
INTRODUCTION
The concept of linking an antibody to a toxin to create a safe and effective agent against cancer cells is not a new one. The magic bullet concept of Paul Ehrlich is over 100 years old,[1] while the first credible experiments linking chemotherapeutic agents to antibodies were performed almost 55 years ago).[2] At this point, despite all the years of research, there have only been four antibody drug conjugates (ADCs) approved by the FDA (Figure 1).[3-6] Brentuximab vedotin (SGN-35; Adcetris™) and Trastuzumab emtansine (T-DM1; Kadcycla™) were recently approved. Both revolutionized treatment for their respective indication (relapsed Hodgkin’s lymphoma (HL)/systemic anaplastic large cell lymphoma (sALC), and Her2-positive breast cancer), while denileukin difitox (Ontak™) and, gemtuzumab ozogamicin (GO; Mylotarg™) have seen limited clinical use. In fact, GO was recently taken off the market in the United States.
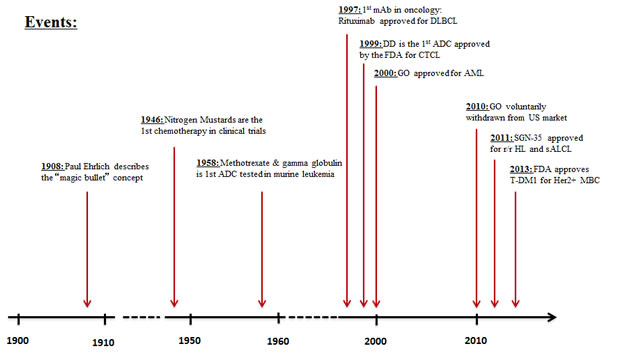
Figure 1: Timeline delineating the evolution of antibody-drug conjugate discovery and therapy in cancer therapeutics [3-6]. ADC: Antibody-drug conjugate, mAb: Monoclonal antibody, DLBCL: Diffuse large B cell lymphoma, DD: Denileukin Difitox, CTCL: Cutaneous T cell lymphoma, GO: Gentuzumab Ozogamicin, AML: Acute myeloid leukemia, r/r: relapsed and/or refractory, HL: Hodgkin’s lymphoma, sALCL: Systemic anaplastic large-cell lymphoma, FDA: Food and Drug Administration, Her2: Human Epidermal Growth Factor Receptor 2, MBC: Metastatic breast cancer.
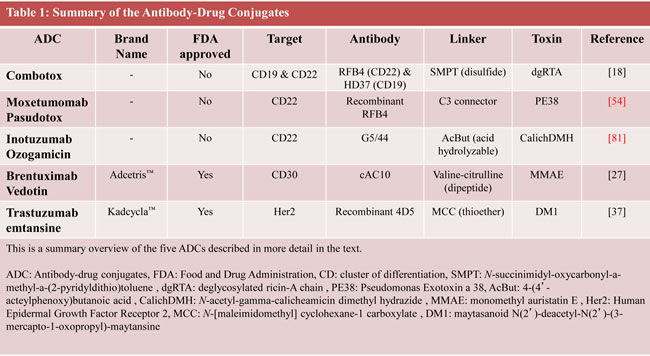
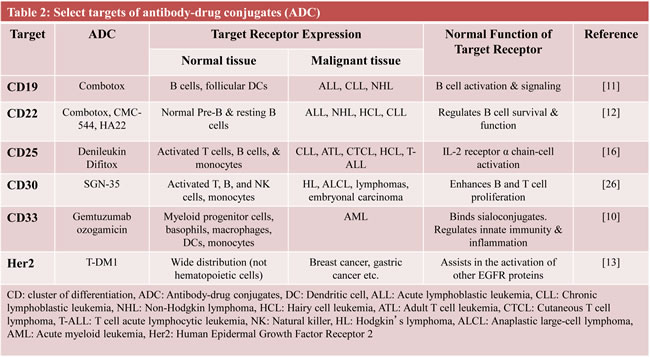
In general, ADCs comprise three components: they are made up of a monoclonal antibody (mAb) conjugated to a toxin via a chemical linker. The mAb allows targeted delivery of a potent cell toxin to specific malignant cells, thereby maximizing drug delivery while limiting bystander effects of traditional cytotoxic agents (Figure 2). In the last few years, it has become apparent that determining the perfect linker may be just as important as the other components of an ADC in increasing efficacy and decreasing toxicity.[7-9] This review describes in detail the development of five unique ADCs, each using a different combination of linker technology and toxin. These examples demonstrate that various components of an ADC are equally important in determining its efficacy and relative safety (Table 1).[10-13]
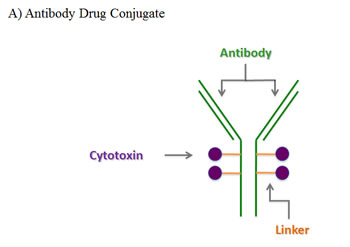
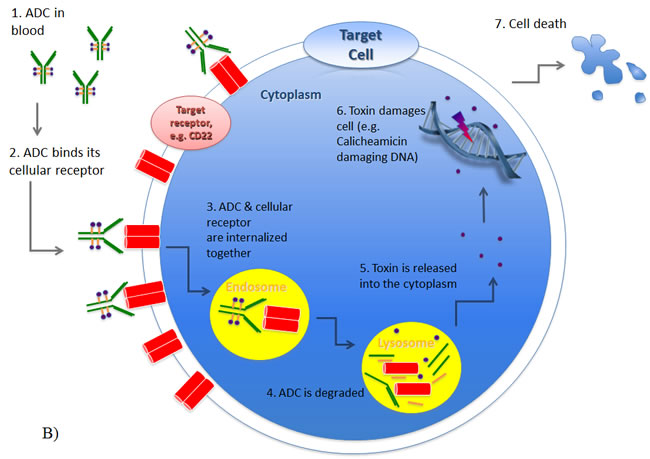
Figure 2: Composition and mode of action of antibody-drug-conjugates (ADC). A) An ADC is composed of a monoclonal antibody directed against a specific epitope on a target cell. A cytotoxic compound is attached to the antibody via a linker. B) Once administered, the antibody component of the ADC binds to the targeted cell receptor, which enables the ADC to be internalized (usually via endocytosis) and subsequently degraded. The released toxin causes cell death via various mechanisms depending on the toxin, such as DNA damage or inhibiting protein translation.
DESIGN AND EFFICACY OF ANTIBODY DRUG CONJUGATES
Combotox is a mixture of ricin-coupled antibodies against CD19 and CD22 and is effective in acute lymphoblastic leukemia
The immunotoxin (IT) Combotox is the 1:1 mixture of two murine monoclonal IgG1 antibodies, both individually linked to a ricin based toxin. It consists of RFB4, a CD22 antibody, and HD37, a CD19 antibody. Both antibodies are coupled to the toxin deglycosylated ricin-A chain (dgRTA), via N-succinimidyl-oxycarbonyl-a-methyl-a-(2-pyridyldithio)toluene (SMPT), a heterobifunctional, thiol-containing cross-linker. The toxin, dgRTA, is the isolated A-chain of the ricin toxic protein, which mediates its activity by disrupting the 60s ribosomal subunit, thereby inhibiting protein translation. It was later deglycosylated to reduce its hepatotoxicity.