
INTRODUCTION
Hepatitis C virus (HCV) infection presents a serious public health problem worldwide [1]. Studies confirm that majority patients of HCV infection will develop chronic infection, which in turn leads to the occurrence of liver cirrhosis and hepatocellular carcinoma (HCC) [2]. The mechanism accounting for the high rate of persistent infection is that this virus has evolved one or multiple mechanisms to evade the host immunity [3-7]. Thus, the molecular mechanisms by which the virus establishes chronic infection are under intense investigation.
HCV core protein (21kDa), synthesized upon virus infection and replication, takes part in activities of virus life cycle and in assembly of virus particles. It is released from infected liver cells [8] and exhibits multiple functions to affect host immunity [9-13]. Many researchers have found that HCV core protein combining with gC1qR can inhibit the proliferation of T lymphocytes by reducing IL-2 and IL-2Rα gene transcription [14], and impair IL-12 secretion by macrophages through induction of negative regulators such as Tim-3, PD-1 and SOCS-1 [15, 16]. It can also reduce the ability of DC cells to present antigen by down-regulating the MHC-I expression [17].
Liver as the main host for HCV infection is composed of hepatocytes, liver stromal cells, and a variety of immunocytes which include DCs, macrophages, NK cells, NKT, T and B lymphocytes [18]. Macrophages and NK cells in the liver function as the first line of defense against infection similar as those in other organs [19]. Kupffer cells, traditionally denote hepatic resident macrophages, represent up to 15~20% of the total number of liver cells, and 80~90% of the total body macrophage pool [20]. Polarization of macrophages to different subtypes with distinct functions depends on the tissue microenvironment and external stimuli [21]. The previous studies in our team demonstrated that the phosphorylation of transcription factor STAT3 and the expression of membrane CD206 are up-regulated in macrophages co-cultured with HCV core protein and liver cells, which increases macrophage tendency to become M2 type macrophages [22]. M2 macrophages have been implicated in inhibiting inflammatory responses and immuno-suppression [23-26], resulting in adverse effects on HCV elimination. In other words, in the settings of persistent HCV infection, macrophage activities transformed in liver pathological microenvironment may be responsible for developing chronic HCV diseases. Macrophages can devour pathogens, meanwhile release large amounts of cytokines that impact on the strength and types of adaptive immune responses [27]. In addition, activated macrophages can also produce multiple negative regulators [28, 29]. The negative regulators contribute to inhibit further activation of inflammatory signaling pathways, thereby, protect the host from excessive immune responses [30, 31].
In this study, the aims are to explore the negative regulator A20 expression in macrophages induced by HCV core protein and the receptors and pathways involved in A20 induction. A20 (tumor necrosis factor α inducible protein 3, TNFAIP3), which is known as a powerful suppressor in cytokine signaling, can inhibit the activity of NF-κB and NF-κB-mediated inflammatory responses [32-34]. The results indicated that the interaction of HCV core protein with gC1qR could upregulate the expression of A20, which down-regulated the secretion of IL-6, IL-1β and TGF-β1 in macrophages. gC1qR, which is the receptor of the globular heads of complement C1q, is involved in the immune response to the inflammation and microbial infection [35, 36]. HCV core protein/gC1qR engagement on macrophages triggered the activation of MAPK, NF-κB and PI3K/AKT signaling pathways, among which P38, JNK and NF-κB pathways activated the A20 expression. This study reveals that HCV core protein causes a low-grade chronic infection mediated by macrophages which promotes the establishment of chronic HCV infection.
RESULTS
HCV core protein induces the expression of negative regulator A20 in macrophages
HCV core protein was successfully expressed and purified, and proved to be a single band by SDS-PAGE and Western blot (Figure 1A). The protein contained 3 EU/mL of endotoxin and was quantified as 0.5 mg/mL by Bradford method. Human monocytes (THP-1) derived macrophages (MΦ-THP-1) were generated by PMA treatment for 48 hours. To examine the expression of A20, MΦ-THP-1 cells were treated with different amounts of HCV core protein for different times. The results showed that the expression of A20 in macrophages was dose-dependently upregulated by HCV core protein (Figure 1B). Next, chose 10μg/mL core protein to treat macrophages (MΦ-THP-1), the expression of A20 began to increase at 30 minutes, and reached the peak at 10 hours (Figure 1C), while in mouse BMDM (bone marrow derived macrophage), it was induced much faster as early as 0.25 hour and peaked at 0.5 hours by core protein (Figure 1D). Meanwhile, His peptide (10μg/mL), a tag used for purification of the HCV core protein, did not induce A20 expression (Figure 1E).
