
INTRODUCTION
Emx2 is a transcription factor gene controlling a variety of brain developmental subroutines [1]. It cooperates with Otx2 and Pax6 in promoting formation and proper rostro-caudal specification of prosomeres 1-4, respectively [2]. Together with Pax6, it specifies dorsal telencephalon as pallium [3]. Within the early developing pallium, it stimulates the expansion of the neural precursor pool by delaying neuronogenesis progression [4] and the proper generation/survival of Cajal-Retzius pioneer neurons [5]. Moreover, it promotes the development of hippocampal and visual territories [6, 7].
Recently, we found that Emx2 also inhibits astrogliogenesis, by decreasing proliferation of astrocyte-committed progenitors via EgfR and Fgf9 downregulation [8, 9]. Furthermore, several groups reported an inverse correlation between EMX2 expression levels and malignancy of a number of non neural tumors [10–14]. These findings led us to hypothesize employing Emx2 as a tool to suppress highly proliferating glioblastoma multiforme tumors (GBM).
GBM, also classified as WHO grade IV glioma, is the most aggressive malignant primary brain tumor in humans. It can affect cerebral cortex, cerebellum, brainstem and spinal cord. It mainly appears around 65-75 years, as a primary tumor or a recurrency of a previous, lower-grade glioma. Neurological symptoms are highly heterogeneous. Final prognosis is very poor. State of the art treatment combines surgery, temozolomide (TMZ) chemotherapy and radiation. Median survival upon this treatment is 14 months (only 4 in the absence of treatment) [15, 16].
GBMs are characterized by high mitotic rates, diminished apoptosis, poorly differentiated astrocytes and rich neoangiogenesis. Despite these commonalities, their origin and genetic features are highly heterogeneous. Nevertheless, GBMs (in particular, advanced/recurrent ones) share specific structural mutations and copy number variations, among which EGFR and PDGFRA amplification, as well as NF1, PTEN and CDKN2A/B loss [17].
Here we show that, in all GBM lines tested, Emx2 overexpression suppresses glioblastoma growth, both in vitro and in vivo. Interestingly, Emx2 activity relies on modulation of a number of malignancy-related genes, including a subset of those affected in GBM by late, oncogenic copy number variations. This may result into an appreciable therapeutic effect on a large variety of GBMs and prevent selection of drug-resistant clones as well as recurrencies. Finally, Emx2 overexpression driven by the stem-cell-specific “neuro-Nestin promoter” [8] is still able to fully suppress tumor cultures. Therefore, this approach might be appropriate to target GBM cells while not damaging differentiated not-cancerous cells, and adequate to eradicate tumor-initiating-cells, so preventing GBM recurrencies.
RESULTS
Emx2 overexpression kills glioblastoma cells in vitro
To assess if Emx2 can antagonize glioblastoma multiforme, we overexpressed its coding sequence in 2 GBM lines (U87MG and T98G) as well as in GBM cell cultures originating from 5 different patients (GbmA, GbmB, GbmC, GbmD and GbmE), via lentiviral vectors and TetON technology. As controls, we employed the corresponding GBM cultures, infected by Egfp- or Emx2-encoding lentiviruses and kept in the presence or absence of doxycycline, respectively (Figure 1A, 1B and Supplementary Figure S1). In all cases, the activation of the Emx2 transgene arrested the expansion of the culture and led to its collapse, usually within 7-8 days, never beyond the 22nd day (Figure 1C-1I). As we detected in Emx2-gain of function (-GOF) GbmA, GbmB and GbmC samples (Figure 1L-1O), this possibly reflected a decrease of the proliferating fraction (up to -34.5±5.3%, p<0.01, in GbmB) (Figure 1L, 1M) and an increase of the apoptotic fraction (up to 539.7±90.5%, p<0.001, in GbmB) (Figure 1N-1O). Intriguingly, G1-to-S phase progression was not affected (Supplementary Figure S2).
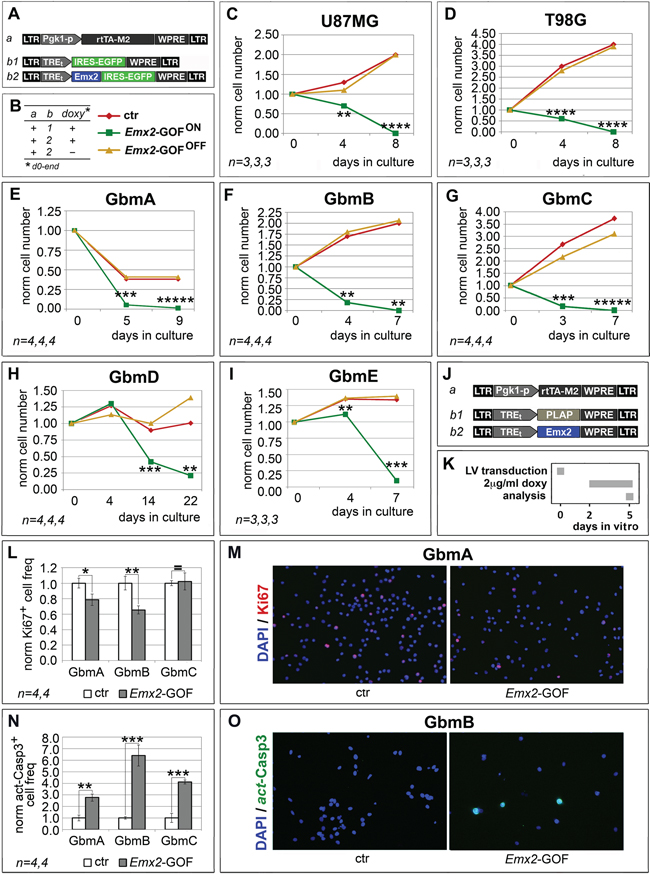
Figure 1: Population dynamics of Emx2 gain-of-function GBM cultures. In vitro kinetic progression of U87MG, T98G, GbmA, GbmB, GbmC, GbmD and GbmE GBM lines C-I., engineered by lentiviral vectors and TetON technology as in A, B., and kept as adherent C, D. or floating cultures E-I., under Fgf2 and Egf. Ki67+ proliferating L, M. and activated-Casp3+ apoptotic N, O. fractions of GbmA, GbmB and GbmC glioblastoma cells, engineered by control (J., a-b1) and Emx2-GOF (J, a-b2) lentiviral sets, and kept as floating cultures according to the timetable in K. Cell numbers were normalized against t=0 values (C-I), or control values L, N. [As for (L, N), absolute average control cell frequencies were: 0.207, 0.155 and 0.131 (Ki67+, in GbmA, GbmB and GbmC cultures, respectively); 0.001, 0.012 and 0.012 (actCasp3+, in GbmA, GbmB and GbmC cultures, respectively)]. n is the number of biological replicates. p-value was calculated by t-test (one-tail, unpaired): *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, *****p<0.00001.