[14] CD22 is a B-cell specific antigen involved in regulating B-cell survival and function. It is an attractive target, in that it is expressed on the majority of B-cell cancers, but not by stem cell precursors (Table 2).[15] CD19 is also a B-cell specific antigen involved in cell signaling. It is expressed in many B-cell malignancies and is downregulated in plasma cells and hematopoetic stem cells, making it a similarly appealing target.[16] It is interesting to note that while there are generally more CD19-positive cancerous B-cells than CD22-positive cells, RFB4-dgRTA is the more potent IT. This could perhaps be explained by the far greater rate of CD22 internalization after RFB4 binding, compared to that of CD19 after HD37 binding.[17]
Combotox was first demonstrated to be effective in a Daudi-Lymphoma (CD19+/CD22+) SCID mouse model, where the use of both antibodies showed a synergistic killing effect, promoting survival to the equivalent killing of ≥5 logs of tumor cells, more than 1 log greater than RFB4-dgRTA alone, and at least 3 logs greater than HD37-dgRTA alone.[18] These results led to a 22 patient Phase I clinical trial for patients with advanced or refractory Non-Hodgkin’s B-Cell Lymphoma (NHL). The results were modestly encouraging, with 9% of patients achieving a partial response (PR) and 23% a minor response (MR). The main side effects from the drug were complications caused by a Vascular Leak Syndrome (VLS). The symptoms of VLS include edema, pulmonary congestion, dyspnea, weight gain, anemia, hypoalbuminemia, hypotension, and in extreme cases multi-organ failure and death. Severe hemolytic uremic syndrome (HUS) was also observed. While three patients died during the trial, only two of these deaths could be directly attributed to Combotox. Additionally, 30-35% of patients developed human anti-mouse and/or anti-RTA antibodies (HAMA and/or HARA). Interestingly, toxic side effects were inversely correlated with the presence of circulating tumor cells (CTCs). Patients with ≥50 CTCs/mm3 tolerated maximum dosing without major toxicities, while those with <50 CTCs/mm3 experienced more serious adverse events. Patients with a history of autologous or allogeneic bone marrow transplant (BMT), or radiation therapy also had a higher mortality.[19]
Since relapsed or refractory precursor-B acute lymphoblastic leukemia (pre-B ALL) usually has circulating blasts cells, Combotox was next tested in this malignancy. Combotox demonstrated its efficacy in in vitro experiments with pre-B ALL lymphoblasts taken from pediatric patients,[20] as well as in vivo early and late disease pre-B ALL murine models.[21] In a Phase I pediatric clinical trial for refractory pre-B ALL (n=17), Combotox treatment resulted in a complete remission (CR) for 18% of patients and hematological improvement (HI) in 35% of patients. The main dose-limiting toxicity (DLT) was Graft Versus Host Disease (GVHD) in two patients with a history of prior stem cell transplantation (SCT). The most common adverse events were mouth sores, rashes, and hyperbilirubinemia. A case of pancreatitis and anaphylaxis each was also reported. Two patients died during the trial, and both deaths were attributed to a high leukemic burden. The maximum tolerated dose (MTD) was determined as 5 mg/m2 per dose for up to three doses. The rate of HAMA/HARA immunogenicity was 18%, about half of that from the NHL adult trial.[22]
