
INTRODUCTION
Bladder cancer is a frequent type of cancer world-wide. In most countries, the majority of bladder cancers are urothelial carcinoma (UC) [1]. Cisplatin (CisPt)-based therapeutic regimen are commonly used both in the perioperative (neoadjuvant, adjuvant) setting for muscle-invasive UC as well as in the palliative setting for recurrent or metastatic UC [2]. CisPt enters cells by passive diffusion as well as by help of transporters [3, 4]. Upon replacement of its chloride ligands by water, DNA adducts are formed by SN2 mechanism (nucleophilic substitution) [5]. The vast majority (60–80%) of DNA adducts generated by CisPt are DNA intrastrand crosslinks (GpG and ApG). Only 1–2% of the DNA crosslinks formed by CisPt are DNA interstrand crosslinks [6, 7]. Platinum-induced DNA crosslinks cause a substantial distortion of the DNA double helix, resulting in transcription and replication blockage [8, 9]. In consequence of stalled replication forks, DNA double-strand breaks (DSBs) can arise as secondary lesions [10]. DSBs are potent triggers of cell death [11] and can be repaired by DNA double-strand break repair (homologous recombination (HR) or non-homologous end joining (NHEJ)). The removal of CisPt-induced DNA crosslinks involves nucleotide excision repair (NER), including transcription-coupled NER (TC-NER) [9]. The relevance of NER mechanisms for the tumor cell response to CisPt is highlighted by the fact that the expression of the NER factor ERCC1 predicts the therapeutic efficacy of CisPt in lung tumors [12, 13] and also seems to be of relevance for UC [14]. The efficacy of platinum-based therapy is limited by intrinsic or acquired drug resistance [15]. Factors that contribute to CisPt resistance are manifold and are poorly characterized for UC [16, 17]. Recently, mechanisms affecting resistance to CisPt have been classified according to their site of action as pre-, on-, post- and off-target [17], with drug transport, DNA repair, apoptosis and signal transduction at membranes, respectively, being representatives of these mechanisms.
Upon induction of DNA damage a highly complex cellular stress response program, known as the DNA damage response (DDR), is activated. It tightly controls cell cycle progression by activation of cell cycle checkpoints and fine-tunes mechanisms of DNA repair and cell death [18, 19]. Activation of the DDR is considered as an inducible barrier against early tumorigenesis [20, 21] and, moreover, to precede genomic instability in bladder cancer [22]. In case of error prone repair of DSBs, genomic instability of bladder carcinomas is favoured [23]. DSBs as well as replication- and transcription-blocking DNA lesions are particular efficient activators of the DDR. The PI3-like kinases ATM and ATR play key roles in the regulation of the DDR [24, 25]. These kinases phosphorylate numerous substrates, among others checkpoint kinases (e.g. Chk1, Chk2) and p53, which eventually affect survival or death of the damaged cell. The relevance of DDR mechanisms for the CisPt sensitivity of UC cells and, most importantly, for CisPt resistant variants, is largely unknown.
Urothelial cancer cells segregate into epithelial and mesenchymal subsets [26]. Therefore, we included both RT-112 and J-82 cells, which are described as UC cells of mainly epithelial- and mesenchymal-like phenotype, respectively [27, 28], in our study. To select CisPt resistant variants we took into account that the therapeutic regimen commonly used in CisPt-based anticancer therapy comprises repetitive treatment cycles, where CisPt is administered by infusion, intermitted by treatment free periods. Therefore, parental RT-112 and J-82 UC cells were selected for resistance by multiple pulse-treatments with CisPt followed by extended recovery periods. The aim of the study was to comparatively analyze CisPt resistant UC cell variants (RT-112R and J-82R) with their respective parental cell types regarding (i) DDR capacity, (ii) the expression of putative CisPt resistance factors as suggested by Galluzzi et al. [17] and (iii) the response to a subset of pharmacological modifiers of the DDR and DNA repair, including inhibitors of checkpoint kinases, which are promising novel anticancer compounds acting by selectively increasing replicative stress and cell death in malignant cells [29]. In doing so, we aimed to identify mechanisms that are of relevance for acquired CisPt resistance of UC cells and, moreover, to figure out therapeutic options to overcome their CisPt resistant phenotype.
RESULTS AND DISCUSSION
Characterization of the CisPt response of the urothelial carcinoma cells RT-112 and J-82
In light of the fact that urothelial cancer cells can segregate into epithelial and mesenchymal subsets [26], RT-112 und J-82 cells, which are representative of epithelial- and mesenchymal-like UC cells, respectively [27, 28], were used in the present study. RT-112 cells differ from J-82 regarding morphology (Figure 1A) and a higher mRNA expression of the epithelial marker E-cadherin as well as a lower expression of the mesenchymal marker vimentin (Figure 1B) as expected. Proliferation rate was higher in RT-112 as compared to J-82 cells (Figure 1C). Analyzing the influence of CisPt on cell viability 24–72 h after CisPt pulse-treatment, we observed that RT-112 cells are 2–3-fold more resistant to moderate doses of CisPt than J-82 cells (Figure 1D–1F). This is reflected by IC50/IC80 values of 10.7 μM / 44.3 μM and 3.9 μM / 13.5 μM for RT-112 and J-82, respectively, as determined after a post-incubation period of 72 h by the Alamar blue assay (Figure 1F). This difference in drug sensitivity is not detectable anymore at very high CisPt doses of ≥ 80 μM (Figure 1D–1G). Measuring cell viability via an alternative method, i.e. the Neutral red assay, similar results were obtained (Figure 1G). Based on a recent report of Galluzzi et al. [17], who has classified putative CisPt resistance factors of tumor cells, we assembled a 96 well-based quantitative real-time (qRT) PCR array to comparatively analyze the mRNA expression of these factors in RT-112 and J-82 cells. The results of this analysis revealed large cell type-specific differences in the basal mRNA expression of both pre-, on-, post- as well as off-target factors [17]. In more detail, we observed a significantly stronger mRNA expression of ATP7A, BRCA1, VDAC, Calpain, p53, Caspase 6 and ERBB2 in RT-112 cells as compared to J-82 cells. By contrast, J-82 cells revealed an enhanced expression of MT1A, XAF1, BCL2, DYRK1VB, HMOX1, GPX1 and HSPA1B as compared to RT-112 cells (Figure 2A, 2B). Analysing gene expression 72 h after treatment with the IC50 of CisPt, we found upregulation of GPX1 and XAF1 concommitantly in both RT-112 and J-82 cells (Figure 2C, 2D). Notably, J-82 cells responded to CisPt treatment with the upregulation of various DNA repair-related factors (i.e. BRCA1, BRCA2, MSH2, XRCC3) (Figure 2D). This response was not found in RT-112 cells (Figure 2C). Taken together, the data show that both basal and CisPt-stimulated mRNA expression of factors affecting CisPt sensitivity [17] considerably vary between the two examined UC cell lines, indicating that the basal defence capacity of epithelial- and mesenchymal-like UC cells against CisPt-induced injury might be different. This hypothesis needs future confirmation by analyzing the CisPt response of additional UC cell lines of epithelial or mesenchymal origin both in vitro and in vivo.