Emx2 antagonizes glioblastoma by a pleiotropic impact on malignancy-related processes
To cast light on molecular mechanisms underlying Emx2 impact on GBM kinetics, we overexpressed its coding sequence in 5 GBM samples and scored mRNA levels of selected genes involved in their malignancy. These genes include: (a) a group implicated in relaying mitogenic signals along RTK cascades (EGFR, PDGF, PDGFRA, PTEN, NF1), (b) a group involved in the control of early G1/late G1 checkpoint (MYC, MYCN, RB1, CDKN2A, CDKN2B, CDK4, CDK6, CCND2), and (c) a more heterogeneous group dealing with a variety of malignancy-related processes, such as stemness, apoptosis, neovasculogenesis (SOX2, HES1, GLI1, TRP53, MDM2, VEGF). In all samples analyzed, Emx2 significantly altered the expression of group (a) genes, consistently with its antioncogenic activity. It downregulated EGFR in all cases. In addition, it decreased PDGF and PDGFRA in 1 and 4 cases, respectively, and increased PTEN, in 1 case (Table 1). In a large subset of samples, Emx2 also modulated mRNA levels of group (b) genes, again in agreement with its antioncogenic activity (Table 1). These genes include - in particular - CDK4 and CDK6, mastering the early G1 checkpoint (decreased in 3 and 2 cases, respectively). Finally, Emx2 downregulated SOX2 in 4 samples and increased TRP53 and HES1 expression, in 2 and 5 samples, respectively (Table 1).
Table 1: Biased mRNA profiling of Emx2 gain-of-function GBM cultures
t |
Gbm A |
Gbm B |
Gbm C |
Gbm D |
U87-MG |
gene function |
|---|---|---|---|---|---|---|
3d |
3d |
3d |
3d |
4d |
||
EGFR |
0.25 ±0.06 |
0.78 ±0.03 |
0.61 ±0.04 |
0.37 ±0.13 |
0.48 ±0.02 |
RTK signaling |
PDGF |
0.40 ±0.004 |
ns |
ns |
1.74 ±0.12 |
ns |
|
PDGFRA |
ns |
0.75 ±0.11 |
0.80 ±0.07 |
0.55 ±0.01 |
0.16 ±0.10 |
|
PTEN |
ns |
1.54 ±0.23 |
ns |
>0.30 ±0.03 |
ns |
|
NF1 |
ns |
ns |
ns |
0.26 ±0.01 |
ns |
|
MYC |
ns |
ns |
ns |
0.83 ±0.04 |
ns |
cell cycle control |
MYCN |
0.55 ±0.03 |
ns |
ns |
0.40 ±0.06 |
ns |
|
RB1 |
ns |
0.45 ±0.16 |
ns |
0.46 ±0.04 |
ns |
|
CDKN2A |
ns |
2.68 ±0.37 |
ns |
0.22 ±0.01 |
ns |
|
CDKN2B |
ns |
2.68 ±0.37 |
ns |
0.22 ±0.01 |
ns |
|
CDK4 |
0.48 ±0.07 |
ns |
ns |
0.81 ±0.06 |
0.72 ±0.08 |
|
CDK6 |
0.30 ±0.02 |
ns |
ns |
0.36 ±0.004 |
ns |
|
CCND2 |
0.16 ±0.06 |
ns |
ns |
1.17 ±0.03 |
ns |
|
SOX2 |
0.30 ±0.04 |
0.60 ±0.06 |
0.70 ±0.11 |
0.19 ±0.01 |
ns |
other malignancy-related |
HES1 |
2.56 ±0.37 |
8.02 ±0.27 |
1.38 ±0.05 |
1.28 ±0.02 |
4.34 ±0.80 |
|
GLI1 |
ns |
2.60 ±0.65 |
ns |
2.90 ±0.25 |
ns |
|
TRP53 |
ns |
2.87 ±0.57 |
4.58 ±0.87 |
0.67 ±0.05 |
0.66 ±0.08 |
|
MDM2 |
ns |
ns |
ns |
0.46 ±0.06 |
ns |
|
VEGF |
ns |
ns |
ns |
ns |
ns |
mRNA levels of presumptive mediators of Emx2 anti-oncogenic activity in GBM cells engineered as in Figure 1A. Seven days after lentiviral transduction, doxycyclin was added at 2 μg/ml. RNA samples were collected at time “t” after doxycyclin addition. qRT-PCR results, normalized against GAPDH and further normalized against their own negative controls, are shown as average ± s.e.m. Values possibly accounting for Emx2 anti-oncogenic activity are highlighted in blue. ns, not significant. n is the number of biological replicates. p-value was calculated by t-test (one-tail, unpaired).