These encouraging results informed a subsequent Phase I trial of single agent Combotox in adults with B-lineage ALL. In this trial (n=17), treatment with Combotox led to specific reductions in peripheral leukemic blasts in most patients: even though only a 13% (n=2) PR rate was observed, 5 patients (31%) experienced a hematological response resulting in an overall response rate (ORR) of 31%. The MTD of Combotox was three doses of 7mg/m2 every other day. The DLT in this trial was VLS. Other serious side effects at grade 3 or higher included elevated liver function tests. HAMA/HARA immunogenicity was 6%, the lowest heretofore reported rate in any Combotox clinical trial.[23] These trials demonstrated that Combotox has specific activity against B-lineage ALL blasts, but single agent treatment may not be sufficient in this rapidly progressive disease. Subsequent in vitro studies were conducted to test its efficacy in combination with chemotherapy in a NOD mouse model of advanced ALL. This murine xenograft experiment revealed that sequential administration of Combotox with cytarabine (Ara-C) is superior to concurrent administration and improved survival over single agent therapy. These results informed the design of a Phase I clinical trial for adults with relapsed or refractory ALL that is presently accruing patients (clinicaltrials.gov NCT01408160).[24]
Brentuximab Vedotin is an immunotoxin against CD30 that is approved for use in Hodgkin’s lymphoma and systemic anaplastic large cell lymphoma
SGN-35 (cAC10-vcMMAE; Seattle Genetics) is the most recent ADC to be approved by the FDA.[25] Going by either the generic name Brentuximab Vedotin, or the trade name, Adcetris™, SGN-35 consists of the chimeric monoclonal IgG1 cAC10 (SGN-30) antibody, which is specific for human CD30, conjugated to a toxin, monomethyl auristatin E (MMAE).[26] CD30, part of the tumor necrosis factor (TNF) receptor family, is an ideal target for antibody-based therapy, as it is highly expressed on HL and sALCL, while restricted to the immune system, specifically to activated lymphocytes (Table 2). MMAE is a synthetic analog of dolastatin 10, a highly potent natural antimitotic agent that inhibits tubulin polymerization. The third component is the stable cathepsin B cleavable valine-citrulline dipeptide linker, with a p-aminobenzylcarbamate spacer put in between the linker and toxin. In vitro cytotoxicity assays on the CD30+ Karpas-299 ALCL and L540cy HL cell lines showed that SGN-35 was both highly potent and antigen-specific. It induced G2/M phase growth arrest quickly followed by apoptosis. Additionally, the drug was shown to be highly stable in human plasma (due to the unique linker), as less than 2% of the drug was released after a 10-day incubation. In vivo experiments in xenograft SCID mouse models of sALCL and HL, and a disseminated sALCL SCID mouse model, all demonstrated dose dependent tumor regression and a high tolerance for the drug.[27] Further in vitro studies showed that the most effective form of SGN-35 would optimally include four molecules of MMAE conjugated to each antibody.[28] The linker in SGN-35 was examined in vivo and was determined to be far more stable than previously used disulfide and hydrazone linkers, with a half-life of almost ten days.[29] The method of MMAE entry into the cell was elucidated via in vitro studies, which showed that both the mAb and MMAE were internalized into the cell via clathrin-dependent mechanisms, while the drug was released via cathepsin B and other lysosomal cysteine proteases.[30] Finally, in an in vivo L540cy HL SCID mouse model, SGN-35 was combined with either ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) or gemcitabine as a combination therapy, and showed synergistic anti-tumor activity.[31]
These results encouraged clinical trials testing the efficacy of SGN-35 in different CD30-positive malignancies. In a Phase I clinical trial with 45 patients with either refractory HL (n=42), sALCL (n=2), or angioimmunoblastic T-cell Lymphoma (n=1), 17 patients had a objective response (OR=CR + PR; 38%), with 11 patients having a CR (24%), while 19 patients (42%) had stable disease (SD). Among patients treated at the MTD (1.8mg/kg), the OR rate was 50%. Additionally, tumor regression was noted in 86% of evaluable patients (36/42), while 81% of patients with disease symptoms at baseline (13/16) experienced symptom resolution. Serious side effects ≥grade 3 included one case each of thrombocytopenia, prostatitis, febrile neutropenia, hyperglycemia, acute renal failure, and presumed sepsis leading to death. The more common grade 1/2 side effects included fatigue, pyrexia, diarrhea, and neutropenia. Peripheral sensory neuropathy (PSN) and associated adverse events were observed in 16 patients (36%).[32]