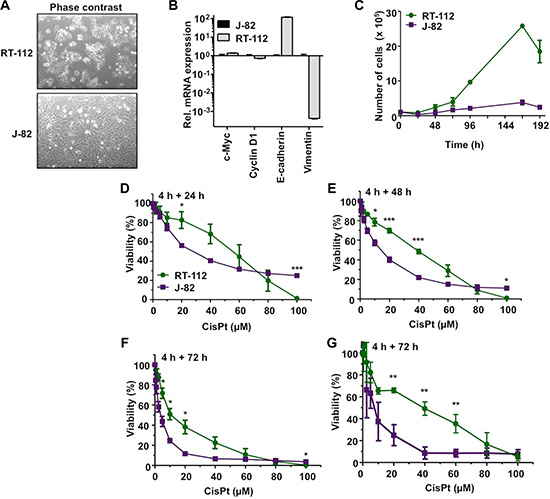
Figure 1: Differential CisPt sensitivity of urothelial carcinoma cells RT-112 and J-82. (A) Different morphology of RT-112 and J-82 cells. (B) Quantitative real-time PCR-based mRNA expression analysis (qRT-PCR) of epithelial (E-cadherin) and mesenchymal (vimentin) markers in J-82 and RT-112 cells. For control, mRNA expression of c-Myc and CyclinD1 was analyzed as well. Relative mRNA expression in J-82 cells was set to 1.0. Data shown are the mean ± SD from one experiment performed in triplicate. (C) Cell growth of RT-112 and J-82 cells was monitored by determining the number of cells over a total period of 8 days. Data shown are the mean ± SD from two to three independent experiments each performed in duplicate. (D–G) Logarithmically growing cells were pulse-treated with different concentrations of cisplatin (CisPt) for 4 h. After post-incubation period of 24 h (D), 48 h (E) or 72 h (F, G) in the absence of CisPt, cell viability was analyzed using the Alamar blue assay (D–F) or the Neutral red assay (G). Data shown are the mean ± SD from three independent experiments, each performed in triplicate. *statistical significance of RT-112 cells vs. J-82 cells. ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05.
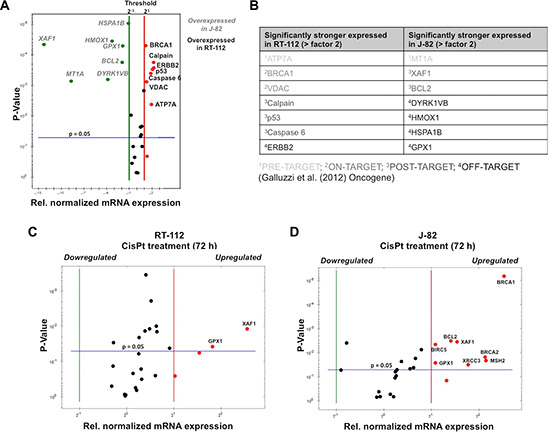
Figure 2: Basal and CisPt-induced mRNA expression of CisPt-related susceptibility factors in UC cells. (A) Basal mRNA expression of CisPt susceptibility factors [17] was analyzed by qRT-PCR analysis. The mean values shown are based on two independent experiments each performed in triplicate. Only differences in mRNA expression of ≤ 0.5 or ≥ 2.0 were considered as biologically relevant. (B) Variations in basal mRNA expression of factors related to CisPt resistance between RT-112 and J-82 cells are classified into mechanisms of pre-, on-, post- and off-target resistance according to Galluzzi et al. [17]. (C, D) mRNA expression of CisPt susceptibility factors was analyzed by qRT-PCR analysis 72 h after treatment with the IC50 of CisPt (according to Figure 1F). The mean values shown are based on a representative experiment performed in triplicate. Only differences in mRNA expression of ≤ 0.5 or ≥ 2.0 were considered as biologically relevant.
Selection of CisPt resistant UC cell variants
In order to elucidate which mechanisms contribute to acquired CisPt resistance of UC cells and having in mind the therapeutic regimen used in the clinic, RT-112 and J-82 cells were repeatedly pulse-treated twice a week (for each 4 h) with the corresponding IC50 of CisPt, followed by a recovery period of one week (Figure 3A). After a total selection time of 10 weeks, CisPt resistant RT-112R und J-82R cells were obtained (Figure 3B–3D). Measuring cell viability by the Alamar blue assay, the resistant variants revealed an about 3-fold increase in the IC50 as compared to the corresponding parental cells (Figure 3B–3D). Similar results were obtained using the Neutral red assay (data not shown). Gain of CisPt resistance was accompanied by morphological alterations, in particular cell enlargement and distinct cell protrusions (Figure 3B–3D). Both RT-112R and J-82R cells showed an increased mRNA expression of the intermediate filament vimentin (Figure 3C–3E) as compared to their respective parental cells. As vimentin expression represents a prototypical marker of mesenchymal cells, we hypothesize that the development of an EMT-like phenotype is favoured in epithelial-like RT-112 cells and is further promoted in J-82 cells during the selection of CisPt resistant UC cell variants. A coherence between EMT and acquired drug resistance was reported by others [26, 30–32]. Flow cytometry-based analyses performed 72 h after CisPt treatment showed a reduction of apoptotic cell death in RT-112R cells as compared to RT-112 (Figure 4A). This effect was only observed in RT-112R cells (Figure 4A, upper panel) but not in J-82R cells (Figure 4A, lower panel). Both RT-112R and J-82R cells were characterized by a more pronounced activation of G2/M checkpoint mechanisms as compared to their corresponding parental counterparts (Figure 4B). The data show that the mechanisms of acquired CisPt resistance differ between individual UC cell lines with protection from CisPt-induced apoptotic mechanisms and alterations in checkpoint control mechanisms being involved.
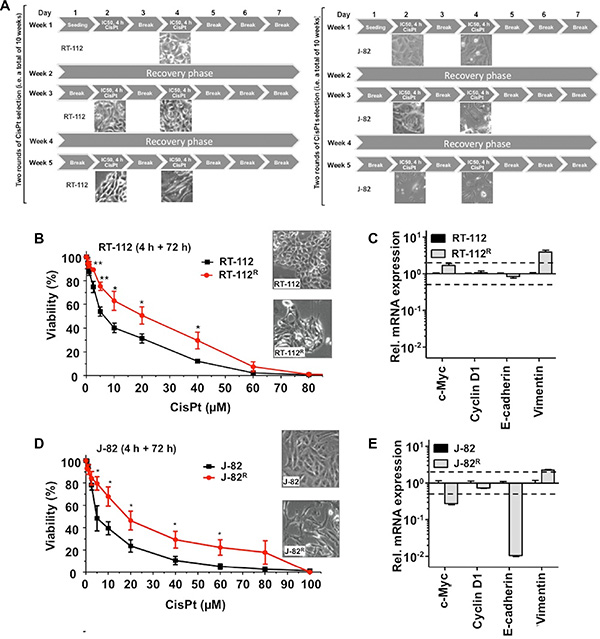
Figure 3: CisPt resistant UC cell variants obtained by long-term selection with CisPt display an intensified mesenchymal phenotype. (A) Schematic representation of the long-term CisPt selection scheme applied to RT-112 and J-82 cells. Cells were pulse-treated with the corresponding IC50 of CisPt (according to Figure 1F) twice a week, followed by a recovery period of one week. This selection scheme was performed over a total time period of 10 weeks (shown are only the first 5 weeks). (B, D) Cell viability of parental RT-112 and CisPt selected RT-112R cells (B) or of parental J-82 and CisPt selected J-82R cells (D) was measured 72 h after a 4 h pulse-treatment with different concentrations of CisPt using the Alamar blue assay. Data shown are the mean ± SD from three independent experiments each performed in quadruplicate. The microscopic pictures illustrate the cell morphology of parental and CisPt resistant cells. *statistical significance of parental cells vs. CisPt resistant cells. **p ≤ 0.01; *p ≤ 0.05. (C, E) Alterations in the mRNA expression of marker genes of epithelial-mesenchymal transition (EMT) in RT-112 versus RT-112R cells (C) or J-82 versus J-82R cells (E). The qRT-PCR based data shown are the mean ± SD from triplicate determinations.E-cadherin is a representative marker of epithelial cells while vimentin is a prototypical marker of mesenchymal cells. For control, mRNA expression of c-Myc and CyclinD1 were also determined.