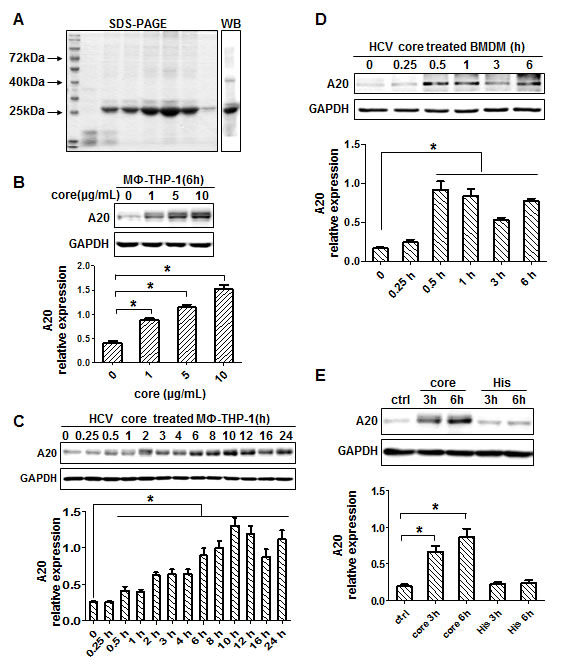
Figure 1: HCV core protein induces A20 expression. A. HCV core protein was analyzed by SDS-PAGE and Western blot. B. A20 was upregulated in MΦ-THP-1 cells treated with different doses of core protein (1, 5, 10μg/mL). MΦ-THP-1 cells C. and BMDM D. were treated with core protein (10μg/mL) for different times, and cell lysates were analyzed by immunoblotting to examine the A20 expression. E. MΦ-THP-1 cells were treated with core protein and His peptide for 3 and 6 hours, and cell lysates were analyzed by immunoblotting to examine the A20 expression.
A20 inhibits the production of IL-6, IL-1β and TGF-β1 in macrophages
To determine the relationship between A20 and cytokine expression, this study used shRNA to knockdown A20 and generated A20 knockdown cells (MΦ-THP-1-sh-A20) and control cells (MΦ-THP-1-sh-luciferase). A20 was markedly suppressed in MΦ-THP-1-sh-A20 cells treated with HCV core protein (p < 0.05) (Figure 2A), indicating a successful knockdown of A20 by shRNA. Interestingly, the levels of IL-6, IL-1β and TGF-β1 in the supernatants of MΦ-THP-1-sh-A20 cells treated with HCV core protein were much higher than those in control cells at different time points (p < 0.05) (Figure 2B, 2C, 2D), demonstrating that IL-6, IL-1β and TGF-β1 were down-regulated by A20. The level of IL-8 was not changed in the supernatants of the two cell lines treated with core protein after 8h, 12h, 24h, however, after 4h, IL-8 in the supernatants of MΦ-THP-1-sh-A20 cells was lower than that in control cells (Figure 2E). The level of TNF was not changed in the supernatants of the two cell lines treated with core protein at different time points (Figure 2F). The result indicates that IL-8 and TNF are not down-regulated by A20.
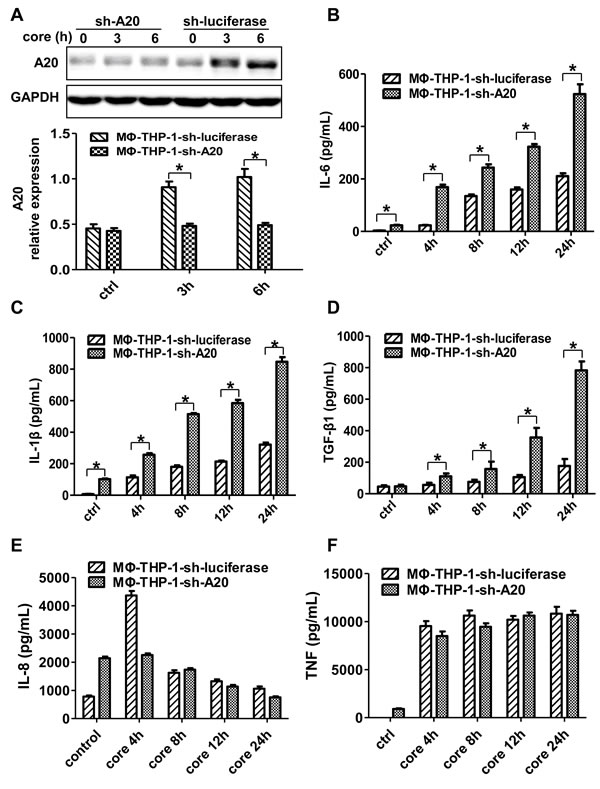
Figure 2: A20 negatively regulates the IL-6, IL-1β and TGF-β1 production. A. MΦ-THP-1 cells expressing A20-specific shRNA or luciferase-specific shRNA were treated with core protein for 3 and 6 hours, cell lysates were analyzed by immunoblotting to examine the A20 expression. The secretion of IL-6 B., IL-1β C., TGF-β1 D., IL-8 E. and TNF F. in cell culture supernatants of MΦ-THP-1 cells expressing A20-specific shRNA or luciferase-specific shRNA treated with core protein (10μg/mL) in different times were analyzed by CBA.
HCV core protein interacting with gC1qR induces A20 expression
To determine whether core protein physically interacts with membrane receptors in MΦ-THP-1, the pull-down experiments were performed. As shown in Figure 3A, HCV core protein could interact with gC1qR but not TLR2, TLR3 and TLR4. The expression of gC1qR was upregulated in macrophages treated by HCV core protein (Figure 3B). Subsequently, knocked-down gC1qR by shRNA and established gC1qR knockdown cell line (THP-1-sh-gC1qR), the gC1qR expression in MΦ-THP-1-sh-gC1qR cells was decreased than that in control cells (MΦ-THP-1-sh-luciferase) (p < 0.05) (Figure 3C). A20 was also decreased in THP-1-sh-gC1qR cells treated with HCV core protein than that in control cells (p < 0.05) (Figure 3D). Meanwhile, A20 expression was increased in macrophages treated by C1q (a gC1qR ligand) (p < 0.05) (Figure 3E). These data indicate that HCV core protein induces A20 expression by interacting with gC1qR.