Since then, two Phase II clinical trials have been reported. One hundred and two patients with refractory HL post autologous SCT were given 1.8 mg/kg SGN-35 every 3 weeks. The ORR was 75% and the CR rate was 34%, while tumor reduction was seen in 94% of patients. Thirty-one of 102 patients were alive and free of documented progressive disease after a median observation time of 18.5 months. Serious side effects ≥grade 3 included PSN, neutropenia, thrombocytopenia, and anemia. Other common adverse events included nausea, fatigue, diarrhea, pyrexia, vomiting, arthralgia, pruritus, myalgia, and peripheral motor neuropathy (PMN).[33] Similarly, 58 patients with refractory sALCL were given 1.8 mg/kg SGN-35 every 3 weeks. The ORR was 86%, with a 57% CR rate while tumor reductions were seen in 97% of patients. In the 17 patients with B-symptoms, 82% achieved resolution in their symptoms. Serious side effects ≥grade 3 were similarly PSN, neutropenia, thrombocytopenia, anemia, fatigue, and pain in the extremities. Interestingly, 4 patients had tumor flares.[34] In an attempt to reduce toxicity and increase potency, 44 patients diagnosed with refractory HL (n=38), sALCL (n=5), or peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) were enrolled in a weekly dosing Phase I clinical trial. SGN-35 elicited a response in 24 patients (59%), CR in 14 patients (34%), and tumor reduction in 93% of evaluable patients (41/44). Of the 7 patients with B-symptoms at baseline, 6 had symptom resolution. Again, serious side effects ≥grade 3 included PSN, anemia, and neutropenia, but also PMN, hyperglycemia, diarrhea, vomiting, hypokalemia, and -magnesemia. Overall, this study showed that SGN-35 might be more effective at a weekly dosing, but also had increased adverse events, especially related to peripheral neuropathy.[35]
Given its impressive activity, the use of SGN-35 is currently being explored in several clinical trials in the upfront setting combined with cytotoxic agents, as single agent and in combination with other drugs for relapsed refractory CD30+ malignancies, as well as maintenance therapy post-induction chemotherapy or following SCT. Preliminary results have been very promising, especially in the upfront treatment of classical HL when combined with doxorubicin, vinblastine and dacarbazine (“A+AVD”), where CR rates approached 95% in a Phase II trial (Ansell SM et al. ASH 2012), and for frontline treatment for patients with CD30+ mature T- and NK-cell lymphomas as “A+CHP” (SGN-35 plus cyclophosphamide, doxorubicin, prednisone) where an ORR of 100% and a CR rate of 88% was observed (Fanale MA et al. ASH 2012). Both combinations are currently explored in ongoing phase III clinical trials (clinicaltrials.gov NCT01712490 and NCT01777152).
Although a great addition to lymphoma therapy, a potential caveat for the future widespread use of SGN-35 might be the increased risk for pulmonary toxicity, especially when used with other agents such as bleomycin (Ansell SM et al. ASH 2012) or gemcitabine, as well as the development of progressive multifocal leukoencephalopathy (PML), for which the FDA has issued a black box warning.[36] An interesting observation is that retreatment with SGN-35 might result in responses even in patients who have previously progressed on therapy with brentuximub vedotin (Bartlett N et al. ASCO 2010). This observation, though, will need confirmation in a larger more systematically conducted clinical trial.