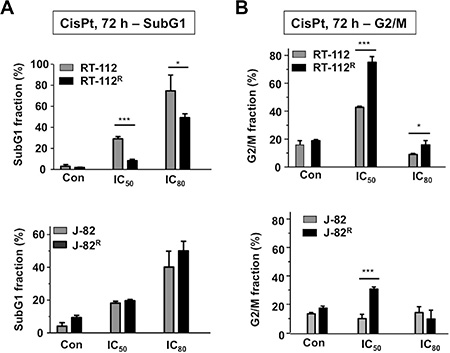
Figure 4: Effects of CisPt on cell cycle distribution of parental and CisPt resistant UC cells. (A, B) Parental (RT-112, J-82) and CisPt resistant (RT-112R, J-82R) UC cells were treated with the IC50 or IC80 of CisPt (according to Figure 1F). After incubation period of 72 h, subG1 fraction (A) and cells present in G2/M phase of the cell cycle (B) were determined by flow cytometry-based analyses. Data shown are the mean ± SD from three independent experiments each performed in duplicate. *statistical significance of parental cells vs. CisPt resistant cells. ***p ≤ 0.001; *p ≤ 0.05.
Induction of DNA damage and activation of the DNA damage response (DDR) in parental and CisPt resistant UC cell variants
In order to measure the induction of DNA damage following CisPt treatment, ATM/ATR-catalyzed S139 phosphorylation of histone H2AX and the recruitment of 53BP1 to sites of damage were monitored by immunocytochemistry (Figure 5A–5B). Moreover, the level of CisPt-induced DNA intrastrand crosslinks was monitored by Southwestern analysis (Figure 5C–5D). The formation of nuclear γH2AX foci and 53BP1 foci is part of the DNA damage response (DDR) and is believed to reflect predominantly the formation of DNA double-strand breaks (DSBs) [19]. Following CisPt treatment, DSBs are believed to be mainly generated as secondary lesions from primary DNA platinum-adducts that stall replication forks [10]. As observed 4 h and 24 h after CisPt pulse-treatment for 4 h, we found a significant reduction in the number of DSBs in J-82R cells, but not in RT-112R cells (Figure 5A–5B). This finding indicates that CisPt resistance of J-82R cells, but not of RT-112R cells, might result from a reduced formation of highly cytotoxic DSBs and/or attenuated DDR following CisPt treatment. Bearing in mind that CisPt-induced DSBs mainly originate from primary Pt-(GpG) DNA adducts, we next monitored the formation of Pt-(GpG) intrastrand crosslinks by Southwestern blot analyses. The data show that DNA intrastrand crosslink formation was significantly lower in the J-82R subline as compared to J-82 parental cells (Figure 5D). Based on these observations we suggest that acquired CisPt resistance of J-82 cells involves a reduced formation of primary (i.e. Pt-(GpG) adducts) and secondary (i.e. DSBs) DNA damage following CisPt treatment. Mechanistically, it is feasible that pre-target resistance mechanisms such as transport or detoxification mechanisms take part [17]. In this context it is noteworthy that the level of CisPt-induced Pt-(GpG) DNA intrastrand crosslinks is higher in parental J-82 cells as compared to RT-112 cells (Figure 5C) if the corresponding IC50 and IC80 were used. This finding indicates that the level of Pt-(GpG) intrastrand crosslinks does not necessarily predicts the level of cytotoxicity.
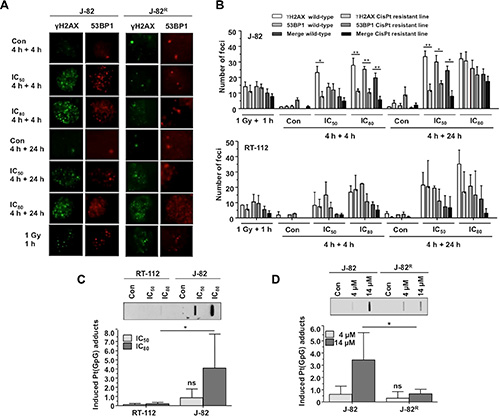
Figure 5: Formation and repair of DNA damage in parental UC cells and CisPt resistant UC variants. (A, B) Parental (RT-112, J-82) and CisPt resistant (RT-112R, J-82R) UC cells were pulse-treated for 4 h with the IC50 or IC80 of CisPt (according to Figure 1F) for 4 h. After a post-incubation period of 4 h or 24 h in the absence of CisPt, the formation of nuclear γH2AX and 53BP1 foci was analyzed by immunocytochemistry. Data shown are the mean ± SD from three independent experiments with each ≥ 50 nuclei being analyzed per experiment. (A) representative microscopic pictures from J-82 and J-82R cells. (B) histograms with quantitative data from J-82/J-82R cells (upper panel) and RT-112/RT-112R cells (lower panel). (C) RT-112 and J-82 cells were pulse-treated for 4 h with the IC50 or IC80 of CisPt (according to Figure 1F). The level of Pt-(GpG) intrastrand crosslinks was determined by Southwestern analyzes using an anti-Pt-(GpG)-specific antibody. Autoradiographies were analyzed densitometrically and the signal intensities of the untreated controls were subtracted. The upper part of the figure shows the result of a representative experiment. In the lower part, mean values ± SD from two independent experiments each performed in triplicate are shown. Con, untreated control. *statistical significance of RT-112 versus J-82. ns, not significant. (D) Parental (J-82) and CisPt resistant cells (J-82R) were pulse-treated for 4 h with the indicated CisPt dose. The level of Pt-(GpG) intrastrand crosslinks was determined by Southwestern analyzes using an anti-Pt-(GpG)-specific antibody. Autoradiographies were analyzed densitometrically and the signal intensities of the untreated controls were subtracted. The upper part of the figure shows the result of a representative experiment. In the lower part, mean values ± SD from two independent experiments each performed in triplicate are shown. Con, untreated control. *statistical significance of J-82 versus J-82R (*p ≤ 0.05). ns, not significant.