To complement mRNA profiling, we also monitored Emx2 overexpressing GBM cells for key phospho-proteins involved in malignancy-related, intracellular signal transduction (Figure 2 and Supplementary Figure S3). We found a significant decrease of p(Thr202/Tyr204)Erk1/2 (-40.3±6.3%, p<0.005, see Figure 2C, 2D). This may stem from depressed EGF and PDGF signalling. It may be a key determinant of the kinetic behaviour of Emx2-GOF GBM cells [16]. Furthermore, we detected a robust increase of p(Ser463/465)Smad1,5,8 (+100%, p<0.003, see Figure 2E, 2F). This is an index of enhanced Bmp signalling, which was shown to be instrumental to Emx2-dependent inhibition of astroblast proliferation [9]. Finally, we found that Stat3 phosphorylation levels in Tyr705 and Ser727, crucial to self-renewing abilities of GBM cells [18], were unchanged (Figure 2G-2J).
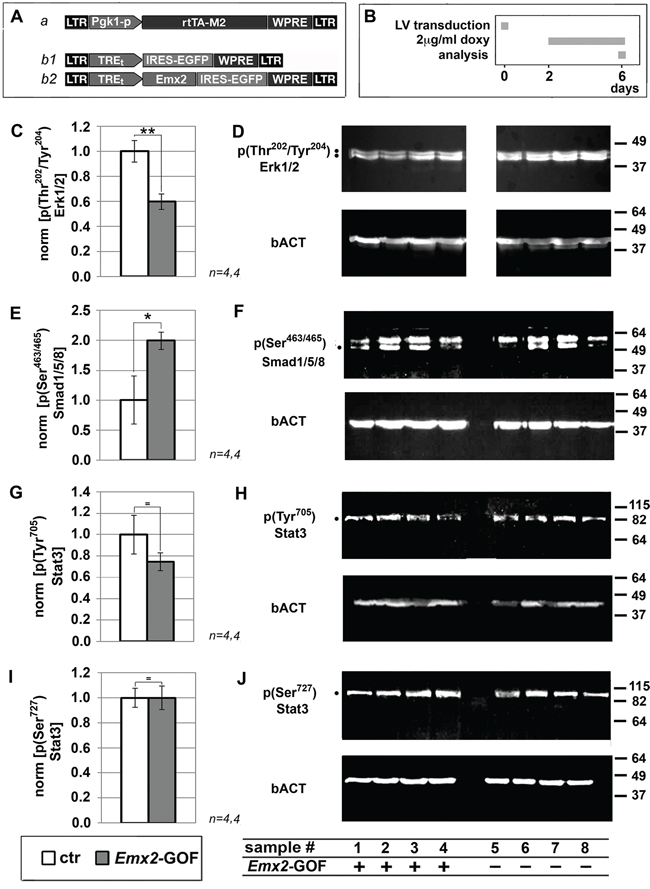
Figure 2: Immunoprofiling of Emx2 gain-of-function GBM cultures for key intracellular signalling transducers. Evaluation of p(Thr202/Tyr204)Erk1/2 C, D., p(Ser463/Ser465)Smad1/5/8 E, F. p(Tyr705)Stat3 G, H. and p(Ser727)Stat3 I,J. levels in U87 cell samples, engineered as in A, B. Values were normalied against controls. n is the number of biological replicates. p-value was calculated by t-test (one-tail, unpaired): *p<0.05, **p<0.01.
Next, we tested the functional relevance of selected mRNA/protein changes described above to the Emx2 antioncogenic activity. For this purpose, we chose a few “X” agents neutralizing such changes and evaluated their capability to rescue the original GBM kinetic profiles (Figure 3A-3C).
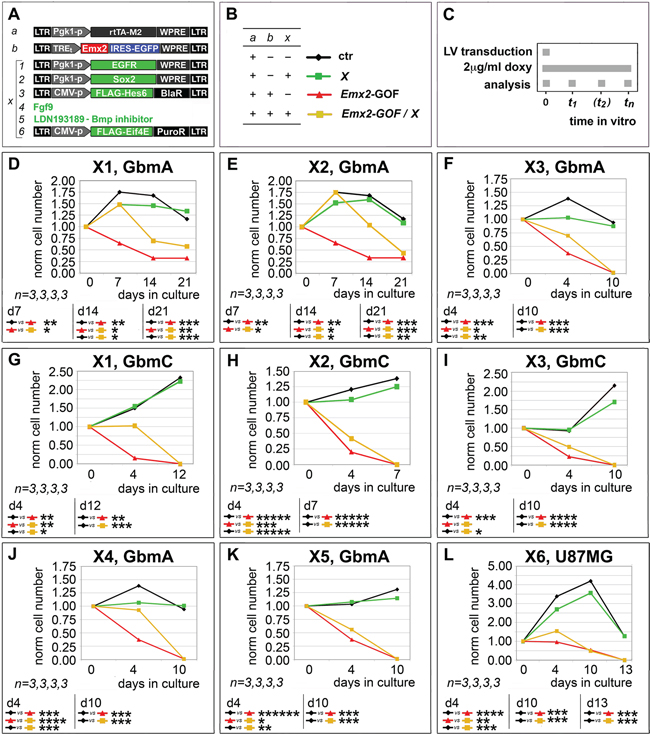
Figure 3: Rescue of Emx2 antioncogenic activity via modulation of its presumptive mediators. GBM cells were engineered and cultured as in A-C. In particular, lentiviruses harboring an IRES-EGFP or a PLAP module under the control of a TREt promoter were used as controls for “b” and “X1, X2, X3, X6”, respectively. Cells were scored for the capability of selected “X” agents (restoring presumptive mediators of Emx2 anti-oncogenic activity) to rescue their control kinetic profiles D-L. n is the number of biological replicates. p-value was calculated by t-test (one-tail, unpaired).
First, we tried to restore the basic expansion rate of GbmA and GbmC cultures, previously made gain-of-function for Emx2, by transducing them with an EGFR-expressing transgene (Figure 3A, rescue X1). This manipulation slowed down the decline of these cultures, however only in a partial and temporary fashion (Figure 3D, 3G). A similar effect was elicited by overexpression of the stemness-factor SOX2 (Figure 3A, rescue X2) in Emx2-GOF GbmA and GbmC (Figure 3E, 3H). Noticeably, neither EGFR nor SOX2 overexpression perturbed GBM kinetics in control conditions (Figure 3D, 3E, 3G, 3H).