Trastuzumab emtansine targets her-2-neu receptors in breast cancer
Trastuzumab emtansine (T-DM1; Kadcycla™) is the most recently FDA approved ADC and indicated for the treatment of Human Epidermal Growth Factor Receptor 2 (HER2) positive metastatic breast cancer. Based off of the HER2 (EGFR2, neu) targeting mAb trastuzumab (Herceptin™), T-DM1 consists of the antibody trastuzumab conjugated to the anti-mitotic maytasanoid N(2’)-deacetyl-N(2’)-(3-mercapto-1-oxopropyl)-maytansine (DM1) via an N-[maleimidomethyl] cyclohexane-1 carboxylate (MCC) linker. DM1 is a powerful microtubule-depolymerizing agent with action thought to be similar to that of the vinca alkaloid class of chemotherapeutics. The linker is connected to DM1 via a nonreducible thioether bond, while it connects to the mAb at ε-lysine side chains. This novel linker was originally seen to be superior to a number of disulfide bond based linkers, as initial reports showed that the MCC linked mAbs had a lower clearance rate, longer half-life, and stronger anti-tumor effects along with reduced toxicity in several in vivo (but not in vitro) models of Her2 positive breast cancer.[37] However, recent research seems to contradict this observation, as comparisons between MCC and N-succinimydyl 4-(2-pyridyldithio)-pentanoate (SPP)-reducible disulfide based linkers have shown the opposite: in vitro, MCC-T-DM1 had a stronger cytotoxic effect compared to SPP-T-DM1, but this did not translate to more potent tumor reduction in in vivo models of breast cancer.[38] To further complicate this scenario, new THIOMAB technologies have been developed that allow for a more homogenous population of ADCs. THIOMABs are antibodies that can be used as intermediates to produce ADCs secondary to an engineered unpaired cysteine residue on each heavy chain.[39]
Most ADCs are in fact a heterogeneous mixture of mAbs, with different numbers of toxic drugs attached to each mAb.[40] For example, MCC-T-DM1 exists in a heterogeneous form, ranging from 0-7 DM1 molecules on each mAb, with a mean of 3.3 per mAb. The thio-T-DM1 is constructed with a nonreducible bis-maleimido trioxyethylene glycol (BMPEO) linker that attaches to the engineered cysteines on the thio-mAb. This form of T-DM1 has an average of 1.8 DM1 drugs per mAb, with 90% of the population having 2 DM1 molecules per mAb. In vitro studies proved the equivalent efficacy of this newer ADC, while in vivo models showed that it was more effective at reducing tumor burdens while having a safer profile in rats and cynomolgous monkeys.[41]
Much preclinical research has been conducted on T-DM1. It has proven to be effective in and ex vivo at killing trastuzumab-resistant breast, and other epithelial tumor cell lines. The dominant mechanism of cytotoxicity appears to be through apoptosis and cell lysis, and not cytostatic G2-M phase arrest as would be expected from its drug class. Protease inhibitors blocked its activity, implicating that T-DM1 is broken down in a lysosomal dependent manner to an active catabolite, lysine-MCC-DM1, which has anti-microtubule activity, but cannot penetrate the cell membrane, thereby lessening the bystander effect.[37] In vitro studies have also shown that DM1 itself is far more potent than commonly used cytotoxic agents in breast cancer such as paclitaxel or doxorubicin, and that T-DM1 retains the same mechanisms of action as trastuzumab alone: they both have equivalent Her2 binding affinities, antibody-dependent cell-meditated cytotoxicity (ADCC) activation, Akt inactivation, and HER2 ectodomain shedding induction, implying that T-DM1 may be able to replace Herceptin™ as a frontline chemotherapy. T-DM1 was also effective against a variety of trastuzumab and lapatinib resistant, and PI3K-pathway activated, cell lines ex and in vivo.[42] Recently, it has been discovered that T-DM1 can also cause cytotoxicity through mitotic catastrophe.[43]
Numerous clinical trials exploring the use of T-DM1 in breast cancer as a single agent or in combination with cytotoxic and Her2-directed therapy have been completed by now. In a Phase I clinical trial in advanced HER2+ breast cancer patients (n=24), increasing doses (0.3-4.8 mg/kg) of T-DM1 were administered once every 3 weeks. The OR rate was 25% without any CRs, while in the 15 patients who received the drug at the MTD of 3.6 mg/kg, 73% had clinical benefit (OR and stable disease) at 6 months. Serious side effects ≥ grade 3 included thrombocytopenia and pulmonary hypertension. The most common adverse events were thrombocytopenia, elevated hepatic transaminases, fatigue, anemia, and nausea. Fortunately, only 1of 22 patients evaluated developed anti-therapeutic antibodies (ATA). The MCC linker was shown to be quite stable as well: patients exhibited a 70-fold difference in T-DM1 versus DM1 plasma concentrations.[44]