In a next step we comparatively analyzed the DDR of UC parental cells and corresponding CisPt resistant variants following CisPt treatment by Western blot analysis. The data obtained uncover large variations in the activation of DDR mechanisms already in J-82 versus RT-112 parental cells, as reflected on the levels of γH2AX, p-Chk1, p-p53 and p-Kap1 (Figure 6A–6B). In general, J-82 revealed a more profound activation of the DDR than RT-112 cells. This is likely related to the higher level of Pt-(GpG) adducts in J-82 cells (see Figure 5C), resulting in stronger activation of DDR mechanisms. Comparative analyses of J-82 cells versus CisPt resistant J-82R cells showed lower phosphorylation levels of H2AX, Chk1, p53 and Kap1 in the CisPt resistant variants (Figure 6A). Again, this is in line with the observed decrease in Pt-(GpG) DNA adducts and DSB formation described in J-82R as compared to J-82 cells (see Figure 5). RT-112R cells revealed a specifically increased phosphorylation of Chk1 as compared to RT-112 parental cells (Figure 6B). This is indicative of a selectively increased potency of RT-112R cells to activate checkpoint control mechanisms that might contribute to protection from CisPt induced apoptotic mechanisms (see Figure 4A).
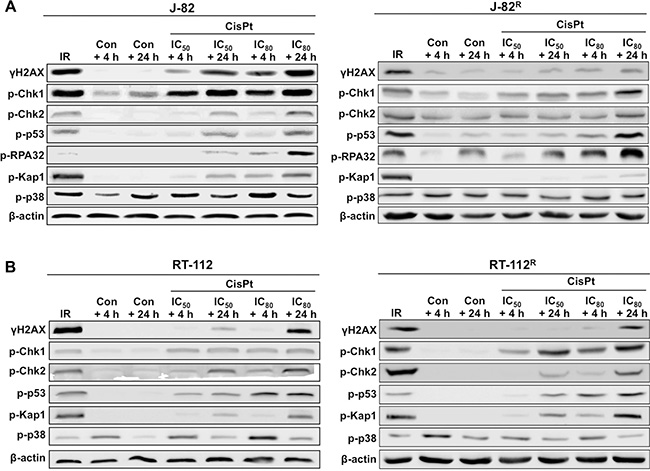
Figure 6: Comparative analyzes of CisPt-induced mechanisms of the DNA damage response (DDR) in parental and CisPt resistant cells. Parental (J-82 (A) and RT-112 (B)) and CisPt resistant (J-82R (A) and RT-112R (B)) cells were treated with the IC50 or IC80 of CisPt (according to Figure 1F) for 4 h. After post-incubation periods of 4 h or 24 h cells were harvested for Western blot analyses using phospho-specific antibodies as indicated. For control, cells were irradiated with 10 Gy (IR) and analysis was performed 1 h later. Data shown are representative of two independent experiments. Expression of β-actin was determined as protein loading control.
Expression of CisPt susceptibility factors in CisPt resistant UC cells
Next, we analyzed the mRNA expression of CisPt-related susceptibility factors reported by Galluzzi et al. [17] in RT-112R and J-82R cells as compared to the corresponding parental cells. Regarding RT-112R cells, we found a significant increase in the mRNA expression of metallothionein (MT1A) and the XIAP-associated factor 1 (XAF1) as compared to RT-112 cells (Figure 7B). In J-82R cells we observed an elevated mRNA expression of the antioxidative factors heme oxygenase 1 (HMOX1) and glutathione S-transerase M1 (GSTM1) as well as of XAF1 as compared to the corresponding controls (Figure 7A). Hence, the two types of CisPt resistant UC cell variants were characterized by an increased mRNA expression of XAF1. In this context we would like to note that selection of CisPt resistant J-82 and RT-112 cells by a selection protocol using continuous treatment with increasing CisPt doses over a time period of 4 month also resulted in increased level of XAF1 mRNA in CisPt resistant J-82 cells but not in RT-112 cells (Supplementary Figure S1). The finding of upregulated XAF1 mRNA expression in drug resistant UC cell variants was unexpected considering that XAF1 is known to inhibit the anti-apoptotic factor XIAP, and hence is anticipated to promote cell death [33]. Correspondingly, high XAF1 level was suggested as predictive marker in pancreatic cancer associated with better overall survival [34]. Therefore, it appears possible that its increased mRNA expression in J-82R cells accidentially correlates with CisPt resistance but is not causative for acquired CisPt resistance of UC cells. Alternatively, XAF1 might have a so far not yet decribed pro-survival function in CisPt resistant UC cells. In this context it is noteworthy that a cell cycle regulatory function has been suggested for XAF1 in gastrointestinal cancer, which rests on its interaction with Chk1 [35]. Interestingly enough induction of XAF1 mRNA expression was also observed in both J-82 and RT-112 parental cells 72 h after CisPt addition (see Figure 2C–2D). So, forthcoming studies are clearly required to dissect the role of XAF1 in the response of UC cells to CisPt. In addition, the data indicate that the improvement of anti-oxidative capacity, as reflected by the upregulation of HMOX1 and GSTM1, and increased expression of metallothionein MT1A might be of particular relevance for acquired CisPt resistance of some subtypes of UC. Bearing in mind that oxidative stress contributes to the cytotoxicity of CisPt [36, 37], upregulation of anti-oxidative mechanisms might be a meaningful cytoprotective strategy of UC cells, as is the upregulation of metallothioneins [38]. Noteworthy, upregulation of the mRNA expression of DNA repair factors (i.e. BRCA1, BRCA2, ERCC1, MLH1, MSH2, XRCC3), which are involved in the repair of CisPt-induced DNA damage, was not observed in the CisPt resistant variants.
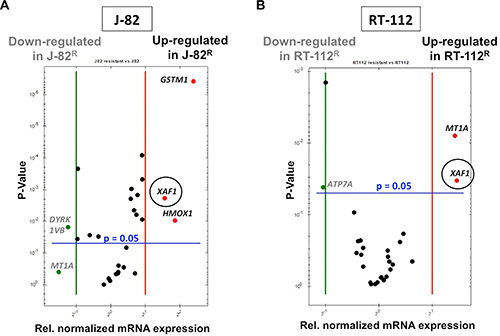
Figure 7: Alterations in gene expression that go along with acquired CisPt resistance of epithelial- and mesenchymal-like UC cells. Alterations in the mRNA expression of selected subset of CisPt-related susceptibility factors [17] was analyzed in drug resistant J-82R (A) and RT-112R cells (B) as compared to the corresponding parental cells by qRT-PCR. Relative mRNA expression in parental J-82 cells was set to 1.0. Only alterations in gene expression of ≤ 0.5 or ≥ 2.0 between wild-type (J-82 and RT-112) and CisPt resistant variants (J-82R and RT-112R) were considered as biologically relevant. Shown are the genes that are either up- or downregulated in CisPt resistant cells as compared to the parental cells.
J-82R cells show enhanced sensitivity to a Chk1 inhibitor
In search of pharmacological approaches to overcome acquired CisPt resistance of J-82R cells, we examined their sensitivity to a selected subset of pharmacological inhibitors. Unfortunately, these analyses could not be performed with RT-112R cells because their CisPt resistant phenotype turned out as not stable and got lost upon freezing. For these analyses inhibitors of the DDR-related kinases ATM/ATR (VE-822) as well as of checkpoint (Chk) kinases (AZD-7762 (Chk1 and Chk2 inhibitor) and LY2603618 (Chk1-specific inhibitor)) and Wee1 kinase (MK-1775) were included. Noteworthy, targeting of ATR/Chk1-regulated replicative stress responses of tumor cells has recently been suggested as a novel therapeutic strategy [29]. As additional candidate inhibitors we analyzed the impact of the cyclin-dependent kinase (CDK) inhibitor roscovitine, the multikinase inhibitor sorafenib, which is frequently used as anticancer drug in the clinic, as well as of inhibitors of the DNA repair proteins RAD51 (RI-1) and PARP-1 (olaparib) on the viability of parental J-82 versus resistant J-82R cells. As a further candidate inhibitor we employed lovastatin, because statins have been shown to exhibit anticancer activity in various preclinical model systems [39] and are discussed to overcome acquired drug resistance to doxorubicin in neuroblastoma cells [40].