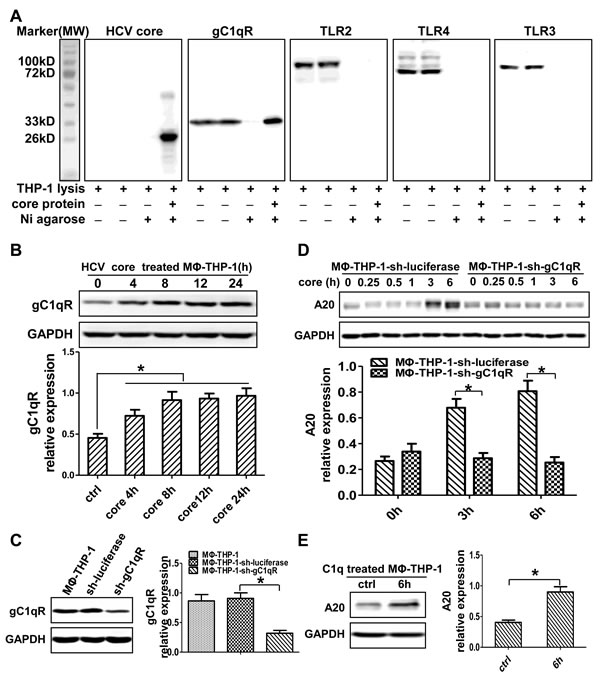
Figure 3: HCV core protein interacting with gC1qR induces A20 expression. A. MΦ-THP-1 cell lysates were incubated with purified core protein coupling Ni agarose, the bound proteins were identified by immunoblotting with indicated antibodies. B. MΦ-THP-1 cells were treated with core protein (10μg/mL) for 4, 8, 12 and 24 hours, cell lysates were analyzed by immunoblotting to examine the gC1qR expression. C. Cell lysates of MΦ-THP-1 and MΦ-THP-1 cells expressing gC1qR-specific shRNA or luciferase-specific shRNA were analyzed by immunoblotting to detect the gC1qR expression. D. MΦ-THP-1 cells expressing gC1qR-specific shRNA or luciferase-specific shRNA were treated with core protein for indicated times. Cell lysates were analyzed by immunoblotting to examine the A20 expression. E. Experiment was carried out to test A20 expression in MΦ-THP-1 cells treated with C1q (70μg/mL) for 6 hours.
JNK and P38 MAPK signaling pathways play pivotal roles in core protein/gC1qR-mediated A20 induction
To identify the effect of MAPK pathways on A20 induction, whether HCV core protein could trigger the activation of MAPK pathways is questioned firstly. As shown in Figure 4A and 4B, P38, JNK and ERK were activated in MΦ-THP-1 cells and BMDM treated with HCV core protein (p < 0.05). Then, THP-1-sh-gC1qR cells were treated with HCV core protein, the expression of p-P38 and p-JNK were significantly lower than control cells (MΦ-THP-1-sh-luciferase) (p < 0.05) (Figure 4C). Furthermore, C1q, the ligand of gC1qR, was used to treat MΦ-THP-1 cells, and found that C1q could also activate P38, JNK and ERK pathways (Figure 4D). The results indicated that HCV core protein acted through gC1qR to activate P38, JNK, and ERK signaling pathways. Next, MΦ-THP-1 cells were pretreated with specific inhibitors for P38 (SB203580), JNK (SP600125), ERK (PD98059), respectively, prior to treatment with core protein, and followed by measuring A20. As shown in Figure 4E, A20 was significantly inhibited after blocking P38 and JNK pathways, but not the ERK pathway. These results suggest that P38 and JNK pathways mediate A20 expression induced by HCV core protein in macrophages.
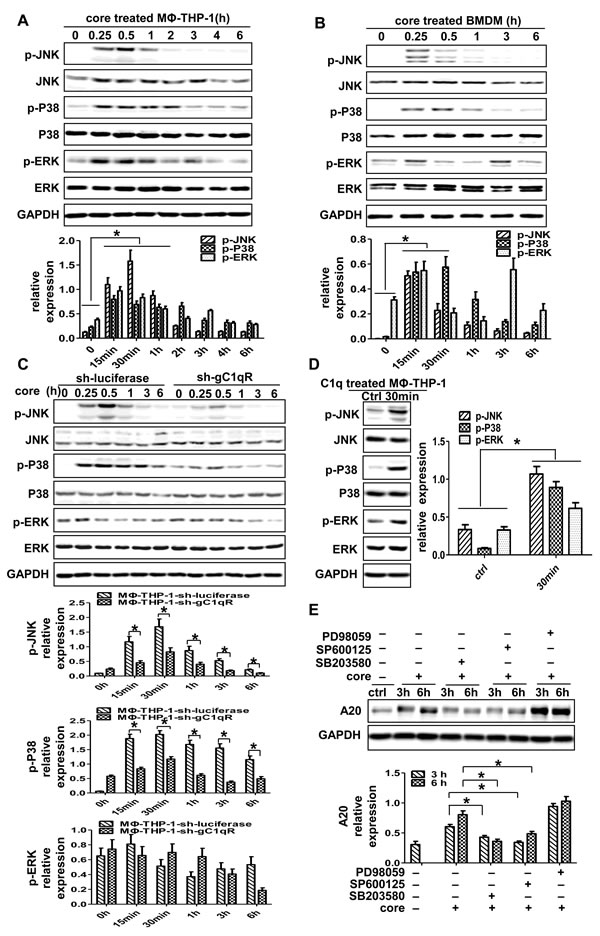
Figure 4: P38 and JNK signaling pathways play pivotal roles in the induction of A20. MΦ-THP-1 cells A., BMDM B. and MΦ-THP-1 cells expressing gC1qR-specific shRNA or luciferase-specific shRNA C. were treated with core protein (10μg/mL) in different times, cell lysates were analyzed by immunoblotting with indicated antibodies. D. MΦ-THP-1 cells were treated with C1q (70μg/mL) for 30 minutes, cell lysates were analyzed by immunoblotting with indicated antibodies. E. MΦ-THP-1 cells were pretreated with SB203580 (30μM), SP600125 (20μM) or PD98059 (50μM) for 30 minutes respectively, then incubated with core protein for 3 and 6 hours, cell lysates were analyzed by immunoblotting to examine the A20 expression.