Based on this data, a Phase II trial was initiated treating 112 patients with HER2 positive metastatic breast cancer (MBC) who had received prior Her2 based therapy, with T-DM1. The ORR was 26%, with no CRs, and a median progression-free survival (PFS) of 4.6 months. Only 74 of the 95 enrolled patients had their Her2 status retroactively confirmed via FISH (fluorescent in-situ hybridization) or IHC (immunohistochemistry). In these 74 patients the ORR was higher (34%) and the median PFS was 8.2 months. Serious side effects ≥grade 3 included hypokalemia, thrombocytopenia, and fatigue. The most common adverse events (AE) were fatigue, nausea, headache, pyrexia, epistaxis, constipation, cough, diarrhea, vomiting, hypokalemia, arthralgia, extremity pain, anemia, and dyspnea. Interestingly, 31% of patients developed ophthalmological AEs. Seven out of 108 evaluable patients tested positive for ATA.[45]
Another recently completed Phase II trial of single agent T-DM1 enrolled 110 heavily pretreated patients. The ORR was 35%, with no CRs, and a median PFS of 6.9 months. Again, by FISH testing Her2 positive status was confirmed for only 80 out of 95 patients. For this subgroup, the ORR was 41% with a median PFS of 7.3 months. Serious side effects ≥grade 3 included thrombocytopenia, fatigue, and cellulitis, with 8% of patients reporting serious hepatic toxicity. Again, the most common AEs were fatigue, thrombocytopenia, nausea, elevated AST, constipation, pyrexia, epistaxis, headache, hypokalemia, decreased appetite, dry mouth, and anemia. Six out of 108 patients developed ATA.[46] In an effort to increase drug exposure, while preserving a similar side effect profile, a weekly dosing schedule of T-DM1 was explored in a Phase I trial (n=28). The MTD was determined to be 2.4 mg/kg, with an ORR of 46% and no CRs. AEs were very similar to the other trials, while no patients developed ATAs.[47]
In a retrospective analysis of the above mentioned 3 trials,[36-38] the effect of prior T-DM1 treatment on subsequent treatment outcomes was explored. Clinical outcomes of 15 patients who had received T-DM1 and were now receiving other therapies were examined to determine if T-DM1 could adversely affect follow-up treatment, especially when giving Herceptin™ or Lapatinib. There did not appear to be any negative consequences in receiving T-DM1, but the interpretation is limited by the small sample size.[48] Additionally, to better understand the mechanism behind T-DM1 induced thrombocytopenia, the main DLT of the drug, a novel high-content, quantitative, live-cell imaging technique was used. The investigators demonstrated that T-DM1 is taken up by megakaryocytes (MKs) via a non-EGFR-dependant pathway (MKs are HER2-). The microtubule targeting of DM1 in MK inhibits pro-platelet production and MK differentiation, induces abnormal tubulin organization, and suppresses microtubule dynamic instability.[49]
The results of the landmark phase III EMILIA trial lead to the FDA approval of the drug for Her2-positive MBC patients previously treated with a trastuzumab and a taxane. In this trial, T-DM1 was compared to combination therapy with lapatinib, an oral Her2 inhibitor, and capecitabine in 991 patients with advanced HER2 positive breast cancer who had previously been treated with trastuzumab and a taxane. Treatment with T-DM1 resulted in significantly improved ORR (43.6% versus 30.8%), prolonged progression-free (median PFS 9.6 months versus 6.4 months; Hazard Ratio [HR] 0.65; p<0.001), and overall survival (median OS 30.9 months versus 25.1 months; HR= 0.68; p<0.001). Additionally, compared to lapatinib plus capecitabine, patients experienced less grade 3 toxicities (41% versus 57%).[50] In the meantime many clinical trials exploring the use of this promising agent in the upfront metastatic and adjuvant setting alone and in combination with other agents are currently undergoing. A recently reported randomized Phase II clinical trial compared T-DM1 to a combination of trastuzumab and docetaxel (HT) as first-line therapy for patients with Her2-positive MBC and appeared better tolerated and more effective.[51] Given these results T-DM1 seems bound to become the preferred treatment for Her2+ MBC.