J-82R cells turned out to be slightly more sensitive to treatment with the pan Chk inhibitor AZD-7762 (Figure 8A) and showed a significantly enhanced sensitivity to the Chk1-specific inhibitor LY2603618 as compared to parental cells (Figure 8B). The J-82R cells also revealed a tendentially enhanced sensitivity to the Wee1 kinase inhibitor MK-1775 (Figure 8C) but not to the CDK inhibitor roscovitine (Figure 8D). The pronounced loss of cell viability of J-82R cells following Chk1 inhibition seems to be specific as it was not observed upon inhibition of ATM/ATR kinase or the DNA repair factors RAD51 and PARP-1 (Table 1). Pre-treatment of J-82R cells with low non-toxic concentration of Chk inhibitors increased their sensitivity to CisPt (Figure 8E–8F), indicating that targeting of Chk might be particular useful to overcome acquired CisPt resistance of some subtypes of UC cells. Whether targeting of Chk is equally effective in epithelial and mesenchymal-like UC cells remains to be elucidated in forthcoming studies. Noteworthy, Chk inhibition was reported to overcome CisPt resistance of head and neck cancer cells [41] as well as of clear cell carcinoma of the ovary [42] in vitro, supporting the hypothesis that targeting of Chk might be a useful approach to deal with acquired CisPt resistance of different types of tumor cells. Taken together, our data support the current view that increasing replicative stress in tumor cells might be a promising therapeutic strategy also in UC [29]. In fact, the antitumor potency of Chk1 inhibitors is currently investigated in clinical trials. Whereas AZD-7762 revealed inacceptable cardiotoxicity [43], SCH-900776 seems to be better tolerated in humans [44].
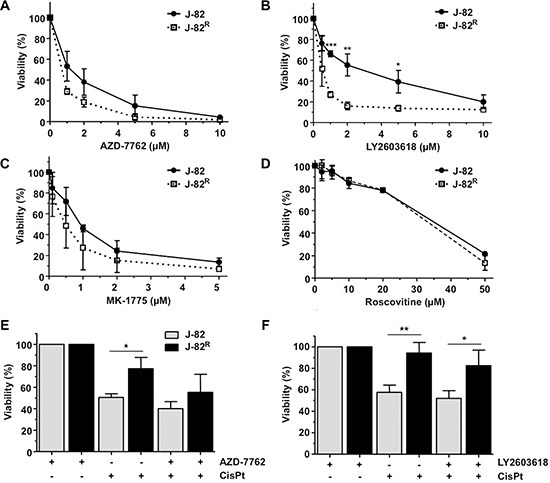
Figure 8: Influence of selected protein kinase inhibitors on the viability of CisPt resistant J-82R cells. (A–D) J-82 cells and CisPt resistant cells (J-82R) were treated with different concentrations of the pan Chk inhibitor AZD-7762 (A), the Chk1-specific inhibitor LY2603618 (B), the Wee1 kinase inhibitor MK-1775 (C) or the cyclin dependent kinase inhibitor roscovitine (D). After an incubation period of 72 h in the presence of the corresponding inhibitor, viability was analyzed using the Alamar blue assay. Data shown are the mean ± SD from three independent experiments, each performed in quadruplicate. *statistical significance of parental cells vs. CisPt resistant cells. ***p ≤ 0.001; **p ≤ 0.01; *p ≤ 0.05. (E–F) J-82 parental and CisPt resistant cells (J-82R) were pre-treated with 0.4 μM of the pan Chk inhibitor AZD-7762 (E) or the Chk1-specific inhibitor LY2603618 (F) for 1 h followed by the addition of CisPt (2 μM). After further incubation period of 72 h in the absence of the Chk inhibitor, cell viability was analyzed using the Alamar blue assay. Relative viability in the corresponding inhibitor-only treated controls was set to 100%. Data shown are the mean ± SD from three independent experiments, each performed in quadruplicate. *statistical significance of parental cells vs. CisPt resistant cells. **p ≤ 0.01; *p ≤ 0.05.
Table 1: Influence of selected pharmacological modulators of the DNA damage response (DDR) and of DNA repair factors on the viability of parental and CisPt resistant J-82 cells
Inhibitor |
Dose |
Cell line |
|
|---|---|---|---|
J-82 |
J-82R |
||
AZD-7762 |
IC50 |
1.2 μM |
0.7 μM |
|
IC80 |
4.4 μM |
1.8 μM |
LY2603618 |
IC50 |
2.82 μM |
0.54 μM |
|
IC80 |
9.85 μM |
1.63 μM |
MK-1775 |
IC50 |
0.92 μM |
0.47 μM |
|
IC80 |
3.1 μM |
1.7 μM |
VE-822 |
IC50 |
~ 10 μM |
> 10 μM |
Roscovitine |
IC50 |
25 μM |
35 μM |
Sorafenib |
IC50 |
9 μM |
> 10 μM |
RI-1 |
IC50 |
150 μM |
140 μM |
Olaparib |
IC50 |
375 μM |
347 μM |
Lovastatin |
IC50 |
26 μM |
> 30 μM |
J-82 cells and the CisPt resistant subline (J-82R) were treated with different concentrations of the pan Chk inhibitor AZD-7762, the Chkl-specific inhibitor LY2603618, the Wee1 kinase inhibitor MK-1775, the ATM/ATR inhibitor VE-822, the cyclin-dependent kinase inhibitor roscovitine, the Raf kinase inhibitor sorafenib, the Rad51 inhibitor RI-1, the PARP-1 inhibitor olaparib or the HMG-CoA reductase inhibitor lovastatin. After an incubation period of 72 h hours, cell viability was analyzed using the Alamar blue assay. Listed are the resulting IC50 and IC80 from 2–3 three independent experiments, each performed in quadruplicate.
Taken together the data show that different molecular mechanims are involved in acquired resistance of different types of UC cells to CisPt. Apparently, molecularly different sets of CisPt defence programs can become activated in individual UC cells. We hypothesize that acquired CisPt resistance in (epithelial-like) RT-112 cells might be preferentially related to protection from pro-apoptotic mechanisms, whereas gain of CisPt resistance in (mesenchymal-like) J-82 UC cells seems to be characterized by a lower level of CisPt formed DNA damage and attenuated DDR. Mechanisms of transport and DNA repair seem to be of minor relevance for aquired CisPt resistance of UC cells. Hence, therapeutic targeting of apoptosis- and/or DDR-related mechanisms are suggested as preferential to overcome acquired CisPt resistance in UC. Importantly, inhibitors of Chk might be useful to handle CisPt resistance in UC cells. Forthcoming in vivo studies are required to scrutinize the potency of Chk1 specific inhibitors to work against the non-responsiveness of urothelial carcinoma cells to CisPt-based anticancer therapy in a clincally relevant setting.