Moreover, we tried to counteract HES1, one of the main Emx2-responders, whose overexpression was previously linked to proliferation arrest in a variety of contexts [19–21]. To this aim, we delivered its established functional antagonist HES6 [22] to GBM cells (Figure 3A, rescue X3). This manipulation slowed down the collapse of GbmA and GbmC cultures overexpressing Emx2, while not fully preventing it (Figure 3F, 3I). Conversely, HES6 overexpression did not promote GBM expansion in control conditions (Figure 3F, 3I).
Next, we tried to restore RTK signalling defects evoked by Emx2 (Figure 2C, 2D), providing GBM cultures with an excess of Fgf9 (Figure 3A, rescue X4). This is a key ligand down-regulated by Emx2 [9] and proven to promote proliferation within the astrocytic lineage [23]. Also, we silenced BMP signalling (Figure 3A, rescue X5), already proposed as a key therapeutic tool against GBM [24, 25]. Both manipulations slowed down the decline of Emx2-GOF GbmA cultures, however only to a partial extent (Figure 3J, 3K). Neither Fgf9 nor BMP-inhibitor delivery promoted an expansion of control GbmA cells (Figure 3J, 3K).
Finally, in addition to Emx2 impact on transcription, we considered the possibility that the anti-oncogenic activity of this protein could be strenghtened by its capability to chelate the translational factor Eif4e [26]. In agreement with this prediction, Eif4e overexpression in Emx2-GOF U87MG cultures (Figure 3A, rescue X6) delayed their decline, while not affecting U87MG controls (Figure 3L).
All that suggests that Emx2 may act by perturbing a number of genes and metabolic nodes crucial to GBM aggressiveness. It points at Emx2 as a promising therapeutic tool to simultaneously attack a variety of key effectors of GBM malignancy.
Emx2 overexpression elicits GBM collapse in vivo
To assess the portability of Emx2 antioncogenic activity in vivo, we transplanted engineered GBM cells (U87MG, GbmA, GbmC and GbmD) into the neocortical parenchyma of P4 wild-type mouse pups. Specifically, we injected a 1:1 mix of cells, made alternatively gain-of-function for Emx2 or a control, and labelled with Egfp and mCherry, respectively. One week later, we sacrificed the animals and scored each brain for the ratio between the number of Emx2-GOF cells (Egfp+) and the number of control cells (mCherry+). This ratio was equal to 0.40±0.05 (p<0.099, n=3), 0.44±0.13 (p<0.049, n=4), 0.34±0.12 (p<0.025, n=4) and 0.29±0.04 (p<0.006, n=4), for U87MG, GbmA, GbmC and GbmD, respectively. This indicates that Emx2 exerts a robust antioncogenic activity even in vivo (Figure 4). (In a previous pilot test run with Egfp+ and mCherry+ GBM cells not harboring additional transgenes, this ratio was close to 1) (Supplementary Figure S4).
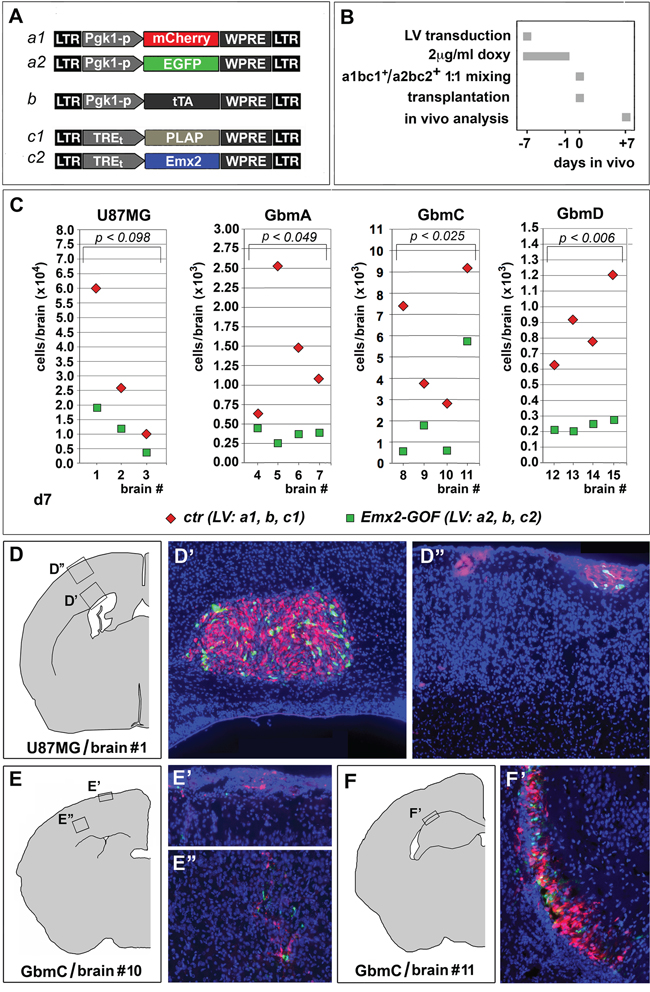
Figure 4: Emx2 antioncogenic activity in vivo. Experimental strategy and employed lentiviral vectors are shown in A, B. A 1:1 mix of differently fluoro-labelled, Emx2-GOF and control engineered GBM cells was transplanted into the cortical parenchyma of P4 wild type mouse pups. One week later, engrafted cells of different genotypes were scored in every single brain C-F'. 3-4 different brains were analyzed for every GBM line tested. p-value was calculated by t-test (one-tail, paired).