Moxetumomab Pasudotox targets CD22-positive lymphoid malignancies
Moxetumomab Pasudotox (HA22, CAT-8015) is a second-generation recombinant Immunotoxin (RIT) which targets CD22 and is derived of the same RFB4 monoclonal antibody used for Combotox. HA22 is a high-CD22 affinity version of BL22 (CAT-3888), another RIT. BL22 itself was based off of an ADC that combined the antibody RFB4 linked to a Pseudomonas Exotoxin (PE). PE exerts its cytotoxic effect on cells by ADP-ribosylating Elongation Factor 2 (EF2), a protein integral for translation, thereby inhibiting protein synthesis and leading to cell death. To reduce non-specific toxicity, the PE toxin was modified by removing its cell-binding domain, leading to the development of PE38.[52] To improve its pharmacokinetic profile, increase its potency, and facilitate production, the IT was turned into an RIT. The variable domain (Fv) portions of the mAb were cloned, and a disulfide linkage was added between the heavy (VH) and light (VL) chain to increase stability. Then the VH domain was genetically fused to the PE38 toxin via the C3 (six amino-acid) connector. This process eliminated the need for a linker. BL22 is highly cytotoxic to CD22+ cell lines and prevents tumor growth in vivo in a Burkitt Lymphoma mouse model.[53] Preclinical mouse and primate models showed strong CD22-specific and dose-dependent anti-tumor activity for an alternate day treatment cycle for three doses, while also being well tolerated.[54] Further in vitro studies with cells from 28 cancer patient samples showed that BL22 was cytotoxic in around half of the patients’ samples. Cytotoxicity and CD22 expression were positively correlated.[55]
The initial results from the first Phase I clinical trial of BL22 in patients with Hairy Cell Leukemia (HCL) were very promising. Sixteen out of the 31 patients on the trial had refractory HCL, and in this cohort of patients, 69% had CRs with an ORR of 81%. Of the 11 CRs, only one had signs of Minimal Residual Disease (MRD) in the bone marrow. Unfortunately some serious side effects associated with BL22 were observed, including Cytokine Release Syndrome (CRS) and Hemolytic Uremic Syndrome (HUS). To prevent CRS, patients were subsequently pre-treated with daily Rofecoxib and Infliximab a week before and after treatment. This strategy appeared to prevent further cases of CRS. Less serious side effects included hypoalbuminemia, elevated aminotransferase levels, nausea, myalgia, edema, and elevated creatinine levels, which were all reversible. Twenty-five percent of the patients developed neutralizing antibodies to the IT.[56] In the meanwhile, HA22 was developed from BL22 as an RIT with a greater than 10 times affinity for CD22. This higher affinity also resulted in greater cytotoxicity toward CD22+ cell lines and enabled researchers to examine its efficacy in hematological malignancies with lower CD22 expression.[57] Further versions of CD22 with even greater affinity and cytotoxicity were developed, although they so far have not been clinically pursued.[58, 59] Additional variants of HA22 have been tested that have increased stability with reduced immunogenicity and antigenicity, including HA22-8X,[60] HA22-LR,[61] HA22-LR-8M,[62] and HA22-LR-L010.[63] Additionally, the mechanisms of BL22 cytotoxicity were further elucidated, with experiments showing that BL22 can induce apoptosis via a caspase-3-like protease, in addition to its ability to inhibit translation.[64] Further research has shown that BL22 leads to PARP (Poly (ADP-ribose) polymerase) cleavage and both caspase-3 and caspase-9 activation in patient CLL (chronic lymphocytic leukemia) samples.[65] The sensitivity of cells to apoptosis, which is inversely proportional to Bcl2 expression, was seen as a key determinant in the efficacy of BL22 on Mantle Cell Lymphoma (MCL) cell lines, instead of being correlated with CD22 expression. This suggests Bcl2 overexpression as a possible method for cells to develop resistance against BL22.[66]
The final results from the first BL22 Phase I clinical trial in B-Cell malignancies were as encouraging as the initial results for HCL patients. A total of 46 patients with various B-cell malignancies (31 with HCL, 11 with CLL, and 4 with other NHL) were enrolled. HCL patients had a high CR rate of 61% and ORR of 80%, however, the response was much more muted in the other patients, with only 3 out of 11 CLL patients having a marginal response. Although originally, prophylactic anti-inflammatory drugs were used to prevent CRS, it soon became apparent that HUS proved to be a more serious safety issue. In order to prevent HUS, prophylactic intravenous fluid was administered to diminish renal toxicity. This measure, along with reducing the dose of BL22, appeared to ameliorate the issue. Otherwise, the safety profile was similar to that of the initial Phase 1 study in HCL, with 35% of HCL, but no CLL or NHL patients developing neutralizing antibodies.[67] In a Phase II trial testing BL22 in 36 patients with refractory HCL, CRs were seen in 47% of patients (with 18% having MRD), and an ORR of 72%. Interestingly, smaller spleen size was highly correlated with an improved response. Serious associated toxicities included transaminitis, hypoalbuminemia, fever, thrombocytopenia, VLS, proteinuria, anemia, and hypoxia. All of these toxicities were reversible and did not constitute a DLT or were associated with HUS. The most common AEs were hypoalbuminemia, transaminitis, edema, myalgia, proteinuria, fatigue, nausea, and fever. Neutralizing antibodies were seen in 11% of patients.[68]