NF-κB signaling pathway involves in HCV core protein-mediated A20 induction
To figure out whether NF-kB involves in A20 induction, we detected the NF-kB p65 and p105 in MΦ-THP-1 and BMDM treated with HCV core protein. As shown in Figure 5A and 5B, p-NF-κB p65 and p-NF-κB p105 were upregulated in macrophages stimulated by core protein (p < 0.05). Furthermore, the expression of p-NF-κB p65 and p105 were measured in THP-1-sh-gC1qR cells treated with HCV core protein, the results showed significantly lower levels of p-p65 and p-p105 in gC1qR knockdown cells than control cells (Figure 5C). In contrast, unlike core protein, C1q failed to activate NF-κB pathway in MΦ-THP-1 (Figure 5D). Next, MΦ-THP-1 cells were pretreated with the inhibitor for IKKβ (IMD 0354) to block NF-κB pathway prior to treatment with core protein, and found that the expression of A20 was significantly inhibited (p < 0.05) (Figure 5E). These results indicate that NF-κB pathway is activated by HCV core protein and participated in A20 induction.
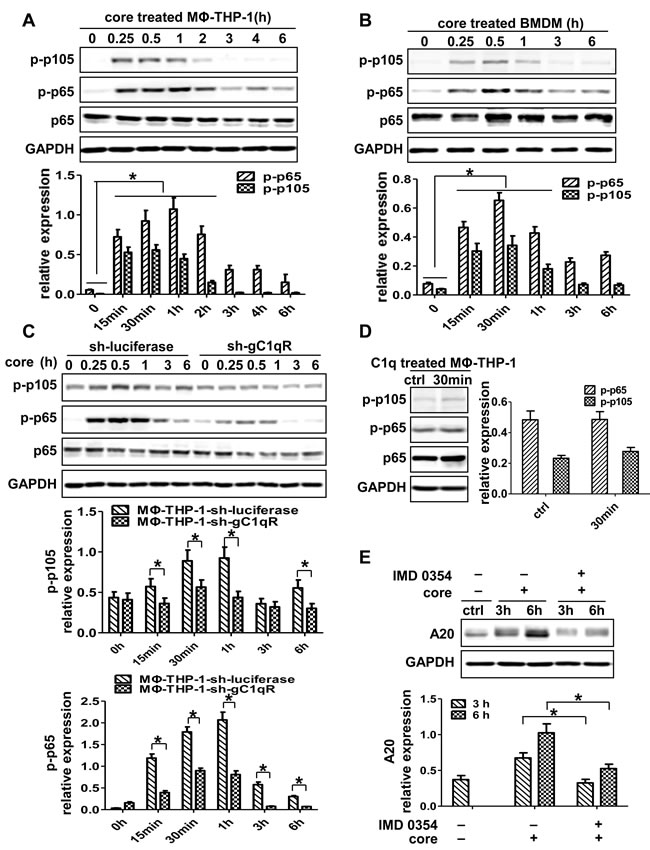
Figure 5: NF-κB signaling pathway plays critical role in the induction of A20. MΦ-THP-1 cells A., BMDM B. and MΦ-THP-1 cells expressing gC1qR-specific shRNA or luciferase-specific shRNA C. were treated with core protein (10μg/mL) in different times, cell lysates were analyzed by immunoblotting with indicated antibodies. D. MΦ-THP-1 cells were treated with C1q (70μg/mL) for 30 minutes, cell lysates were analyzed by immunoblotting with indicated antibodies. E. MΦ-THP-1 cells were pretreated with IMD 0354 (3μM) for 30 minutes, then incubated with core protein for 3 and 6 hours, cell lysates were analyzed by immunoblotting to examine the A20 expression.
PI3K/AKT pathway is not directly involved in A20 induction by HCV core protein in macrophages
To define the role of PI3K/AKT pathway in core protein-mediated A20 induction, the activity of p-AKT was measured. As shown in Figure 6A and 6B, p-AKT was increased in MΦ-THP-1 and BMDM upon core protein treatment (p < 0.05). The expression patterns of p-AKT were different between two cells, in that it reached peak at 30 min in MΦ-THP-1 cells, whereas its expression in BMDMs had two peaks with the first peak at 15 min and the second peak at 6 h, suggesting a secondary effect on p-AKT expression at the late time point in BMDMs. HCV core protein-induced p-AKT expression was abolished in the THP-1 cells knocking-down gC1qR (Figure 6C), while C1q induced p-AKT expression in THP-1 cells by interacting with gC1qR (Figure 6D). To determine whether PI3K/AKT pathway mediates A20 induction, the PI3K and AKT pathways were blocked with Wortmaninn and MK 2206 2HCl, respectively, prior to core protein treatment, and found that the expression of A20 did no decrease after blocking PI3K/AKT pathway (Figure 6E). These results indicate that though the PI3K/AKT pathway can be activated by core protein through binding to the gC1qR on macrophages, it is not involved in core protein-induced A20 expression.