MATERIALS AND METHODS
Materials
RT-112 and J-82 urothelial carcinoma cells originate from the German Collection of Microorganisms and Cell Culture (DSMZ) (Braunschweig, Germany). Cisplatin was obtained from the pharmaceutical department of the University Hospital Düsseldorf and originates from TEVA (Ulm, Germany). The following antibodies were used: antibodies detecting Ser139 phosphorylated histone H2AX (γH2AX), H2AX (Millipore (Billerica, MA, USA)), β-actin (Santa Cruz Biotechnology (Santa Cruz, CA, USA)), 53BP1, p-p53, p-Chk1, p-p38 (Cell Signaling (Denvers, MA, USA)), p-Chk2 (Abcam (Cambridge, UK)), p-RPA32 and p-KAP1 (Bethyl Laboratories (Montgomery, AL, USA)). The antibody detecting GpG intrastrand crosslinks induced by CisPt was generously provided by J. Thomale (Essen, Germany) and has been described before [30]. The fluorescent antibodies Alexa Flour 488 and 546 were obtained from Life Technologies (Carlsbad, CA, USA). Horseradish peroxidase-conjugated secondary antibodies were purchased from Rockland (Gilbertsville, PA, USA). ATM/ATR inhibitor VE-822 (CatNo:S7102) and Wee1 kinase inhibitor MK-1775 (CatNo: S1525) were obtained from Selleckchem (Munich, Germany), lovastatin (CatNo: M2147), cyclin-dependent kinase inhibitor roscovitine (CatNo: R7772) and the pan (i.e. Chk1 and Chk2) checkpoint kinase (Chk) inhibitor AZD-7762 (CatNo: SMLO350) from Sigma Aldrich Life Science (Darmstadt, Germany), Rad51 inhibitor RI-1 (CatNo: 553514) from Calbiochem (San Diego, CA, United States), Chk1-specific inhibitor LY2603618 (CatNo: A8638) and PARP-1 inhibitor olaparib (CatNo: A4154) are from Apexbio (Houston, TX, USA) and the Raf kinase inhibitor sorafenib was obtained from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany) (CatNo: Sc-220125).
Cell culture
RT-112 and J-82 cells were grown in DMEM (Sigma (Steinheim, Germany)) containing 10% of fetal calf serum (FCS) (PAA Labratories (Cölbe, Germany) and 1% penicillin/streptomycin (Sigma (Steinheim, Germany)) at 37°C in an atmosphere containing 5% CO2. If not stated otherwise, treatments of logarithmically growing cells were performed 24 h after seeding.
Determination of cell viability
Cell viability was determined using the Alamar blue assay [45]. In this assay, viable cells are detectable by their ability to effectively metabolize the non-fluorescent dye resazurin (Sigma, Steinheim (Germany)) to fluorescent resorufin. Cells were incubated for 1.5 h with the resazurin solution (final concentration 40 μM) before fluorescence was measured (excitation: 535 nm, emission: 590 nm, 5 flashes, integration time: 20 μs). Relative viability in the untreated controls was set to 100%. In addition, cell viability was also determined by use of the Neutral red assay [46]. In this assay, viable cells accumulate the red dye 2-methyl-3-amino-7-dimethylaminophenazine in lysosomes, whereas dead cells are unable to do so. Thus, the staining intensity is directly proportional to the number of viable cells. For this assay, cells were incubated for 1.5 h with the neutral red solution (Sigma Aldrich (Steinheim, Germany)) (final concentration 0.1 mg/ml) before fixation with 1% formaldehyde. Afterwards the dye was extracted with 50% ethanol and absorption was measured at 550 nm. Relative viability in the untreated controls was set to 100%.
Flow cytometry-based analysis of cell cycle distribution and cell death
Cell cycle distribution was analyzed by flow cytometry. Adherent cells were trypsinized and combined with the medium that contains floating cells. After centrifugation (800 × g, 5 min, RT), cell pellet was washed and resuspended in PBS. Afterwards, the cells were fixed with ice-cold ethanol (–20°C, ≥ 20 min). After centrifugation (800 × g, 5 min, 4°C) the supernatant was discarded. The cells were resuspended in PBS containing RNase A (Serva Electrophoresis GmbH (Heidelberg, Germany)) (1 μg/μl) and incubated for 1 h at RT. After adding of propidium iodide (Sigma (Steinheim, Germany)) cells were subjected to flow cytometric analysis (Becton Dickinson (Heidelberg, Germany)). The SubG1 fraction was considered as a measure of dead (apoptotic) cells.
Analysis of DNA damage induction
The formation of DNA double-strand breaks (DSBs) was investigated by measuring the level of S139 phosphorylated H2AX (γH2AX), which is a surrogate marker of DNA damage [47, 48], by Western blot analysis or by immunocytochemistry-based detection of nuclear γH2AX and 53BP1 foci. For immunocytochemical analysis, the cells were seeded onto coverslips. After treatment the cells were fixed with 4% formaldehyde in phosphate buffered saline (PBS) (MERCK (Darmstadt, Germany)) (15 min, RT) and incubated with ice-cold methanol (over night, –20°C). After blocking (1.5 h, RT; blocking solution: 5% BSA in PBS/0.3% Triton X-100, incubation with γH2AX antibody (mouse) and 53BP1 antibody (rabbit) was performed (1:500, over night, 4°C), followed by further washing with PBS/0.3% Triton X-100 and addition of the secondary fluorescence-labelled antibody (1:500, 1 h, RT, in the dark). After washing, the cells were mounted in Vectashield (Vector Laboratories (Burlingame, CA, USA)) containing DAPI. NuclearγH2AX and 53BP1 foci were counted by microscopical analysis using an Olympus BX43 fluorescence microscope and the number of co-localized γH2AX and 53BP1 foci was calculated.
The level of Pt-(GpG) DNA intrastrand crosslinks was monitored by Southwestern blot analysis. To this end, genomic DNA was isolated using the “DNeasy Blood and Tissue” kit (Qiagen (Hilden, Germany)). The concentration and purity of the DNA was measured photometrically (NanoVue™Plus (GE Healthcare, UK)). 0.5 μg of the DNA was diluted in 100 μl of TE buffer, denatured by heating (10 min, 95°C) and cooled on ice. Afterwards, 100 μl ice-cold ammonium acetate (2 M) was added. A nitrocellulose membrane was soaked in 1 M ammonium acetate and fixed into a slot-blot apparatus (Roth (Karlsruhe, Germany)). The DNA was transferred onto the membrane by use of a vacuum pump. After washing with 1 M ammonium acetate and water, the membrane was incubated with 5 × SSC (10 × SSC: 1.5 M NaCl, 150 mM sodium citrate, pH 7.0) for 5 min and baked for 2 h at 80°C before it was blocked in 5% non-fat milk in TBS/0.1% Tween 20 over night at 4°C. Incubation with the primary antibody directed against Pt-(GpG) intrastrand crosslinks (1:200) [49] was conducted for 1 h at RT, followed by incubation with peroxidase-conjugated anti-rat IgG secondary antibody (1:2000, 2 h, RT). Visualization of the Pt-(GpG) intrastrand crosslinks was done by chemiluminescence and autoradiographies were densitometrically analyzed. Additionally, the membrane was stained with methylene blue (MP Biomedicals (Santa Ana, CA, USA)) to ensure equal DNA loading.
