatic melanoma patients analyzed in this study. A, B. Geographical distribution and gender of the patients whose melanomas were analyzed for mutations using Sequenom MassARRAY (MelaCarta Panel v1.0). C. Distribution of the anatomical sites of the analyzed metastatic tumour samples. D. Histogram showing the age distribution of the patients.
BRAF mutation analysis in New Zealand melanomas
Similar to previous studies [9], mutations in the BRAF oncogene were the most frequent in our cohort of NZ melanomas. For all melanomas analyzed (from all localities in NZ), BRAF mutations were identified in 33.1% of melanomas (175/529), and were detected by either MelaCarta and/or Sanger sequencing. Among BRAF mutations characterized by high resolution melting analysis (HRM) and Sanger sequencing we identified one novel BRAF mutation, BRAFL597H (Table 1). The majority of BRAF mutations were identified in both MelaCarta and Sanger sequencing platforms (Supplementary data Table S1), although 18/175 (10.3%) of all the BRAF mutations detected were only identified by Sanger sequencing, and were not detected by the MelaCarta platform. Of the exon 15 BRAF mutations not detected by MelaCarta, 11/18 (61.1%) were not included in the MelaCarta panel (Table 1). In contrast the MelaCarta panel detected an additional 4% of all BRAF mutations including BRAFG466A and BRAFG469E amino acid substitutions which lay outside of the exon 15 region sequenced by the Sanger method (Table 1).
Table 1: Predicted amino acid mutations in NZ melanomas identified in BRAF outside codon 600 detected using Sanger sequencing and/or Sequenom MelaCarta
BRAF Mutation |
Geographical Location |
Site of primary |
Morphology of primary |
Method of Detection |
Present in MelaCarta |
|---|---|---|---|---|---|
S607P |
Wellington |
Skin |
NOS |
Sanger |
No |
G606R |
Tauranga |
Trunk |
NOS |
Sanger |
No |
S605N |
Auckland |
Lower limb |
NOS |
Sanger |
No |
R603STOP |
Christchurch |
Lower limb |
NOS |
Sanger |
No |
K601E |
Auckland |
Upper limb |
SSM |
Sanger & Sequenom |
Yes |
K601E |
Wellington |
Trunk |
nodular |
Sanger & Sequenom |
Yes |
K601E |
Christchurch |
Upper limb |
nodular |
Sanger & Sequenom |
Yes |
K601N |
Wellington |
Scalp/Neck |
nodular |
Sanger |
No |
L597H** |
Wellington |
Upper limb |
NOS |
Sanger |
No |
L597S |
Tauranga |
Skin |
NOS |
Sanger |
Yes |
L597Q |
Wellington |
Spine |
NOS |
Sequenom |
Yes |
V600E and L597Q |
Wellington |
Trunk |
SSM |
Sanger |
Yes |
V600E and G596D |
Tauranga |
Trunk |
NOS |
Sanger |
G596D is not in Melacarta |
G596R |
Auckland |
unavailable |
unavailable |
Sanger |
No |
F595L |
Auckland |
Scalp/Neck |
nodular |
Sanger |
No |
D594A |
Wellington |
Lower limb |
SSM |
Sanger |
No |
D594E |
Auckland |
Ear |
NOS |
Sanger |
No |
D594N |
Wellington |
Scalp/Neck |
nodular |
Sanger |
No |
V590A* |
Tauranga |
Groin |
NOS |
Sanger |
No |
H585Y* |
Wellington |
Skin |
amelanotic |
Sanger |
No |
H585Y* |
Wellington |
Skin |
NOS |
Sanger |
No |
L584F |
Wellington |
Upper limb |
NOS |
Sanger |
No |
G469E |
Auckland |
unavaliable |
NOS |
Sequenom |
Yes |
G469E |
Christchurch |
Lymph Node |
superficial spreading |
Sequenom |
Yes |
G469E |
Wellington |
Urinary Bladder |
NOS |
Sequenom |
Yes |
G466A |
Wellington |
Small Bowel |
nodular |
Sequenom |
Yes |
Using Sanger sequencing we identified a previously unreported BRAF mutation at codon 597 (**L597H c.1790_1791TA>AT) and two additional mutations that have not been associated with melanoma in earlier reports*
Of the mutations identified in BRAF, BRAFV600E substitutions were the most common mutation type, comprising 73.7% of all BRAF mutations. The frequency of V600E mutations was 23.4% (109/466) of all melanomas identified by the MelaCarta panel alone or 24.4% (129/529) of all melanomas analyzed by MelaCarta plus Sanger sequencing. BRAFV600K mutations were detected in 4.3-4.5% of melanoma samples (MelaCarta, 21/466; or MelaCarta plus Sanger, 23/529) and these comprised 13.2%-15.0% of BRAF mutations detected. Patient age at the time of diagnosis of the first distant metastasis was available for 391 of the 466 patients (for MelaCarta assays). Patients with BRAF mutant metastatic melanoma were significantly younger at diagnosis of the first distant metastasis (mean = 59.1 years; SD=14.9) compared to the patients with BRAF wild-type melanoma (mean= 65.4 years; SD= 14.5; P= 0.0001, Student's t-test). Patients with BRAFV600E mutant metastatic melanoma were also associated with a significantly younger age (median= 57.0 years) compared to patients with BRAF wild-type melanoma (median= 67.0, P= 2.5 x 10-7, Mann-Whitney test). By comparison to BRAFV600E mutant metastatic melanomas, the BRAFV600K mutation was significantly associated with older age (median= 66.5) at diagnosis of the first distant metastasis (P= 0.0053, Mann-Whitney test). Similar associations with age and BRAF mutation status were previously reported [14, 15]. Patient gender was not associated with any BRAF mutant genotype.