Disappontingly, Emx2 overexpression in pyramidal neurons is highly toxic (our unpublished data). Therefore, generalized Emx2 delivery to the diseased brain of GBM patients would not be a suitable approach. To circumvent this issue, we thought to restrict therapeutic Emx2 overexpression to tumor precursor cells, by putting it under the control of a cis-active element selectively firing in neural stem cells (Figure 5A, “Nes-p”; Supplementary Figure S5). Remarkably, this design turned out to be feasible, as it successfully replicated the kinetic outcome elicited upon generalized Emx2 overexpression (compare Figure 5C, 5D and Figure 1E, 1F).
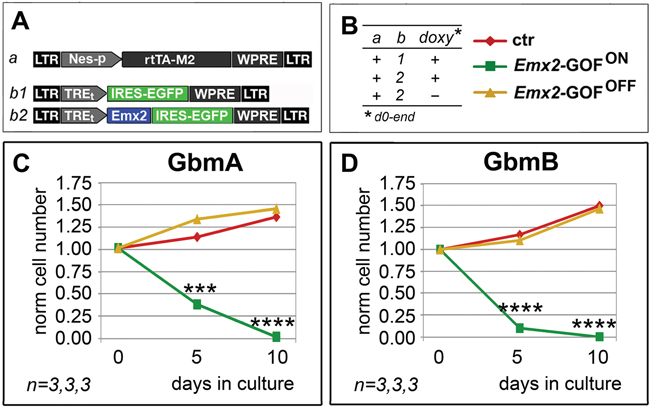
Figure 5: Persisting antioncogenic efficacy of Emx2 upon neural nestin enhancer-restricted overexpression. In vitro kinetic progression of GbmA and GbmB lines C, D., engineered by lentiviral vectors and TetON technology as in A, B., and kept as floating cultures, under Fgf2 and Egf. Cell numbers were normalized against t=0 values. n is the number of biological replicates. p-value was calculated by t-test (one-tail, unpaired): ***p<0.001, ****p<0.0001.
DISCUSSION
Here we showed that Emx2 overexpression in a number of GBM cultures forced them to collapse, by promoting cell death and inhibiting cell proliferation. Emx2 impact on GBM metabolism was complex and a number of genes and pathways sensitive to its overexpression were co-involved in its antitumoral activity. Remarkably, such activity was confirmed in vivo, upon transplantation of conditionally engineered tumor cells into the neocortical parenchyma of mouse neonates. Last but not least, restricting Emx2 overexpression to presumptive tumor stem cells replicated the outcome of generalized gene overexpression.
Multiple bodies of correlative data suggest that EMX2 downregulation could contribute to the genesis of GBMs. The COSMIC database (http://cancer.sanger.ac.uk/cosmic) reports two distinct homozygous EMX2 deletions occurring in 2 out of 801 gliomas (data not shown). Analysis of Allen Brain - Ivy Glioblastoma Atlas data showed us that EMX2-mRNA levels are specifically reduced by ≥2-folds in GBM tumors with respect to surrounding tissue (Supplementary Figure S6). Next, in the majority of glioblastoma cultures analyzed in the present study, endogenous EMX2-mRNA was undetectable. When it was present, its level was lower than in astrogenic, fetal cortico-cerebral NSCs (Supplementary Figure S7). Finally, a consistent scenario was found in acutely immunopanned GBM astrocytes, compared to astrocytes purified from the surrounding healthy tissue [27]. Albeit intriguing, all these correlative data are obviously not sufficient to draw firm conclusions about EMX2 role in GBM etiopathogenesis, which was out of the aims of the present study. Nevertheless, our data suggest that, regardless of its role in the oncogenic process, Emx2 may be a powerful tool for counteracting GBM tumors.
Subject of this study were two established GBM cell lines (U87MG and T98G) and five tumor cultures derived from operated GBM patients. Promisingly, all of them robustly responded to Emx2 and rapidly collapsed, because of defective proliferation and exaggerated cell death (Figure 1 and Supplementary Figure S2).
Noticeably, their molecular responses were complex and not stereotyped (Table 1), possibly reflecting GBM etiopathogenetic heterogeneity [28, 17, 29]. The vast majority of molecular changes evoked by Emx2 were consistent with its antitumor activity (Table 1 and Figure 2), a selection of them was proven to be instrumental to it (Figure 3). However, in no case, counteracting each of these changes could fully restore original kinetic properties of the cultures (Figure 3). All this means that Emx2 antioncogenic efficacy may emerge as a consequence of its ability to attack a variety of metabolic nodes crucial to malignancy. In addition to a robust inhibition of GBM expansion, this ability might help preventing selection of drug-resistant clones and recurrencies. Intriguingly, a number of Emx2-repressed and -stimulated genes reported above are the very same affected by duplications and deletions in late stage glioma cancers, respectively [17]. This further suggests that our manipulation could be therapeutically effective on a variety of high grade gliomas, regardless of their primary molecular origin.
Remarkably, Emx2 overexpression elicited a pronounced anti-GBM activity even in vivo (Figure 4). This was found by cotransplanting conditionally engineered tumor cells, alternatively expressing Emx2 or a control transgene, into the cortical parenchyma of wild type mouse pups and scoring the outcome one week later. This is a novel experimental setup, allowing to preliminarly assess antioncogenic power in vivo, quickly, in an immunocompetent environment, and in the presence of strong progliogenic cues. Finally, of particular therapeutic interest is the fact that a collapse of engineered GBM cultures also occurred when Emx2 overexpression was restricted to Nes-p+ precursor cells (Figure 5). These cells, in fact, are likely to include tumor initiating cells (TICs), from which tumor recurrencies are supposed to originate [38]. Such cells may escape even the attack by the most advanced oncolytic vectors developed against GBM [39].
All these results point to Emx2 as a novel, promising tool for GBM therapy. However, for this purpose long-term survival tests as well as the selection of a more appropriate, not-genotoxic vectors for gene delivery, are mandatory. Moreover, an in depth exploration of mechanisms mediating Emx2 activity, by unbiased GBM transcriptome profiling, is also due. These issues will be hopefully subject of a dedicated follow-up study.
MATERIALS AND METHODS
Lentiviral vectors packaging and titration
All lentiviruses were generated and titrated as previously described [40]. The full list of genomic plasmids used for lentiviruses packging is reported in Supplementary Materials.