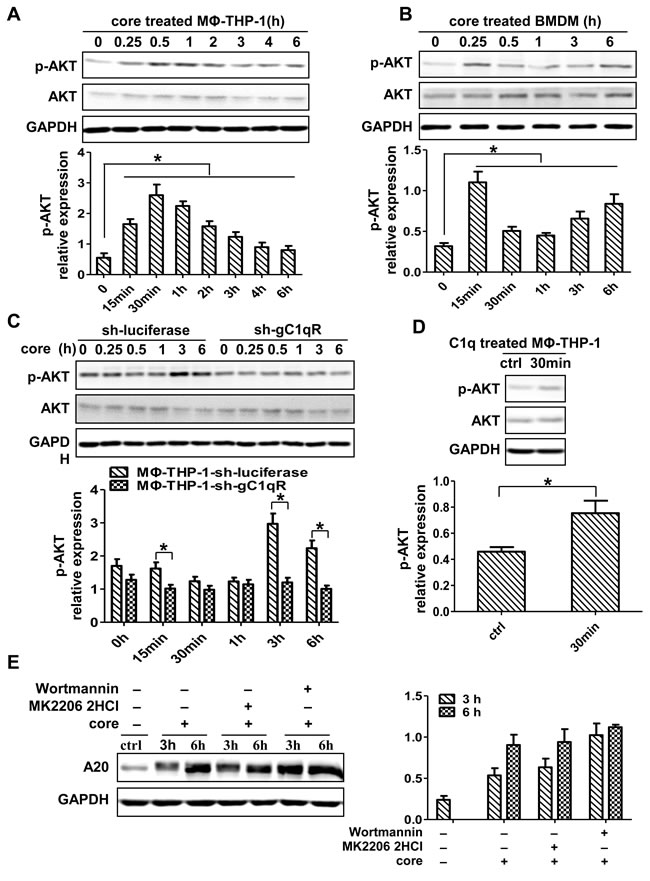
Figure 6: PI3K/AKT signaling pathway has no direct effect on the induction of A20. MΦ-THP-1 cells A., BMDM B. and MΦ-THP-1 cells expressing gC1qR-specific shRNA or luciferase-specific shRNA C. were treated with core protein (10μg/mL) in different times, cell lysates were analyzed by immunoblotting with indicated antibodies. D. MΦ-THP-1 cells were treated with C1q (70μg/mL) for 30 minutes, cell lysates were analyzed by immunoblotting with indicated antibodies. E. MΦ-THP-1 cells were pretreated with MK 2206 2HCl (5μM) or Wortmannin (200nM) for 30 minutes respectively, then incubated with core protein for 3 and 6 hours, cell lysates were analyzed by immunoblotting to examine the A20 expression.
DISCUSSION
A20, an important negative regulator for cytokine expression, one of molecules induced by TNF, was discovered in human umbilical vein endothelial cells [37]. Studies reported that A20 could protect cells from TNF-α-mediated cytotoxic effect and apoptosis [38, 39], and could also inhibit NF-κB-mediated inflammatory responses [32, 33]. This study mainly focused on the negative regulatory mechanisms induced by HCV infection in macrophages. It was showed that exogenous core protein induced the negative regulator A20 expression in macrophages. In line with our observation, Jia et al recently reported that the HCV core protein enhanced A20 mRNA expression, which contributed to inefficient M1 macrophage polarization during Hepatitis C virus infection [40]. Another report from Ma et al showed that A20 played an important role in negative regulation of mDC functions during chronic viral infection [41]. Our current study demonstrates that A20 induction in macrophages has differential effects on cytokine expression, namely inhibits IL-1β, IL-6 and TGF-b1 but not IL-8 and TNF. This differential effect leads to dysregulation of cytokine balance, which may result in a state of low grade inflammatory responses and HCV chronic infection.
To identify the mechanisms of A20 expression, the receptors and signaling pathways induced by HCV core protein were analyzed. It had been reported that HCV core protein could be a ligand combining with gC1qR to regulate the host immune responses [16, 42, 43]. In addition, Chung et al suggested that HCV core protein could induce TLR tolerance in macrophages through interacting with TLR2 [44]. However, our study showed that HCV core protein could interact with gC1qR but not TLR2, TLR3 and TLR4. In addition, the study demonstrated that exogenous HCV core protein upregulated the expression of gC1qR in macrophages, which might increase the susceptibility to HCV core protein mediated immune dysregulation. It should be noted that this study only tested the interaction between core protein and membrane receptors such as gC1qR, TLR2, TLR3 and TLR4, whether HCV core protein interacts with other membrane receptors and proteins needs to be further studied.