The Cancer Genome Atlas (SKCM data in TCGA) (Figure 2B) currently includes mutation information for 368 metastatic melanoma patients derived from exome sequencing data. Comparing BRAF mutation rates in NZ melanomas with the melanoma mutation data from TCGA, we found that BRAFV600E mutations occurred in a relatively smaller percentage of NZ melanomas than in TCGA, although this was not significant (Figure 2B).
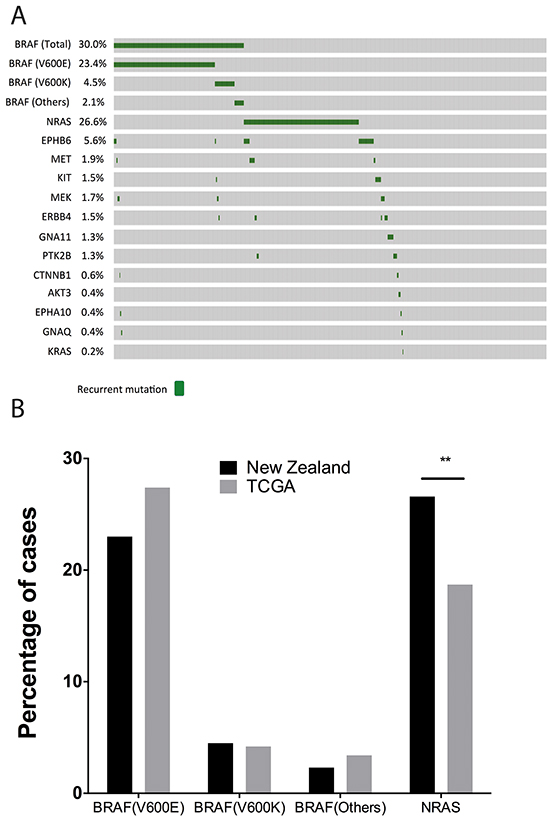
Figure 2: Overview of the mutational landscape in New Zealand population. A. Oncoprint of mutations identified with the Sequenom MassARRAY (MelaCarta Panel) in 466 patients. The oncoprint was generated using cBioportal tools. B. Comparison of BRAF and NRAS mutations in the NZ population with the TCGA patient cohort.
Analysis of mutations in an additional 19 genes in the MelaCarta panel in New Zealand melanoma patients
The MelaCarta panel v1.0 [16] includes a further 59 somatic mutations, which are frequently altered in melanoma in a further 19 genes in addition to BRAF. We analyzed these mutations in 466 melanomas, consisting of 333 melanoma patients from the North Island of NZ, from Auckland, Tauranga and Wellington, and 133 melanoma patients from South Island, from Christchurch (see Figure 1A). Mutations in NRAS were identified in 26.6% of melanomas (124/466) (Figure 2). The NRASQ61 mutation was identified in 24.5% (115/466), and NRASG12/13 mutations were identified in 2.0% (9/466) of melanomas. NRAS mutations were significantly associated with older age (Table 2; median age of patients with NRAS mutation = 69; median age of patients with BRAF mutation = 60; median age of patients wild type for BRAF and NRAS mutations = 66, P = 0.0000274, Kruskal Wallis test), which is consistent with previously published data from USA, Australian and European populations [17-19]. Mutation status was also significantly associated with the site of the primary tumor (P= 0.004, Chi-square test, Table 2); BRAF and NRAS mutations were relatively more common in melanomas from the trunk (23.6% and 21.0%) compared to tumors without mutations detected (Wild Type) (15.3%). In contrast, NRAS mutations were more common in melanomas arising from the arm/leg extremities (45.2%) compared with BRAF mutant (30.0%) or melanomas without mutations detected (33.7%) for this location. Melanomas with no detectable BRAF or NRAS mutations were more common in head and neck melanomas (19.8%) compared to either BRAF (10.0%) or NRAS (8.9%) mutant melanomas. These associations are consistent with previous reports [17, 19].
Table 2: Clinical and pathological characteristics, and their association with four genotypes: BRAF mutation, NRAS mutation, BRAF and NRAS mutations not detected (Wild Type, WT), and EPHB6 mutation.