Western blot analysis
The activation status of the DNA damage response (DDR) machinery was investigated by Western blot analysis employing a set of phospho-specific antibodies, which detect prototypical factors that become activated by phosphorylation in the course of the DDR. Total cell extracts were obtained by lysing an equal number of cells in Roti®-Load buffer (Carl Roth GmbH (Karlsruhe, Germany)) (5 min, RT). After sonication (EpiShear™ Probe sonicator, Active Motif (La Hulpe, Belgium)) proteins were denatured by heating (5 min, 95°C) and separated by SDS-PAGE (12.5% gel). Subsequently, proteins were transferred onto a nitrocellulose membrane (GE Healthcare (Little Chalfont, UK)) via the Protean Mini Cell System (BioRad (München, Germany)). After blocking in 5% non-fat milk in TBS/0.1% Tween 20 (MERCK (Darmstadt, Germany)) (2 h, RT), the membrane was incubated with the corresponding primary antibody (1:1000, over night, 4°C). After washing with TBS/0.1% Tween 20 the secondary (peroxidase-conjugated) antibody was added (1:2000, 2 h, RT). For visualization of the bound antibodies the Fusion FX7 imaging system (PeqLab (Erlangen, Germany)) was used.
Quantitative real-time PCR-based mRNA expression analyses
Putative markers of CisPt susceptibility were selected on the basis of a recent review by Galluzzi et al. [17] who has classified putative CisPt resistance factors of tumor cells into mechanisms of pre-, on-, post- and off-target. Based on this report we assembled a 96 well-based quantitative real-time (qRT) PCR array to analyze the mRNA expression of these factors in RT-112 und J-82 cells. In addition, mRNA expression of the epithelial marker E-cadherin as well as the mesenchymal marker vimentin and the proliferation factors c-Myc and cyclinD1 was analyzed by qRT-PCR. Total RNA was purified using the RNeasy Mini Kit (Qiagen (Hilden, Germany)). The reverse transcriptase (RT) reaction was performed by use of the OmniScript Kit (Qiagen) with 2000 ng of mRNA. For each PCR reaction 40 ng of cDNA and 0.25 μM of the corresponding primers (Eurofins MWG Synthesis GmbH (Ebersberg, Germany)) were used. Quantitative real-time PCR analysis was performed in triplicates employing the QPCR-SYBR Green Fluorescein Mix (Thermo Fisher Scientific (Dreieich, Germany)) and a CFX96 Real-Time System (BioRad (Munich, Germany)) with the Bio-Rad CFX Manager 3.1 software. PCR runs (35–40 cycles) were done as follows: 95°C – 10 min; 95°C – 15 s; 60°C – 30 s; 72°C – 40 s; 72°C – 10 min. At the end of the runs, melting curves were analyzed to ensure the specificity of the amplification reaction. mRNA levels of β-actin, GAPDH, PPIA, RPL32 and 18S were taken for normalization. If not stated otherwise, relative mRNA expression of untreated control cells was set to 1.0.
Statistical analysis
For statistical analysis the unpaired two-tailed Student’s t-test was applied using the GraphPad Prism 5.01 software. p-Values ≤ 0.05 were considered as significant and were marked with an asterisk.
HIGHLIGHTS
Expression of CisPt specific resistance factors differs between urothelial carcinoma cells lines
Selection of CisPt resistant UC cell variants promotes an EMT-like phenotype
Aquired CisPt resistance of epithelial-like RT-112 UC cells is related to a lower frequency of apoptosis
CisPt resistant mesenchymal-like J-82 UC cells are characterized by reduced formation of DNA damage and attenuated DDR
Acquired CisPt resistance is reversible by pharmacological inhibition of Chk1.
Abbreviations
53BP1, 53 binding protein 1; ApG, adenine-guanine; ATM, ataxia telangiectasia mutated; ATP7A, copper-extruding P-type ATPase; ATR, ataxia telangiectasia mutated and Rad3-related; BCL2, B-cell/lymphoma 2; BIRC5, Survivin; BRCA, breast cancer; Chk, checkpoint kinase; CisPt, cisplatin; DDR, DNA damage response; DSBs, DNA double-strand breaks; DYRK1VB, dual specificity tyrosine-phosphorylation-regulated kinase 1B; ERBB2, oncogenic EGFR-like receptor; Ercc1, excision repair cross complementing gene 1; EMT, epithelial mesenchymal transition; GpG, guanine-guanine; GPX1, glutathione peroxidase 1; GSTM1, glutathion S-transferase type M1; HMOX1, heme oxygenase type 1; H2AX, histone H2AX; γH2AX, S139 phosphorylated H2AX; HSPA1B, heat shock protein 1B; IR, ionizing radiation; Kap1, KRAB-associated protein 1; MSH2, mutS homolog 2; MT1A, metallothionein 1A; NER, nucleotide excision repair; RT, reverse transcriptase; TC-NER, transcription-coupled NER; PARP-1, poly (ADP-ribose) polymerase 1; RPA32, replication protein A2; UC, urothelial carcinoma; UC, urothelial carcinoma; VDAC, voltage-dependent anion channel; Wee1, nuclear protein tyrosine kinase regulating G2 checkpoint; XAF1, Xiap-associated factor 1; XRCC3, X-ray repair cross-complementing gene 3.
ACKNOWLEDGMENTS
This work was supported by the „Strategischer Forschungsfond (SFF)“ of the Heinrich Heine University Düsseldorf. M. Skowron was supported by a fellowship of the Duesseldorf School of Oncology (funded by the Comprehensive Cancer Centre Düsseldorf/ Deutsche Krebshilfe and the Medical Faculty of the Heinrich-Heine-University Düsseldorf). We thank Lena Schumacher for excellent technical support, Jürgen Thomale for providing the Pt-(GpG) adduct specific antibody and Christian Henninger for optimizing quantitative real-time PCR.
CONFLICTS OF INTEREST
There are no potential conflicts of interest.
REFERENCES
1. Knowles MA. Molecular subtypes of bladder cancer: Jekyll, Hyde or chalk and cheese? Carcinogenesis. 2006; 27:361–373.
2. Stenzl A, Cowan NC, De Santis M, Kuczyk MA, Merseburger AS, Ribal MJ, Sherif A, Witjes JA, European Association of U. [Treatment of muscle-invasive and metastatic bladder cancer: update of the EAU guidelines]. [Article in Spanish]. Actas urologicas espanolas. 2012; 36:449–460.
3. Pabla N, Murphy RF, Liu K, Dong Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am J Physiol Renal Physiol. 2009; 296:F505–511.
4. Ciarimboli G, Ludwig T, Lang D, Pavenstadt H, Koepsell H, Piechota HJ, Haier J, Jaehde U, Zisowsky J, Schlatter E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol. 2005; 167:1477–1484.
5. Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969; 222:385–386.
6. Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature. 1995; 377:649–652.
7. Fichtinger-Schepman AM, van der Veer JL, den Hartog JH, Lohman PH, Reedijk J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry. 1985; 24:707–713.
8. Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Progress in nucleic acid research and molecular biology. 2001; 67:93–130.