GBM cell culture
Human U87MG GBM cell line (derived from a malignant glioma from a female patient by explant technique; PCR-validated as follows: Amelogenin: X; CSF1PO: 10,11; D13S317: 8,11; D16S539: 12; D5S818: 11,12; D7S820: 8,9; THO1: 9.3; TPOX: 8; vWA: 15,17) and T98G GBM cell line (derived from glioblastoma multiform tumour from a 61-year-old Caucasian male; PCR-validated as follows: Amelogenin: X,Y; CSF1PO: 10,12; D13S317: 13; D16S539: 13; D5S818: 10,12; D7S820: 9,10; THO1: 7,9.3; TPOX: 8; vWA: 17,20) were purchased from SIGMA (#89081402 and #92090213, respectively). Low passage criopreserved samples of them were employed to run this analysis. They were kept as adherent cultures in DMEM/Glutamax medium (ThermoFisher, Waltham - MA, #31966), supplemented with 10% FBS and 1X Pencillin/Streptomycin. Cells were cultured at 500 cells/μl and passaged by trypsin on days of counting or, at most, every 4 days.
GbmA-E cells originate from GBM surgical samples collected at IRCCS A.O.U. San Martino-IST, Dept of Neuroscience and Sense organs, Unit of Neurosurgery and Neurotraumatology, with patients' informed consent and in compliance with pertinent Italian law. They were derived in AD laboratory. Low passage criopreserved samples of them were employed in AM laboratory to run this analysis. GbmA cells were cultured in “DMEM / F12 / glutamax / NeurobasalA” (ThermoFisher #10888-022). GbmB-E were cultured as spheres in NeuroCult™ NS-A Proliferation Kit (Human) (StemCell Technologies, Vancouver - Canada, #05751). In both cases, mediums were supplemented with 1X Penicillin/Streptomycin, 2 μg/ml human heparin (StemCell Technologies #07980), 20 ng/ml recombinant human EGF (ThermoFisher #PHG0311), 20 ng/ml recombinant human FGF2 (ThermoFisher #PHG0261). All the cells were cultured under normoxic conditions (5% CO2, 21% O2, 74% N2). Cells were cultured at 500 cells/μl and passaged by Accutase (Sigma, Milan - Italy, #A6964) on days of counting or, at most, every 4 days.
When required, cells were acutely infected, at a concentration of 500 cells/μl, by a mix containing lentiviral vectors, each one at a multiplicity of infection (m.o.i) = 6. This moi is sufficient to infect the almost totality of GBM cells in such conditions. Where required, they were subsequently transferred to polylysinated coverslips. In specific cases, medium was supplemented by 0.7 μM LDN193189 (Stemgent, Lexington - MA, #130-096-226), 20 ng/ml Fgf9 (Sigma #SRP4057-10UG), or 10μg/ml BrdU. TetON-controlled transgenes were switched on by 2 μg/ml doxycyclin (Sigma #D9891-10G). n is the number of biological replicates. p-value was calculated by t-test (one-tail, unpaired).
Cell growth curves
After sphere dissociation, 2x105 GBM cells/well were seeded in a 24-well plate and infected with LV_Pgk1p-rtTA-M2-WPRE and LV_TREt-IRES-EGFP-WPRE or LV_TREt-Emx2-IRES-EGFP-WPRE, each at m.o.i. 6. Viable cells (trypan blue-excluding) were counted at fixed days on a hemocytometer. After every cell count, differently engineered cells were plated at the same concentration (2 × 105 GBM cells/well). Cell counting was performed on 1/200 of each biological sample (in case of Figure 1, 3 and 5 data, at t=0, each sample included 200,000 cells). Growth curves were interrupted when Emx2-GOF cell cultures had collapsed.
mRNA profiling
Aliquots of 2*105 GBM cells were infected with a lentivector mix containing LV_Pgk1p-rtTA-M2-WPRE and LV_TREt-IRES-EGFP-WPRE or LV_TREt-Emx2-IRES-EGFP-WPRE, each at m.o.i. 6. Seven days after infection, 2 μg/ml doxycycline was added. Three or four days later, cell pellets were processed for RNA extraction by Trizol™ (ThermoFisher #15596-026). Agarose gel electrophoresis and spectrophotometric measurements (NanoDrop ND-1000) were employed to estimate quantity, quality and purity of the resulting RNA. Prior to analysis, samples were processed by the TurboDNAfree kit (ThermoFisher #AM1907), according to manufacturer's instructions.
At least 1.0 μg RNA from each sample was retrotranscribed by SuperScriptIII™ (ThermoFisher #18080044) in the presence of random hexamers, according to manufacturer's instructions. RT-minus samples were scored as controls, in the case of intronless transcripts.
1/50 of the resulting cDNA was used as substrate of any subsequent qPCR reaction. PCR reactions were performed by the SybrGreen™ platform (Biorad, Milan - Italy, #1725270), according to manufacturer’s instructions.
The full list of primers is reported in Supplementary Materials and Methods.
Each biological replicate was scored at least in technical triplicate and data were normalized against GAPDH. Results were averaged and further normalized on their controls. Averages ± s.e.m.'s were reported in Table 1. Statistical significance of results was evaluated by the t-test (one-tail; unpaired). “n” is the number of samples.
Western blots
Western analysis was performed according to standard methods. Total cell lysates in CHAPS buffer were quantified by BCA protein assay kit (ThermoFisher #10678484) and denatured at 95°C for 5 min, prior to loading. Thirty micrograms of proteins were loaded per each lane and run on a 12% acrylamide − 0.1% SDS gel.
Full details about antibodies employed are reported in Supplementary Materials and Methods. Different antigens and bACT were sequentially revealed by an ECL kit (GE Healthcare, Milan - Italy, #GERPN2109). Images were acquired by an Alliance LD2–77.WL apparatus (Uvitec, Cambridge) and analyzed by Adobe Photoshop CS2 software™ and Microsoft Excel 11 software™.