Clinical and pathological factors |
All patients |
BRAF |
NRAS |
WT |
3 group |
EPHB6 |
|---|---|---|---|---|---|---|
No. of patients |
466 |
140 (30.0) |
124 (26.6) |
202 (43.3) |
26 |
|
Age at diagnosis (years) |
||||||
Median |
66 |
60 |
69 |
66 |
2.74 × 10-5 |
64 |
Gender |
||||||
Male |
243 |
78 (55.7) |
64 (51.6) |
101 (50.0) |
0.7564 |
9 (34.6) |
Female |
162 |
45 (32.1) |
46 (37.1) |
71 (35.1) |
14 (53.8) |
|
Unknown |
61 |
17 (12.1) |
14 (11.3) |
30 (14.9) |
3 (11.5) |
|
Primary tumor site |
||||||
Trunk |
90 |
33 (23.6) |
26 (21.0) |
31 (15.3) |
0.004 |
5 (19.2) |
Extremity |
166 |
42 (30.0) |
56 (45.2) |
68 (33.7) |
11 (42.3) |
|
Head/Neck |
65 |
14 (10.0) |
11(8.9) |
40 (19.8) |
3 (11.5) |
|
Unknown |
123 |
51 (36.4) |
31(25.0) |
63 (31.2) |
7 (26.9) |
|
Primary tumor histology |
||||||
Superficial Spreading |
103 |
33 (23.6) |
26 (20.1) |
44 (21.8) |
0.034 |
6 (23.1) |
Nodular |
67 |
12 (8.6) |
24 (19.4) |
31 (15.3) |
6 (23.1) |
|
Acral Lentiginous |
4 |
1 (0.7) |
0 |
3 (1.5) |
1 (4.3) |
|
Lentigo Maligna |
15 |
4 (2.9) |
2 (1.6) |
9 (4.5) |
0 |
|
Spindle |
5 |
0 |
1 (0.8) |
4 (2.0) |
0 |
|
Desmoplastic |
3 |
0 |
0 |
3 (1.5) |
0 |
|
NOS |
196 |
70 (50.0) |
54 (43.5) |
72 (35.6) |
11 (42.3) |
|
Other* |
10 |
3 (2.1) |
5 (4.0) |
2 (1.0) |
1 (4.3) |
|
Unknown |
41 |
17 (12.1) |
12 (9.7) |
34 (16.8) |
1 (4.3) |
|
No. of patients |
403 |
123 |
112 |
168 |
25 |
|
Breslow thickness (mm) |
||||||
≤2 |
265 |
72 (58.5) |
75 (67.0) |
118 (70.2) |
0.1183 |
16 (64.0) |
2.1-4.0 |
52 |
15 (12.2) |
17 (15.2) |
20 (11.9) |
3 (12.0) |
|
>4 |
86 |
36 (29.3) |
20 (17.9) |
30 (17.9) |
6 (24.0) |
|
*Other consists of blue naevus, and epithelioid cell melanoma.
The gene mutation status was also significantly associated with the histology of the primary tumor (P=0.034). Thirty-three tumors (23.6%) harboring BRAF mutations were identified as superficial spreading melanomas (SSM) compared with 26 (20.1%) melanomas with NRAS mutations (Table 2). In contrast, NRAS mutations were more commonly found in nodular melanomas (19.4%) compared to either BRAF mutant (8.6%) or WT tumors (15.3%). No statistically significant mutational associations with Breslow thickness were observed, although a slightly higher percentage of BRAF mutations occurred in melanomas >4mm thick. These findings are consistent with reports from previous studies [17].
Melanomas from South Island patients have a significantly higher incidence of NRAS mutations, while melanomas from North Island patients have a significantly higher incidence of EPHB6G404S mutations
Melanomas from patients in the North and South Islands of NZ were analyzed to assess mutation incidence in association with different geographic locations (Figure 3A, 3B). We found that while the BRAF mutation (BRAFV600E/K) frequencies between the North and South Island were similar (Figure 3C), the prevalence of NRAS mutations in the South Island cohort (38.3%; 51/133) was significantly higher than in North island patients (21.9%; 73/333, P= 0.0005, Figure 3C). In addition, the NRAS mutation frequency in South Island was significantly elevated when compared to the NRAS mutation rate in TCGA data (22.0%; 81/368, P= 0. 0004). In contrast, the North Island NRAS mutation frequency was not significantly different from TCGA (21.9% versus 22.0%). Using logistical modeling, the expected odds ratio of NRAS mutations in melanomas in the South Island was 2.35 times that of the North Island with a 95% confidence interval ([1.50, 3.70], P=0.00016.)
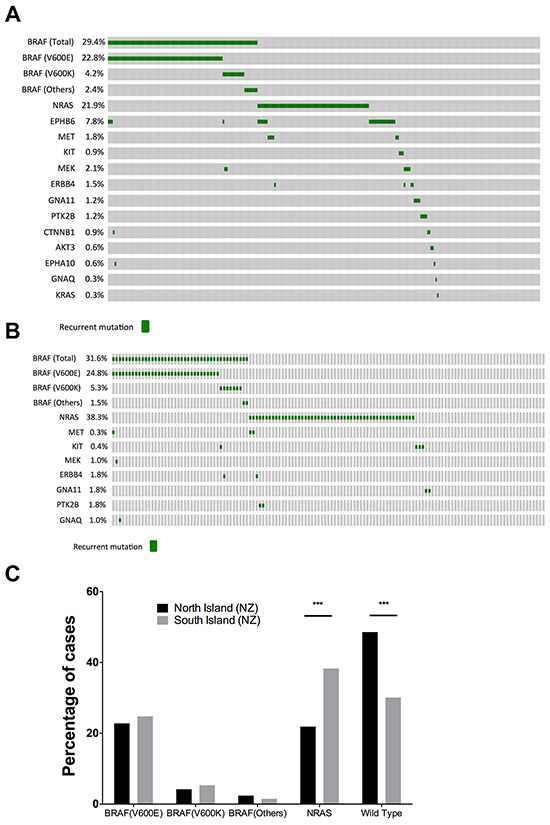
Figure 3: Comparison of BRAF and NRAS mutation profiles between North and South island melanomas. Oncoprints of mutations in A. North and B. South Island melanomas. C. Comparison of the mutation frequencies of BRAF and NRAS mutations in North and South Islands. South Island melanomas had a significantly higher prevalence of NRAS mutant melanomas (P= 0.0004, Fishers exact test).