9. Ljungman M. The transcription stress response. Cell Cycle. 2007; 6:2252–2257.
10. Olive PL, Banath JP. Kinetics of H2AX phosphorylation after exposure to cisplatin. Cytometry B Clin Cytom. 2009; 76:79–90.
11. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013; 332:237–248.
12. Olaussen KA, Mountzios G, Soria JC. ERCC1 as a risk stratifier in platinum-based chemotherapy for nonsmall-cell lung cancer. Curr Opin Pulm Med. 2007; 13:284–289.
13. Olaussen KA, Dunant A, Fouret P, Brambilla E, Andre F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, Stahel R, Sabatier L, Pignon JP, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med. 2006; 355:983–991.
14. Kim KH, Do IG, Kim HS, Chang MH, Kim HS, Jun HJ, Uhm J, Yi SY, Lim do H, Ji SH, Park MJ, Lee J, Park SH, et al. Excision repair cross-complementation group 1 (ERCC1) expression in advanced urothelial carcinoma patients receiving cisplatin-based chemotherapy. Acta pathologica, microbiologica, et immunologica Scandinavica. 2010; 118:941–948.
15. Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007; 33:9–23.
16. Drayton RM, Catto JW. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev Anticancer Ther. 2012; 12:271–281.
17. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012; 31:1869–1883.
18. Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000; 408:433–439.
19. Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007; 28:739–745.
20. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005; 434:864–870.
21. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078.
22. Schepeler T, Lamy P, Hvidberg V, Laurberg JR, Fristrup N, Reinert T, Bartkova J, Tropia L, Bartek J, Halazonetis TD, Pan CC, Borre M, Dyrskjot L, et al. A high resolution genomic portrait of bladder cancer: correlation between genomic aberrations and the DNA damage response. Oncogene. 2013; 32:3577–3586.
23. Bentley J, Diggle CP, Harnden P, Knowles MA, Kiltie AE. DNA double strand break repair in human bladder cancer is error prone and involves microhomology-associated end-joining. Nucleic Acids Res. 2004; 32:5249–5259.
24. Shiloh Y. ATM, ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001; 11:71–77.
25. Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Current opinion in cell biology. 2001; 13:225–231.
26. McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, Siefker-Radtke A, Dinney C. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009; 28:335–344.
27. Tran MN, Choi W, Wszolek MF, Navai N, Lee IL, Nitti G, Wen S, Flores ER, Siefker-Radtke A, Czerniak B, Dinney C, Barton M, McConkey DJ. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem. 2013; 288:3275–3288.
28. Earl J, Rico D, Carrillo-de-Santa-Pau E, Rodriguez-Santiago B, Mendez-Pertuz M, Auer H, Gomez G, Grossman HB, Pisano DG, Schulz WA, Perez-Jurado LA, Carrato A, Theodorescu D, et al. The UBC-40 Urothelial Bladder Cancer cell line index: a genomic resource for functional studies. BMC Genomics. 2015; 16:403.
29. Dobbelstein M, Sorensen CS. Exploiting replicative stress to treat cancer. Nature reviews Drug discovery. 2015; 14:405–423.
30. Liu Y, Du F, Zhao Q, Jin J, Ma X, Li H. Acquisition of 5-fluorouracil resistance induces epithelial-mesenchymal transitions through the Hedgehog signaling pathway in HCT-8 colon cancer cells. Oncology letters. 2015; 9:2675–2679.
31. Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015; 6:10697–10711. doi: 10.18632/oncotarget.4037.
32. Bourlon MT, Flaig TW. The Epidemiological, Mechanistic and Potential Clinical Role of AR in Urothelial Carcinoma. Curr Drug Targets. 2015.
33. Plenchette S, Cheung HH, Fong WG, LaCasse EC, Korneluk RG. The role of XAF1 in cancer. Current opinion in investigational drugs. 2007; 8:469–476.
34. Huang J, Yao WY, Zhu Q, Tu SP, Yuan F, Wang HF, Zhang YP, Yuan YZ. XAF1 as a prognostic biomarker and therapeutic target in pancreatic cancer. Cancer science. 2010; 101:559–567.
35. Wang J, Gu Q, Li M, Zhang W, Yang M, Zou B, Chan S, Qiao L, Jiang B, Tu S, Ma J, Hung IF, Lan HY, Wong BC. Identification of XAF1 as a novel cell cycle regulator through modulating G(2)/M checkpoint and interaction with checkpoint kinase 1 in gastrointestinal cancer. Carcinogenesis. 2009; 30:1507–1516.
36. Gomez-Sierra T, Molina-Jijon E, Tapia E, Hernandez-Pando R, Garcia-Nino WR, Maldonado PD, Reyes JL, Barrera-Oviedo D, Torres I, Pedraza-Chaverri J. S-allylcysteine prevents cisplatin-induced nephrotoxicity and oxidative stress. The Journal of pharmacy and pharmacology. 2014; 66:1271–1281.
37. Brozovic A, Ambriovic-Ristov A, Osmak M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit Rev Toxicol. 2010; 40:347–359.
38. Siegsmund MJ, Marx C, Seemann O, Schummer B, Steidler A, Toktomambetova L, Kohrmann KU, Rassweiler J, Alken P. Cisplatin-resistant bladder carcinoma cells: enhanced expression of metallothioneins. Urol Res. 1999; 27:157–163.
39. Graaf MR, Richel DJ, van Noorden CJ, Guchelaar HJ. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat Rev. 2004; 30:609–641.
40. Sieczkowski E, Lehner C, Ambros PF, Hohenegger M. Double impact on p-glycoprotein by statins enhances doxorubicin cytotoxicity in human neuroblastoma cells. Int J Cancer. 2010; 126:2025–2035.
41. Gadhikar MA, Sciuto MR, Alves MV, Pickering CR, Osman AA, Neskey DM, Zhao M, Fitzgerald AL, Myers JN, Frederick MJ. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013; 12:1860–1873.
42. Itamochi H, Nishimura M, Oumi N, Kato M, Oishi T, Shimada M, Sato S, Naniwa J, Sato S, Kudoh A, Kigawa J, Harada T. Checkpoint kinase inhibitor AZD7762 overcomes cisplatin resistance in clear cell carcinoma of the ovary. Int J Gynecol Cancer. 2014; 24:61–69.
43. Sausville E, Lorusso P, Carducci M, Carter J, Quinn MF, Malburg L, Azad N, Cosgrove D, Knight R, Barker P, Zabludoff S, Agbo F, Oakes P, et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014; 73:539–549.
44. Karp JE, Thomas BM, Greer JM, Sorge C, Gore SD, Pratz KW, Smith BD, Flatten KS, Peterson K, Schneider P, Mackey K, Freshwater T, Levis MJ, et al. Phase I, pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin Cancer Res. 2012; 18:6723–6731.
45. Liedert B, Pluim D, Schellens J, Thomale J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res. 2006; 34:e47.
46. O‘Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000; 267:5421–5426.
47. Lowik CW, Alblas MJ, van de Ruit M, Papapoulos SE, van der Pluijm G. Quantification of adherent and nonadherent cells cultured in 96-well plates using the supravital stain neutral red. Anal Biochem. 1993; 213:426–433.
48. Olive PL. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004; 75:355–373.
49. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273:5858–5868.