Furthermore, this study confirmed that interaction between HCV core protein and gC1qR triggered the activation of MAPK, NF-κB and PI3K/AKT signaling pathways in macrophages, among which P38, JNK and NF-κB pathways played major roles in the induction of A20. Meanwhile, C1q, a ligand of gC1qR, activated the MAPK and PI3K/AKT signaling pathways but not NF-κB signaling in macrophages. These results indicate that exogenous HCV core protein is mainly binding to the gC1qR, however, we could not exclude other receptors and proteins in macrophages that might be involved in A20 induction by core protein. Interestingly, gC1qR knockdown abrogated the expression of A20 induced by core protein, moreover, the expression of A20 was also induced by gC1qR ligand C1q, which further confirmed that exogenous HCV core protein interacting with gC1qR induced A20 expression.
In summary, we reveal that HCV core/gC1qR engagement triggers the activation of P38, JNK and NF-κB pathways and induces high level of A20 expression. A20 down-regulates many cytokines in macrophages, resulting in low-grade inflammation in liver that may cause a constant state of damage and repair to liver cells and hepatic stellate cells, which eventually leads to liver fibrosis and cancer [45]. Therefore, exploring the mechanisms of HCV core protein-induced A20 expression in macrophages will be favorable in understanding the HCV pathogenesis and provides a novel insight for the mechanisms of HCV chronic infection.
MATERIALS AND METHODS
Antibodies and reagents
The following antibodies and reagents were used in this study: anti-phospho-JNK (Cell Signaling 4668); anti-phospho-P38 (Cell Signaling 4511); anti-phospho-ERK (Cell Signaling 4370); anti-phospho-NF-κBp65 (Cell Signaling 3033); anti-phospho-NF-κBp105 (Cell Signaling 3035); anti-phospho-AKT (Ser473; Cell Signaling 4060); anti-JNK (bioworld BS3630); anti-P38 (bioworld BS3567); anti-ERK (bioworld BS1112); anti-NF-κBp65 (Cell Signaling 4764); anti-AKT (bioworld MB0052); anti-TLR2(Cell Signaling 12276); anti-TLR3 (GeneTex GTX113022); anti-TLR4 (GeneTex GTX125909); anti-A20/TNFAIP3 (Cell Signaling 5630); anti-gC1qR/p33 (Millipore MAB1161); Human C1q were obtained from Sigma (C1740); Inhibitors specific for P38 (SB203580), JNK (SP600125), ERK (PD98059), IKKβ (IMD 0354), PI3K (Wortmaninn) and AKT (MK 2206 2HCl) were obtained from selleckchem. CBA Kit was obtained from BD. Ni-NTA Agarose were obtained from QIAGEN. His peptide was obtained from Chinese Peptide Company.
Cell lines and cell culture
The human monocytic leukemia cell line THP-1 (resource center of Peking Union Medical College Hospital, China) and HEK 293T cells (Shanghai stem cell bank) were cultured in RPMI 1640 (Gibco) containing penicillin-streptomycin (100mg/mL for each drug, Solarbio), 10mM HEPES (Solarbio), 0.05mM 2-mercaptoethanol, and 10% fetal bovine serum (Gibco) at 37oC with 5% CO2 in a humidified atmosphere. Before each test, monocytes (THP-1) were differentiated into macrophages (MΦ-THP-1) after treated with phorbol 12-myristate 13-acetate (PMA, 15ng/mL, multiscience) for 48h. Mouse BMDMs were isolated and cultured using standard protocols [46, 47]. Bone marrow cells from BALB/c mice were collected by flushing the femurs from six-to eight-week-old mice with phosphate buffered saline (PBS) and then cultured in RPMI 1640 (Gibco) containing 10% fetal bovine serum (BI) and macrophage-colony-stimulating factor (M-CSF) (Peprotech) at 37oC with 5% CO2 for 7 days. After 7 days of culturing, non-adherent cells were eliminated. Adherent cells, which were highly enriched in BMDM, were recovered for studies. All the animal procedures were vetted and approved by the Hebei Medical University Ethical Committee for Animal Experimentation.
Expression and purification of HCV core protein and pull-down assay
HCV core protein was expressed and purified as previously described [22]. This fused protein was analyzed by SDS-PAGE and Western blot with antibody (mouse anti HCV core monoclonal antibody, Santa Cruz sc-57800). HCV core protein was treated with Endotoxin Removal Kit to remove endotoxin, and then detected endotoxin in Limulus reagents assay. It was quantified by Bradford method before used in cell experiments. Pull-down assay was conducted by incubating cell lysates with purified core protein coupling Ni agarose at 4°C overnight. Agarose was washed and examined by Western blot.
ShRNA knockdown of gC1qR or A20 and generation of cell lines
Short hairpin RNA (shRNA) expressing stable gC1qR, A20 and luciferase transformant were generated by infecting the cells with lentiviral expressing specific shRNA in pSIF1-H1-copGFP vector (System Biosciences). The sequences used in the shRNA targeting gC1qR, A20, and luciferase (the control shRNA) were as follows: 5′-GCTGAGAGTGACATCTTCTCT-3′, 5′-GGATCTGCAGTACTTGCTTCA-3′, and 5′-CTTACGCTGAGTACTTCGA-3′. Western blot was performed to determine the knockdown efficiency.
Quantification of cytokine production by cytometric bead assay
Cytometric Bead Assay (CBA) was used to detect the levels of cytokines (TNF, IL-8, IL-6, IL-1β and TGF-β1) in cell culture supernatants of MΦ-THP-1-sh-gC1qR and MΦ-THP-1-sh-luciferase cells treated with HCV core protein (10μg/mL) for indicated times. The cells treated with PBS alone served as control groups. Data were acquired using a FACS calibur flow cytometer. The results were based on standard concentration curves and expressed as pg/mL.