In contrast, 5.6% (26/466) of melanoma samples contained an EPHB6G404S mutation, and when divided between North and South Islands, 7.8% (26/333) of North Island melanoma samples contained an EPHB6G404S mutation, making this the third most commonly mutated protein-coding gene after BRAF and NRAS. Remarkably, all 26 EPHB6G404S mutation were observed in North Island melanomas, and EPHB6G404S mutations were not identified in South Island melanomas (P=0.0002). Closer inspection of MelaCarta data revealed that two EPHB6G404S mutations were present at very low levels in South Island melanomas, but were not identified by the MelaCarta software. Melanomas containing EPHB6G404S mutations in the North Island were identified from the following centres; Auckland n=6, Tauranga n=9, Wellington n=11. Overall, EPHB6G404S mutations were more frequently observed in melanomas without BRAF or NRAS mutations (16/202 melanomas, 7.9%), than co-occurring with either BRAF or NRAS mutations (10/264 melanomas, 3.8%, p-value = 0.04).
In addition to the BRAF, NRAS and EPHB6 mutations, a small proportion of samples (<2% each, but in total comprising ~11.2% of patients) harbored mutations in MET, KIT, MEK, ERBB4, GNA11, PTK2B, CTNNB1, AKT3, EPHA10, GNAQ and KRAS genes (Figures 2A, 3A, 3B). Furthermore, 33/529 melanomas (6.2%) carried mutations in two or more genes in the MelaCarta panel. The co-detected additional mutations identified using the Melacarta Sequenom panel included six melanomas containing NRAS together with EPHB6G404S mutation, and six melanomas containing NRAS together with METT992I mutation (Table 3).
Table 3: Additional mutations identified by Sequenom Melacarta analysis in NZ melanomas
First mutation: |
BRAFV600E/K |
NRASQ61K/R/H/L |
ERBB4E452K |
Second mutation: |
EPHB6G404S (4) |
EPHB6G404S (6) |
MEKP124L (1) |
MEKP124L (4) |
METT992I (6) |
||
METT992I (1) |
ERBB4E452K (2) |
||
EPHA10E124K (1) |
PTK2BR429C (1) |
||
GNAQR183Q (1) |
PTK2BG414V (1) |
||
CTNNB1S45F (1) |
|||
ERBB4E452K (1) |
|||
KITV559A (1) |
The amino acid substitutions (such as BRAFV600E or BRAFV600K) shown in the first line of the table indicate the initial mutation identified by the Melacarta analysis. The additional mutation is shown in the lines below the respective initial mutation. The number in brackets indicates the number of times the respective mutation combination was observed.
DISCUSSION
Here we report the frequencies of recurrent mutations in NZ melanomas, including mutations in BRAF, NRAS, and EPHB6. Recurrent mutations in BRAF and NRAS in melanoma constitutively activate the mitogen-activated protein kinase (MAPK) signal transduction pathway, and NRAS mutations can also strongly activate the PI3/AKT pathway [20], leading to the activation of multiple downstream signaling pathways to promote cell proliferation, growth and survival. Although the frequencies of gene mutations have been reported in several international studies, mutation frequencies in NZ melanomas have not been reported to date.
We analyzed a large series of melanomas (n = 529) from different locations in NZ. Overall, BRAF mutations were observed in approximately one third of melanomas in NZ (175/529; 33.1%). BRAF mutations are usually detected in approximately 40-50% of melanomas [21]. Mutations in BRAFV600E (comprising 73.7% of BRAF mutations), and BRAFV600K (comprising 15.0% of BRAF mutations) made up most of the BRAF mutations. Sequenom analysis detected BRAF mutations (25.3%) at almost the same rate as Sanger Sequencing (25.7%) within codon 600. The concordance of BRAFV600 mutation detection between the two methods was 91.9% (Supplementary data Table S1). Our data suggest that 22.1% of BRAF mutations in NZ melanomas were non-V600E mutations, which is similar to the 25% figure obtained in a study of 1,112 cases of melanoma using pyrosequencing or Sequenom MassARRAY [22]. Sanger sequencing detected more mutations at other locations in exon 15 of BRAF than was detected using Sequenom MassARRAY, an observation that is highly relevant to mutation testing for clinical diagnosis. In addition, the BRAF COBAS test (Roche Diagnostics) was carried out on ~200 of the patients analyzed in this study, and the concordance with Sanger sequencing was >95% (data not shown). While the BRAFV600E mutation rate was lower than that reported in previous mutation studies in melanoma [5, 23], BRAFV600K mutations were associated with chronic UV exposure and are more prevalent in geographical areas with the highest levels of UV radiation, including Australia and Texas where BRAFV600K mutations have been reported in 19% to 22.5% of total BRAF mutations, respectively [15, 19]. However we did not find an elevated incidence of the BRAFV600K mutation in NZ.