Animal handling
Animal handling and subsequent procedures were in accordance with European [European Communities Council Directive of November 24, 1986 (86/609/EEC)] and Italian laws (D.L. 04.03.2014, n°26) and were approved by SISSA Board for Animal Welfare. Wild type mice (strains CD1 purchased from Harlan-Italy) were maintained at the SISSA mouse facility. P0 stands for the animal birthday.
Cell transplantation
P4 pups were anaesthetized on ice for 40-60s. 2.0 μl of a solution containing 200,000 cells (100,000 Emx2-GOF-Egfp+ and 100,000 control-mCherry+ cells, pre-engineered by lentiviral transduction 1 week before), mixed with 0.02% fast-green dye in NeuroCult™ NS-A Proliferation (Human) medium (StemCell Technologies #05751), were injected through the skull into the corticocerebral parenchyma, by free hands, using a sharp pulled micropipette (hole external diameter about 40 μm) with the help of light fibers. Animals were left to recover in a warm clean cage. Then they were transferred to their mother and were finally sacrificed 7 days later.
Sample preparation for immunofluorescence
As for the brains, they were fixed by immersion in 4% PFA overnight at 4°C, washed by 1X PBS 3 times, equilibrated in 30%sucrose-1XPBS at 4°C, included into OCT (Killik) and frozen at -80°C. Finally they were coronally sliced at 16 μm, by a Microm cryostat.
As for dissociated cell cultures, they were acutely attached on coverslips, fixed in 4% PFA for 15 min at room temperature and washed by 1X PBS 3 times.
Immunofluorescence
Immunocytofluorescence was performed as previously described [8, 41]. Specifically, BrdU unmasking was performed by 0.2 M HCl, for 15 min at RT. The full list of antibodies employed is reported in Supplementary Materials and Methods.
Immunos were photographed on a Nikon Eclipse TI microscope equipped with a 20X objective and a Hamamatsu camera. Images were imported and analyzed by Photoshop CS2 (Adobe) software. Where not otherwise stated, each experiment was performed at least in biological triplicate.
For each dissociated cell culture sample, cells were counted from at least 20 randomly assorted photographic fields, by an operator blind of sample identity. As for Ki67 and BrdU quantification, 2,000 cells/sample were scored. As for actCasp3 quantification, 10,000, 2,000 and 2,000 cells/sample were scored, in case of GbmA, -B and -C cultures, respectively. Frequencies of immunoreactive cells were averaged and s.e.m.'s were calculated. Results were normalized against controls. Their statistical significance was evaluated by the t-test (one-tail; unpaired). “n” is the number of samples.
Pictures of transplanted brains were electronically encrypted by random inversion of green and red signals and analyzed by an operator blind of sample identity. For each brain, tumor cells were counted from 1 every 5 sections and total, Egfp+ and mCherry+ cell numbers were plotted. Statistical significance of results was evaluated by the t-test (one-tail; paired). “n” is the number of samples.
ACKNOWLEDGMENTS
We thank Minh Do Duc, who built LV_Pgk1p-mCherry-WPRE, Marco Brancaccio, who built LV_TREt-IRES-PLAP-WPRE, Carol Filippis, who performed early preliminary kinetic assays on U87MG and T98G cells, and Clara Grudina for human neural stem cells preparations.
We thank the following Colleagues for the kind gift of plasmids in parentheses: Gord Fishell (pCLE), Rossella Galli (LV_Pgk1p-EGFR-WPRE), Anders Strom (LV_Pgk1p-FLAG-Hes6-BlaR and LV_pLenti6V5D(TOPO)).
CONFLICTS OF INTEREST
CF and AM declare competing financial interests (patent pending).
FUNDING
This work was fully sustained by SISSA intramurary funding.
Authors’ contributions
CF and AM designed this study, analyzed the experimental results and wrote the paper; CF also performed all the experiments; AD provided GbmA, -B, -C, -D, and -E tumor lines; GL provided technical help and assistance with GBM in vivo transplantations.
REFERENCES
1. Gangemi RMR, Daga A, Muzio L, Marubbi D, Cocozza S, Perera M, Verardo S, Bordo D, Griffero F, Capra MC, Mallamaci A, Corte G. Effects of Emx2 inactivation on the gene expression profile of neural precursors. Eur J Neurosci. 2006;23:325–34.
2. Kimura J, Suda Y, Kurokawa D, Hossain ZM, Nakamura M, Takahashi M, Hara A, Aizawa S. Emx2 and Pax6 function in cooperation with Otx2 and Otx1 to develop caudal forebrain primordium that includes future archipallium. J Neurosci. 2005;25:5097–108.
3. Muzio L, DiBenedetto B, Stoykova A, Boncinelli E, Gruss P, Mallamaci A. Conversion of cerebral cortex into basal ganglia in Emx2(-/-) Pax6(Sey/Sey) double-mutant mice. Nat Neurosci. 2002;5:737–45.
4. Heins N, Cremisi F, Malatesta P, Gangemi RM, Corte G, Price J, Goudreau G, Gruss P, Götz M. Emx2 promotes symmetric cell divisions and a multipotential fate in precursors from the cerebral cortex. Mol Cell Neurosci. 2001;18:485–502.
5. Mallamaci A, Mercurio S, Muzio L, Cecchi C, Pardini CL, Gruss P, Boncinelli E. The lack of Emx2 causes impairment of Reelin signaling and defects of neuronal migration in the developing cerebral cortex. J Neurosci. 2000;20:1109–18.
6. Muzio L, DiBenedetto B, Stoykova A, Boncinelli E, Gruss P, Mallamaci A. Emx2 and Pax6 control regionalization of the pre-neuronogenic cortical primordium. Cereb Cortex. 2002;12:129–39.
7. Mallamaci A, Muzio L, Chan CH, Parnavelas J, Boncinelli E. Area identity shifts in the early cerebral cortex of Emx2-/- mutant mice. Nat Neurosci. 2000;3:679–86.