Western blot assay
MΦ-THP-1 cells were stimulated with HCV core protein (10μg/mL) for indicated times, and were lysed by Protein Extraction Reagent supplemented with PMSF and phosphatase inhibitors. Protein concentrations of the whole cell lysates were measured by Nanodrop. Samples were separated by 12% SDS-PAGE, and transferred onto the PVDF membrane, and then blotted with indicated antibodies. Immunoblot signals were quantified by densitometry using image J. The relative expression of phospho-JNK, phospho-P38, phospho-ERK, phospho-NF-κB p65, phospho-NF-κB p105 and phospho-AKT, were calculated as: the intensity of phospho-protein/the intensity of identical total protein. The relative expression of A20 was calculated as: intensity of A20/the intensity of GAPDH.
Statistical analysis
The experiments described in this study were performed in triplicate. Results are expressed as mean ± standard deviation. Differences between variables were tested for significance using the Student’s t test. Statistical analyses for multiple comparisons were conducted using the one-way analysis of variance (ANOVA) function. If data were not normally distributed, before applying ANOVA they were transformed in rank. All analyses mentioned above were fulfilled using the SPSS version 16.0 software program. A value of p < 0.05 was considered significant.
Conflicts of interest
The authors declare that they have no conflict of interest.
Grant Support
This work was supported by grants from the National Science Foundation of China (81500143), the Science and Technology Research Projects of the Colleges and Universities of Hebei Province (QN2016025, QN2015016) and the Science and Technology Planning Project of Hebei Province (162777104D).
REFERENCES
1. Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014; 61:S45-57.
2. Lavanchy D. The global burden of hepatitis C. Liver Int. 2009; 29 Suppl 1:74-81.
3. Qian F, Bolen CR, Jing C, Wang X, Zheng W, Zhao H, Fikrig E, Bruce RD, Kleinstein SH, Montgomery RR. Impaired toll-like receptor 3-mediated immune responses from macrophages of patients chronically infected with hepatitis C virus. Clin Vaccine Immunol. 2013; 20:146-55.
4. Granato M, Lacconi V, Peddis M, Di Renzo L, Valia S, Rivanera D, Antonelli G, Frati L, Faggioni A, Cirone M. Hepatitis C virus present in the sera of infected patients interferes with the autophagic process of monocytes impairing their in-vitro differentiation into dendritic cells. Biochim Biophys Acta. 2014; 1843:1348-55.
5. Varchetta S, Mele D, Mantovani S, Oliviero B, Cremonesi E, Ludovisi S, Michelone G, Alessiani M, Rosati R, Montorsi M, Mondelli MU. Impaired intrahepatic natural killer cell cytotoxic function in chronic hepatitis C virus infection. Hepatology. 2012; 56:841-9.
6. Park SHRehermann B. Immune Responses to HCV and Other Hepatitis Viruses. Immunity. 2014; 40:13-24.
7. Tian ZG. Outflanking HCV. Nature Immunology. 2014; 15:6-8.
8. Bouvier-Alias M, Patel K, Dahari H, Beaucourt S, Larderie P, Blatt L, Hezode C, Picchio G, Dhumeaux D, Neumann AU, McHutchison JG, Pawlotsky JM. Clinical utility of total HCV core antigen quantification: a new indirect marker of HCV replication. Hepatology. 2002; 36:211-8.
9. Tu Z, Hamalainen-Laanaya HK, Nishitani C, Kuroki Y, Crispe IN, Orloff MS. HCV core and NS3 proteins manipulate human blood-derived dendritic cell development and promote Th 17 differentiation. Int Immunol. 2012; 24:97-106.
10. Zhai N, Chi X, Li T, Song H, Li H, Jin X, Crispe IN, Su L, Niu J, Tu Z. Hepatitis C virus core protein triggers expansion and activation of CD4(+)CD25(+) regulatory T cells in chronic hepatitis C patients. Cell Mol Immunol. 2015; 12:743-9.
11. Yao ZQ, Prayther D, Trabue C, Dong ZP, Moorman J. Differential regulation of SOCS-1 signalling in B and T lymphocytes by hepatitis C virus core protein. Immunology. 2008; 125:197-207.
12. Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem. 2011; 286:10847-55.
13. Feng HS, Xie Y, Ping Y, Pu C, Ping ZL. HCV core protein-induced down-regulation of microRNA-152 promoted aberrant proliferation by targeting Wnt1 in HepG2 cells. Hepatology. 2013; 58:402a-403a.
14. Yao ZQ, Nguyen DT, Hiotellis AI, Hahn YS. Hepatitis C virus core protein inhibits human T lymphocyte responses by a complement-dependent regulatory pathway. J Immunol. 2001; 167:5264-72.
15. Zhang Y, Ma CJ, Ni L, Zhang CL, Wu XY, Kumaraguru U, Li CF, Moorman JP, Yao ZQ. Cross-Talk between Programmed Death-1 and Suppressor of Cytokine Signaling-1 in Inhibition of IL-12 Production by Monocytes/Macrophages in Hepatitis C Virus Infection. Journal of Immunology. 2011; 186:3093-3103.
16. Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Jia ZS, Moorman JP, Yao ZQ. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS One. 2011; 6:e19664.