In a previous study from the USA, one of seventy-nine melanomas analyzed was reported to contain an EPHB6G404S mutation [24]. Mutations in EPHB6 have been observed recurrently in other cancer types such as non-small cell lung cancer [25], but to our knowledge our study is the first to identify recurrent EPHB6G404S mutations in melanoma. The EPHB6G404S mutation involves substitution of glycine with serine in the Fibronectin Type III (FN3) domain of the EPHB membrane-bound protein tyrosine kinase, although the functional consequence of this mutation may be negligible, as structure/function analysis programs predicted it is benign [25]. Although we cannot rule out the possibility that the EPHB6G404sequence variant could be a germline polymorphism, this variant was not identified using Melacarta software in South Island patients, and it has not been previously reported as a polymorphic variant in four SNP databases (dbSNP/Uniprot, SNPper, ALFRED, or SNPedia). Interestingly, the EPHB6G404S mutation occurred significantly more frequently in melanomas without detectable BRAF or NRAS mutations.
Overall, NRAS mutations were detected in slightly less than a third (26.6%) of melanomas, and we found that NRAS mutations were much more frequent in melanomas of South Island patients (38.3%) than in North Island melanoma patients (21.9%), or in TCGA data (22%). The higher NRAS mutation frequency in South Island versus North Island was largely attributed to mutations occurring in codon 61 of NRAS exon 2. Therefore, NRAS exon 2 and BRAF exon 15 variants represented the two most frequently mutated exons in NZ melanomas. Further, in 6.2% of cases, the Melacarta Sequenom panel also identified a second mutation in addition to BRAF or NRAS mutations, with NRAS plus EPHB6G404S being one of the most frequent (Table 3).
Differences in NRAS mutation rates in other population groups have been reported previously. For example, markedly different NRAS mutation rates were reported between Middle-South Italy and Sardinia (21% vs. 2%; P<0.0001), and were suggested to be due to differences in genetic background [26], but no conclusive supporting evidence was presented for how the NRAS mutation rate became repressed in Sardinia.
Approximately 74% of the NZ population classify themselves as being of European decent, while only 14.9% and 11.8% of the population classify themselves as being Maori or Asian ethnicity respectively (http://www.stats.govt.nz/Census/2013-census/profile-and-summary-reports/quickstats-culture-identity/ethnic-groups-NZ.aspx). The proportion of Maori or Asian individuals in the NZ population influences melanoma incidence statistics in NZ, because individuals who are of Maori or Asian decent have markedly lower rates of melanoma than those of European decent in NZ (Wellington: Ministry of Health. 2015. www.health.govt.nz/publication/cancer-new-registrations-and-deaths-2012). Although almost 90% of people of Maori or Asian ethnicity in NZ reside in the North Island (www.stats.govt.nz/census), ethnicity was not considered a major contributory factor to differences in oncogene mutation rates observed in this study. The proportion of individuals who were of Maori or Asian decent was relatively small, and only very small differences in the numbers of Maori or Asian patients occurred between North and South Islands.
The main epidemiological risk factor contributing to NZ’s high incidence of melanoma is annual UV radiation exposure. NZ has a 40% higher average UV exposure than North America [27] or central Europe [28]. Moreover, as discussed above, NZ has a mainly fair-skinned Caucasian population. However, the difference in the NRAS mutation frequency between North and South Islands may not directly be associated with DNA damage caused by UVB-radiation [4, 5, 23, 29]. This is because, while several recurrent oncogenic mutations in melanoma have been associated with UVB-radiation [3, 30], BRAFV600E and NRASQ61R mutations do not themselves contain UVB-radiation mutation signatures. The conclusion that direct chronic UVB-induced DNA damage is not a key feature in NZ melanomas is also supported by the observation that BRAFV600K mutations were relatively infrequent in both North and South Island NZ melanoma cohorts.
UV radiation levels during summer in the South Island are on average lower than in the North Island, or in Queensland, Australia (~12°S to 27°S), but are nevertheless higher than similar latitudes in the Northern hemisphere [27]. Clear skies and a high UV index in South Island summers frequently lead to sunburn-associated erythema in fair skinned individuals. However, in winter UV radiation levels are relatively low in the South Island, and also in the Australian southern states, as compared to the rest of Australia, or to the North Island of NZ [27]. A relatively higher incidence of vitamin D deficiency occurs in Southern latitudes than the Northern latitudes of Australia and NZ [31-33], which raises the question of whether high UV-radiation exposure in South Island during Spring, when vitamin D levels are lowest [33], could lead to a high frequency of NRASQ61 mutations versus other mutation types. DNA mutations accumulate for up to 12 hours following acute sunburn, even in the dark, and vitamin D has been suggested to protect DNA from UV damage [34, 35]. Moreover, vitamin D deficiency at diagnosis is linked to higher Breslow thickness in melanoma [36]. In this study we found that NRAS mutations were associated with nodular melanomas, which on average had a higher Breslow thickness than other melanoma types. Regarding relative “skin age” in the South Island versus North Island, living in the South Island has the same effect as increasing skin age by 27 years.