8. Brancaccio M, Pivetta C, Granzotto M, Filippis C, Mallamaci A. Emx2 and Foxg1 inhibit gliogenesis and promote neuronogenesis. Stem Cells. 2010;28:1206–18.
9. Falcone C, Filippis C, Granzotto M, Mallamaci A. Emx2 expression levels in NSCs modulate astrogenesis rates by regulating EgfR and Fgf9. Glia. 2015;63:412–22.
10. Okamoto J, Hirata T, Chen Z, Zhou H-M, Mikami I, Li H, Yagui-Beltran A, Johansson M, Coussens LM, Clement G, Shi Y, Zhang F, Koizumi K, et al. EMX2 is epigenetically silenced and suppresses growth in human lung cancer. Oncogene. 2010;29:5969–75.
11. Okamoto J, Kratz JR, Hirata T, Mikami I, Raz D, Segal M, Chen Z, Zhou HM, Pham P, Li H, Yagui-Beltran A, Ray MR, Koizumi K, et al. Downregulation of EMX2 is associated with clinical outcomes in lung adenocarcinoma patients. Clin Lung Cancer. 2011;12:237–44.
12. Qiu H, Yan Q, Luo X, Zhang H, Bao W, Wan X. EMX2 is downregulated in endometrial cancer and correlated with tumor progression. Int J Gynecol Pathol. 2013;32:193–8.
13. Giroux Leprieur E, Hirata T, Mo M, Chen Z, Okamoto J, Clement G, Li H, Wislez M, Jablons DM, He B. The homeobox gene EMX2 is a prognostic and predictive marker in malignant pleural mesothelioma. Lung Cancer. 2014;85:465–71.
14. Liu Z, Ren YA, Pangas SA, Adams J, Zhou W, Castrillon DH, Wilhelm D, Richards JS. FOXO1/3 and PTEN Depletion in Granulosa Cells Promotes Ovarian Granulosa Cell Tumor Development. Mol Endocrinol. 2015;29:1006–24.
15. Shirazi HA, Grimm S, Raizer J, Mehta MP. Combined modality approaches in the management of adult glioblastoma. Front Oncol. 2011;1:36.
16. Veliz I, Loo Y, Castillo O, Karachaliou N, Nigro O, Rosell R. Advances and challenges in the molecular biology and treatment of glioblastoma-is there any hope for the future? Ann Transl Med. 2015;3:7.
17. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, et al. The Somatic Genomic Landscape of Glioblastoma. Cell. 2013;155:462–77.
18. Park DM, Jung J, Masjkur J, Makrogkikas S, Ebermann D, Saha S, Rogliano R, Paolillo N, Pacioni S, McKay RD, Poser S, Androutsellis-Theotokis A. Hes3 regulates cell number in cultures from glioblastoma multiforme with stem cell characteristics. Sci Rep. 2013;3:1095.
19. Baek JH, Hatakeyama J, Sakamoto S, Ohtsuka T, Kageyama R. Persistent and high levels of Hes1 expression regulate boundary formation in the developing central nervous system. Development. 2006;133:2467–76.
20. Hartman J, Müller P, Foster JS, Wimalasena J, Gustafsson J-A, Ström A. HES-1 inhibits 17beta-estradiol and heregulin-beta1-mediated upregulation of E2F-1. Oncogene. 2004;23:8826–33.
21. Noda N, Honma S, Ohmiya Y. Hes1 is required for contact inhibition of cell proliferation in 3T3-L1 preadipocytes: Role of Hes1 in preadipocytes. Genes to Cells. 2011;16:704–13.
22. Gratton MO, Torban E, Jasmin SB, Theriault FM, German MS, Stifani S. Hes6 promotes cortical neurogenesis and inhibits Hes1 transcription repression activity by multiple mechanisms. Mol Cell Biol. 2003;23:6922–35.
23. Seuntjens E, Nityanandam A, Miquelajauregui A, Debruyn J, Stryjewska A, Goebbels S, Nave KA, Huylebroeck D, Tarabykin V. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat Neurosci. 2009;12:1373–80.
24. Piccirillo SGM, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–5.
25. Li Q, Wijesekera O, Salas SJ, Wang JY, Zhu M, Aprhys C, Chaichana KL, Chesler DA, Zhang H, Smith CL, Guerrero-Cazares H, Levchenko A, Quinones-Hinojosa A. Mesenchymal stem cells from human fat engineered to secrete BMP4 are nononcogenic, suppress brain cancer, and prolong survival. Clin Cancer Res. 2014;20:2375–87.
26. Nédélec S, Foucher I, Brunet I, Bouillot C, Prochiantz A, Trembleau A. Emx2 homeodomain transcription factor interacts with eukaryotic translation initiation factor 4E (eIF4E) in the axons of olfactory sensory neurons. Proc Natl Acad Sci USA. 2004;101:10815–20.
27. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2015;89:37–53.
28. Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110.
29. Sturm D, Bender S, Jones DTW, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, Iavarone A, Aldape K, Brennan CW et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nature Reviews Cancer. 2014;14:92–107.
30. Kelly JJ, Stechishin O, Chojnacki A, Lun X, Sun B, Senger DL, Forsyth P, Auer RN, Dunn JF, Cairncross JG, Parney IF, Weiss S. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27:1722–33.
31. Seymour T, Nowak A, Kakulas F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Frontiers in Oncology. 2015;5:159.
32. Raciti M, Granzotto M, Duc MD, Fimiani C, Cellot G, Cherubini E, Mallamaci A. Reprogramming fibroblasts to neural-precursor-like cells by structured overexpression of pallial patterning genes. Mol Cell Neurosci. 2013;57:42–53.
33. Puzzolo E, Mallamaci A. Cortico-cerebral histogenesis in the opossum Monodelphis domestica: generation of a hexalaminar neocortex in the absence of a basal proliferative compartment. Neural Dev. 2010;5:8.