17. O’Beirne J, Mitchell J, Farzaneh F, Harrison PM. Inhibition of major histocompatibility complex Class I antigen presentation by hepatitis C virus core protein in myeloid dendritic cells. Virology. 2009; 389:1-7.
18. Wang HChen L. Tumor microenviroment and hepatocellular carcinoma metastasis. J Gastroenterol Hepatol. 2013; 28 Suppl 1:43-8.
19. Bang BRSaito T. Hepatic macrophage regulation of Hepatitis C virus infection. Cytokine. 2015; 76:111-111.
20. Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol. 2012; 3:56.
21. Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE. Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood. 2009; 113:3139-46.
22. Yao Z, Song X, Cao S, Liang W, Lu W, Yang L, Zhang Z, Wei L. Role of the exogenous HCV core protein in the interaction of human hepatocyte proliferation and macrophage sub-populations. PLoS One. 2014; 9:e108278.
23. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPAR gamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metabolism. 2007; 6:137-143.
24. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res. 2012; 53:11-24.
25. Gordon SMartinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010; 32:593-604.
26. Sica AMantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012; 122:787-95.
27. Dornand J, Gross A, Lafont V, Liautard J, Oliaro J, Liautard JP. The innate immune response against Brucella in humans. Vet Microbiol. 2002; 90:383-94.
28. Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012; 33:449-58.
29. Xiong YMedvedev AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. 2011; 90:1141-8.
30. Murphy MB, Xiong Y, Pattabiraman G, Manavalan TT, Qiu F, Medvedev AE. Pellino-3 promotes endotoxin tolerance and acts as a negative regulator of TLR2 and TLR4 signaling. J Leukoc Biol. 2015; 98:963-74.
31. Blok DC, Kager LM, Hoogendijk AJ, Lede IO, Rahman W, Afroz R, Bresser P, van der Zee JS, Ghose A, Visser CE, de Jong MD, Zahed AS, Husain MA, et al. Expression of inhibitory regulators of innate immunity in patients with active tuberculosis. BMC Infect Dis. 2015; 15:98.
32. Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol. 2000; 60:1143-51.
33. Beyaert R. Regulation of inflammatory signaling by A20. Immunology. 2014; 143:38-38.
34. Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappa B Signaling by A20 Through Disruption of Ubiquitin Enzyme Complexes. Science. 2010; 327:1135-1139.
35. Watthanasurorot A, Jiravanichpaisal P, Soderhall K, Soderhall I. Retraction of: A calreticulin/gC1qR complex prevents cells from dying: a conserved mechanism from arthropods to humans. J Mol Cell Biol. 2015; 7:588.
36. Svensson D, Wilk L, Morgelin M, Herwald H, Nilsson BO. LL-37-induced host cell cytotoxicity depends on cellular expression of the globular C1q receptor (p33). Biochem J. 2016; 473:87-98.
37. Opipari A, Boguski M, Dixit V. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. Journal of Biological Chemistry. 1990; 265:14705-14708.
38. Opipari A, Hu HM, Yabkowitz R, Dixit V. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. Journal of Biological Chemistry. 1992; 267:12424-12427.
39. Yamaguchi N. The seventh zinc finger motif of A20 is required for the suppression of TNF-alpha-induced apoptosis. FEBS Lett. 2015; 589:1369-75.
40. Fan C, Zhang Y, Zhou Y, Li B, He Y, Guo Y, Jia Z. Up-regulation of A20/ABIN1 contributes to inefficient M1 macrophage polarization during Hepatitis C virus infection. Virol J. 2015; 12:147.
41. Ma L, Zhou Y, Zhang Y, Li Y, Guo Y, He Y, Wang J, Lian J, Hao C, Moorman JP, Yao ZQ, Jia Z. Role of A20 in interferon-alpha-mediated functional restoration of myeloid dendritic cells in patients with chronic hepatitis C. Immunology. 2014; 143:670-8.
42. Zhang Y, Ma CJ, Ni L, Zhang CL, Wu XY, Kumaraguru U, Li CF, Moorman JP, Yao ZQ. Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. The Journal of Immunology. 2011; 186:3093-3103.
43. Eisen-Vandervelde AL, Waggoner SN, Yao ZQ, Cale EM, Hahn CS, Hahn YS. Hepatitis C virus core selectively suppresses interleukin-12 synthesis in human macrophages by interfering with AP-1 activation. Journal of Biological Chemistry. 2004; 279:43479-43486.
44. Chung H, Watanabe T, Kudo M, Chiba T. Hepatitis C virus core protein induces homotolerance and cross-tolerance to Toll-like receptor ligands by activation of Toll-like receptor 2. J Infect Dis. 2010; 202:853-61.
45. Mitchell JK, Lemon SM, McGivern DR. How do persistent infections with hepatitis C virus cause liver cancer? Current Opinion in Virology. 2015; 14:101-108.
46. Park KRBryers JD. Effect of macrophage classical (M1) activation on implant-adherent macrophage interactions with Staphylococcus epidermidis: A murine in vitro model system. J Biomed Mater Res A. 2012; 100:2045-53.
47. Cabrera-Fuentes HA, Lopez ML, McCurdy S, Fischer S, Meiler S, Baumer Y, Galuska SP, Preissner KT, Boisvert WA. Regulation of monocyte/macrophage polarisation by extracellular RNA. Thromb Haemost. 2015; 113:473-81.