Loss of a sun tan, lower vitamin D levels, and susceptibility to sunburns, are common following winter in the South Island of NZ, due to extreme seasonal difference in UV radiation [33]. In contrast, in many climates outdoor workers exposed to the sun all year do not have markedly increased melanoma rates [3]. If low levels of vitamin D potentially lead to higher NRAS mutation rates, this would involve a mechanism as yet not understood. NRAS mutations have previously been associated with UV-associated DNA damage [37-39], but are not frequently observed in melanomas containing high UV-induced mutation loads [40]. Although there is little or no available evidence to suggest that UVA-radiation can induce NRAS mutations in association with pheomelanin-induced reactive oxygen species (ROS) [41], absence of a UVB mutation signature in NRAS does not exclude this possibility.
High and low NRAS and BRAF mutation frequencies, respectively, have implications for the diagnosis, and treatment of melanoma in NZ. A lower rate of BRAFV600E mutations in NZ suggests that other BRAF mutation types may constitute a relatively higher proportion of the total BRAF mutations, and that mutation screening and treatment options should therefore take this into consideration. Also, the higher NRAS mutation rates in the South Island suggest that South Island melanoma patients could benefit more often from the use of immune checkpoint blockade therapy, which has higher response rates in NRAS mutant melanomas [13].
In conclusion, we show for the first time, in one of the largest cohorts of melanoma patients reported, that NRAS and EPHB6 mutation frequencies in NZ melanomas are significantly variable between populations comprised of individuals of the same ethnic group, who have similar genetic backgrounds, and who have similar lifestyle practices and choices. We propose the differences in oncogene mutation frequencies observed in melanomas depends primarily on environmental risk factors, which we speculate could be associated with differences in intermittent exposure, or type of UV radiation of North and South Island NZ individuals. Understanding the reasons for variable mutation frequency will lead to precise interventions to effectively target oncogenic mutations, such as in NRAS or EPHB6 in melanoma.
MATERIALS AND METHODS
Approval by ethics committee
Ethical approval for this study was obtained from the NZ Health and Disability Ethics Committee (HDEC reference numbers 13/CEN/46 and NZ/13/B7B408).
Patient sample and clinical information collection
529 Formalin Fixed Paraffin Embedded (FFPE) samples of melanoma tissue from 529 patients and related clinical information were collected in the period 2000 to 2014 from five different laboratories in NZ, in (North Island) Auckland, Tauranga, Wellington, and (South Island) Christchurch. A total of 466 metastatic melanoma samples (one sample per patient) yielded Sequenom MassARRAY MelaCarta results, and 467 metastatic melanoma samples yielded Sanger sequencing results. Of these, 333 metastatic melanoma samples were from North Island, and 133 samples were from the South Island. Of the 333 samples from the North Island, 55 samples were from Auckland, 78 samples were from Tauranga and 105 samples were from Wellington. Patient clinical information was retrieved from the NZ Cancer Registry.
DNA preparation
FFPE tissues were cut into 10 μm sections and mounted onto glass slides. Experienced pathologists histologically evaluated and determined areas with a large proportion of tumor cells on corresponding H&E slide for all melanoma samples. Tissues were macrodissected manually using a sterile blade in accordance with the marked areas of tissue on the H&E slides. Genomic DNAs were isolated using either a QIAmp DNA FFPE Tissue Kit or DNeasy kit (Qiagen) according to manufacturers instructions. The quantity and quality of DNA was assessed using a Nanodrop spectrophotometer and a Quantus Fluorometer (Promega).
Gene mutations analyses
MelaCarta mass spectrometric analysis
466 of 529 melanoma samples yielded results for 72 somatic mutations in 20 melanoma-related genes using mass spectrometry genotyping on the Sequenom MassARRAY MelaCarta Panel v1.0 (Agena Bioscience, San Diego, CA, USA) and all assays were carried out by the Liggins Institute, University of Auckland, New Zealand. Mutation detection using an optimized mass spectrometric genotyping platform has been suggested to be more sensitive, but not necessarily more specific, than Sanger sequencing [42], although we also used high resolution melting analysis, which greatly improves the sensitivity of Sanger sequencing.
High resolution melting analysis
Approximately 85% of the samples analyzed by Sanger sequencing were also analyzed high resolution melting (HRM) analysis. HRM analysis was set up using 10 ng of genomic DNA and performed in duplicate on the LightCycler 480 (Roche) using previously published primers and amplification conditions [43]. Each run included a wildtype control, a homozygous mutant (BRAFV600E) and a low mutant control (40% mutant 60% wild-type) for normalization. The HRM results were analyzed by Gene Scanning software with normalized, temperature-shifted melting curves displayed as difference plots. Samples were considered mutated when significant difference of fluorescence level for all duplicates fell outside of the range of variation detected for the wild-type control.
Sanger sequencing of BRAF exon 15
Sanger sequencing remains the gold standard for mutation detection in clinical samples [44]. Bi-directional Sanger sequencing of all melanoma samples was carried out using the following protocol at the Capital and Coast District Health Board diagnostic laboratory, Wellington, New Zealand, to detect mutations in exon 15 of the BRAF gene with the following primers
BRAF 15-forward, 5’- TCATAATGCTTGCTCTGA TAGGA
BRAF 15-reverse, 3’-GGCCAAAAATTTAATCAG TGGA.
PCR was performed in a 25μl volume containing 50ng of genomic DNA, 0.5μl of polymerase (5U/μl Bioline), 2.5μl of reaction buffer (10x containing 1.5mmol/L MgCl2 (Bioline) 0.25μl of dNTPs (25μmol/L stock solution) and 0.5μl of each primer (20pmol each). PCR amplification was carried out under the following conditions: 95°C for 15 minutes followed by 40 cycles at 95°C for 30 seconds, 56°C for 30 seconds, 72°C for 30 seconds and a final extension step at 72°C for 10 minutes. Amplified products were purified using ExoSAP-IT (Affymetrix) and sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer’s protocol. Sequences were run on an eight capillary ABI3500 genetic sequencer (Life Technologies).
Data and statistical analyses
MelaCarta Panel outputs were processed using an excel spreadsheet. The TCGA mutation data was downloaded using the cBioportal tool on 8th August 2015. The downloaded data was level 3 and contained information (in text format) about the mutation status of each patient. From this information we segregated primary and metastatic melanomas and compared only metastatic melanoma mutations, as described.
EU cohort
Fishers exact test was used to evaluate the association between mutational prevalence and between different populations. Statistical analysis was performed using the R studio (version 3.2.2) statistical software.
ACKNOWLEDGMENTS
The authors thank Dr Phillip Shepherd for Sequenom technical assistance, Dr Matthew Parry for statistical advice, and Betsy Marshall as well as MelNet NZ, and the Division of Health Sciences, University of Otago, for helping to coordinate and support meetings of the main investigators during the research project.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This research was supported by funds from the Lottery Health Board New Zealand (MRE), the Wellington Medical Research Foundation, Capital and Coast District Health Board, and the Cancer Society of New Zealand Wellington Division (PF and AJ), Hoffman La Roche Ltd (MRE), the New Zealand Institute for Cancer Research Trust (MRE), and the Maurice Wilkins Centre for Molecular Biodiscovery (PRS).
REFERENCES
1. Whiteman DC, Green AC and Olsen CM. The Growing Burden of Invasive Melanoma: Projections of Incidence Rates and Numbers of New Cases in Six Susceptible Populations through 2031. J Invest Dermatol. 2016; 136:1161-1171.
2. Kanavy HE and Gerstenblith MR. Ultraviolet radiation and melanoma. Seminars in Cutaneous Medicine and Surgery. 2011; 30:222-228.
3. Whiteman DC, Pavan WJ and Bastian BC. The melanomas: a synthesis of epidemiological, clinical, histopathological, genetic, and biological aspects, supporting distinct subtypes, causal pathways, and cells of origin. Pigment Cell & Melanoma Research. 2011; 24:879-897.
4. Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, Zhang H, Zeid R, Ren X, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012; 485:502-506.
5. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150:251-263.
6. Vidwans SJ, Flaherty KT, Fisher DE, Tenenbaum JM, Travers MD and Shrager J. A melanoma molecular disease model. PloS One. 2011; 6:e18257.
7. Zhang T, Dutton-Regester K, Brown KM and Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016; 29:266-283.
8. Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, Baboud J, Balasundaram M, Balu S, et al. Genomic Classification of Cutaneous Melanoma. Cell. 2015; 161:1681-1696.
9. Tsao H, Chin L, Garraway LA and Fisher DE. Melanoma: from mutations to medicine. Genes & Development. 2012; 26:1131-1155.
10. Klein O, Clements A, Menzies AM, O’Toole S, Kefford RF and Long GV. BRAF inhibitor activity in V600R metastatic melanoma. European Journal of Cancer. 2013; 49:1073-1079.
11. McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, Garbe C, Testori A, Maio M, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. The Lancet Oncology. 2014; 15:323-332.
12. Sharma A, Shah SR, Illum H and Dowell J. Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs. 2012; 72:2207-2222.
13. Johnson DB and Puzanov I. Treatment of NRAS-mutant melanoma. Current Treatment Options in Oncology. 2015; 16:15.
14. Bucheit AD, Syklawer E, Jakob JA, Bassett RL, Jr., Curry JL, Gershenwald JE, Kim KB, Hwu P, Lazar AJ and Davies MA. Clinical characteristics and outcomes with specific BRAF and NRAS mutations in patients with metastatic melanoma. Cancer. 2013; 119:3821-3829.
15. Menzies AM, Haydu LE, Visintin L, Carlino MS, Howle JR, Thompson JF, Kefford RF, Scolyer RA and Long GV. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clinical Cancer Research. 2012; 18:3242-3249.
16. Dutton-Regester K, Irwin D, Hunt P, Aoude LG, Tembe V, Pupo GM, Lanagan C, Carter CD, O’Connor L, O’Rourke M, Scolyer RA, Mann GJ, et al. A high-throughput panel for identifying clinically relevant mutation profiles in melanoma. Molecular Cancer Therapeutics. 2012; 11:888-897.
17. Devitt B, Liu W, Salemi R, Wolfe R, Kelly J, Tzen CY, Dobrovic A and McArthur G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell & Melanoma Research. 2011; 24:666-672.
18. Edlundh-Rose E, Egyhazi S, Omholt K, Mansson-Brahme E, Platz A, Hansson J and Lundeberg J. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma research. 2006; 16:471-478.
19. Jakob JA, Bassett RL, Jr., Ng CS, Curry JL, Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim KB, Lazar AJ, Hwu P and Davies MA. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012; 118:4014-4023.
20. Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? International Journal of Cancer. 2003; 104:527-532.
21. Guan J, Gupta R and Filipp FV. Cancer systems biology of TCGA SKCM: efficient detection of genomic drivers in melanoma. Scientific Reports. 2015; 5:7857.
22. Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, Yao H, Lazar AJ, Aldape KD, Medeiros LJ and Luthra R. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. The Journal of Molecular Diagnostics. 2013; 15:220-226.
23. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010; 463:191-196.
24. Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA and Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nature Genetics. 2009; 41:1127-1132.
25. Bulk E, Yu J, Hascher A, Koschmieder S, Wiewrodt R, Krug U, Timmermann B, Marra A, Hillejan L, Wiebe K, Berdel WE, Schwab A and Muller-Tidow C. Mutations of the EPHB6 receptor tyrosine kinase induce a pro-metastatic phenotype in non-small cell lung cancer. PloS One. 2012; 7:e44591.
26. Colombino M, Lissia A, Capone M, De Giorgi V, Massi D, Stanganelli I, Fonsatti E, Maio M, Botti G, Caraco C, Mozzillo N, Ascierto PA, Cossu A and Palmieri G. Heterogeneous distribution of BRAF/NRAS mutations among Italian patients with advanced melanoma. Journal of Translational Medicine. 2013; 11:202.
27. McKenzie R, Bodeker G, Scott G, Slusser J and Lantz K. Geographical differences in erythemally-weighted UV measured at mid-latitude USDA sites. Photochemical & Photobiological Sciences. 2006; 5:343-352.
28. Seckmeyer G and Mckenzie RL. Increased Ultraviolet-Radiation in New-Zealand (45-Degrees-S) Relative to Germany (48-Degrees-N). Nature. 1992; 359:135-137.
29. Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature Genetics. 2012; 44:1006-1014.
30. Pimiento JM, Larkin EM, Smalley KS, Wiersma GL, Monks NR, Fedorenko IV, Peterson CA and Nickoloff BJ. Melanoma genotypes and phenotypes get personal. Laboratory Investigation. 2013; 93:858-867.
31. Gill TK, Hill CL, Shanahan EM, Taylor AW, Appleton SL, Grant JF, Shi Z, Dal Grande E, Price K and Adams RJ. Vitamin D levels in an Australian population. BMC Public Health. 2014; 14:1001.
32. Lucas RM, Ponsonby AL, Dear K, Valery PC, Taylor B, van der Mei I, McMichael AJ, Pender MP, Chapman C, Coulthard A, Kilpatrick TJ, Stankovich J, Williams D and Dwyer T. Vitamin D status: multifactorial contribution of environment, genes and other factors in healthy Australian adults across a latitude gradient. The Journal of Steroid Biochemistry and Molecular Biology. 2013; 136:300-308.
33. Wheeler BJ, Dickson NP, Houghton LA, Ward LM and Taylor BJ. Incidence and characteristics of vitamin D deficiency rickets in New Zealand children: a New Zealand Paediatric Surveillance Unit study. Australian and New Zealand journal of Public Health. 2015; 39:380-383.
34. Gordon-Thomson C, Gupta R, Tongkao-on W, Ryan A, Halliday GM and Mason RS. 1 alpha,25 Dihydroxyvitamin D-3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photoch Photobio Sci. 2012; 11:1837-1847.
35. Song EJ, Gordon-Thomson C, Cole L, Stern H, Halliday GM, Damian DL, Reeve VE and Mason RS. 1alpha,25-Dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. The Journal of Steroid Biochemistry and Molecular Biology. 2013; 136:131-138.
36. Wyatt C, Lucas RM, Hurst C and Kimlin MG. Vitamin D deficiency at melanoma diagnosis is associated with higher Breslow thickness. PloS One. 2015; 10:e0126394.
37. Ball NJ, Yohn JJ, Morelli JG, Norris DA, Golitz LE and Hoeffler JP. Ras mutations in human melanoma: a marker of malignant progression. The Journal of Investigative Dermatology. 1994; 102:285-290.
38. van’t Veer LJ, Burgering BM, Versteeg R, Boot AJ, Ruiter DJ, Osanto S, Schrier PI and Bos JL. N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Molecular and Cellular Biology. 1989; 9:3114-3116.
39. van Elsas A, Zerp SF, van der Flier S, Kruse KM, Aarnoudse C, Hayward NK, Ruiter DJ and Schrier PI. Relevance of ultraviolet-induced N-ras oncogene point mutations in development of primary human cutaneous melanoma. The American Journal of Pathology. 1996; 149:883-893.
40. Mar VJ, Wong SQ, Li J, Scolyer RA, McLean C, Papenfuss AT, Tothill RW, Kakavand H, Mann GJ, Thompson JF, Behren A, Cebon JS, Wolfe R, et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clinical Cancer Research. 2013; 19:4589-4598.
41. Nishigori C. Cellular aspects of photocarcinogenesis. Photochemical & Photobiological Sciences. 2006; 5:208-214.
42. MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, Davis M, Yao K, Hanna M, Mondal C, Luongo L, Emery CM, Baker AC, et al. Profiling critical cancer gene mutations in clinical tumor samples. PloS One. 2009; 4:e7887.
43. Lyon E and Wittwer CT. LightCycler technology in molecular diagnostics. The Journal of Molecular Diagnostics. 2009; 11:93-101.
44. Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, Kanagal-Shamanna R, Greaves WO, Medeiros LJ, Aldape KD and Luthra R. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. The Journal of Molecular Diagnostics. 2013; 15:607-622.