BL22 was less successful, however, in treating pediatric pre-B ALL. In a Phase I clinical trial for pediatric patients with refractory B-cell malignancies (n=23), 21 of whom had pre-B ALL, there were no CRs or PRs, although there was transient clinical activity seen in 70% of patients. Serious side effects at ≥grade 3 included transaminitis and myelosuppression. The most common toxicities were hypoalbuminemia, transaminitis, and proteinuria. No cases of HUS or VLS were observed, and no DLT was seen. Although neutralizing antibodies were found in 13% of patients, the drug appeared to be much better tolerated in children than adults.[69] To increase efficacy, the use of Bryostatin I, a weak chemotherapeutic agent that upregulates CD22 expression, was explored. In in vitro CLL and MCL patient samples,[70] Bryostatin I was administered before BL22 and resulted in increased efficacy. Sequential administration of Bryostatin I followed by BL22 may be a useful method to improve results for the treatment of hematological malignancies that have lower levels of CD22 expression. Another way to increase efficacy of CD22-directed therapies may be by using HA22, which has a much higher affinity for CD22. HA22 has been tested in an in vivo mouse model of primary intraocular lymphoma (PIOL), where a single dose injection of HA22 into the eye was enough to cause complete tumor regression, with minimal associated eye toxicity.[71] In pre-clinical studies, HA22 was significantly more cytotoxic than BL22 toward CD22+ cell lines, and more effective in reducing tumor volumes in a Burkitt Lymphoma mouse model.[72] This improved efficacy for HA22 compared with BL22 was also demonstrated in cell cultures of pediatric pre-B ALL patient samples.[73] A potential mechanism for HA22 resistance in ALL patients has been described. In vitro studies on resistant ALL cell lines revealed that a CpG island in the promoter region of DPH4, which codes for the diphthamide biosynthesis protein and is crucial for PE toxin mediation, is hypermethylated, leading to downregulated levels of the protein. This DPH4 downregulation is transient and dependent on the presence of HA22, and can be overcome by pretreatment with the methylation inhibitor azacytidine.[74] More recently, a second mechanism for resistance has been discovered in a resistant Burkitt NHL cell line, where a mutation has led to the deletion of WDR85, another gene that is necessary for dipthamide synthesis (Wei H et al. ASH 2012).
In a Phase I clinical trial with 28 refractory HCL patients, every other day HA22 treatment for 3 doses led to CRs in 46% of patients, with an ORR of 86%. Out of 9 patients who achieved a CR, only one was positive for MRD. Like before, clinical success was inversely proportional to spleen size, with patients having a prior splenectomy suffering worse outcomes. Serious side effects ≥grade 3 included lymphopenia, γ-glutamyltransferase (GGT) elevation, and leukopenia, although none were considered serious enough to be considered a DLT. The most common AEs were hypoalbuminemia, transaminitis, limb/head and neck edema, headache, hypotension, and nausea. Neutralizing antibodies to the toxin were reported in 38% of patients.[75] A recent update to this study involving 20 more patients (n=42) confirms the above findings, with 55% achieving a CR, and an ORR of 88%. Of the 21 CRs evaluable for MRD, only 4 were positive (Kreitman RJ et al. ASCO 2012). A phase I trial treating pediatric ALL with HA22 at 6 doses every other day per 3-weekly cycle is still ongoing, but preliminary results appear promising. ORs were reported for 5 of 17 evaluable patients (29%), with 4 (24%) achieving a CR. Hematological activity (HA), as defined by a ≥50% reduction in blasts and/or improvement in neutrophil and/or platelet counts, was seen in 7 patients (41%). Serious VLS was reported in two of the first seven patients, although this toxicity has been ameliorated when dexamethasone was added as pre-treatment. Anti-moxetumomab pasudotox neutralizing antibodies were reported in 14% of patients (Wayne AS et al. ASH 2011). HA22 is currently being evaluated in at least 2 ongoing phase I clinical trials (ClinicalTrials.gov Identifier: NCT00586924 and NCT00659425).
Inotuzumab Ozogamicin is showing promise in treating CD22+ ALL and NHL
Inotuzumab Ozogamicin (INO; CMC-544) is another anti-CD22 directed ADC. Its antibody is based off of the humanized IgG4 G5/44 mAb (not RFB4). CMC-544 is a close relative of Gemtuzumab Ozogamicin (GO; Mylotarg™), an anti-CD33 mAb with the same linker and toxin as CMC-544. GO was the first ADC approved by the FDA for the treatment of AML,[76] but was later voluntarily withdrawn from the US market because of concern about its toxicity (mainly sinusoidal obstruction syndrome).[77] Of note, recent findings support GO’s usefulness and reduced toxicity when given in lower doses during induction therapy, by increasing disease-free survival specifically in older patients.[78, 79] The IgG4 G5/44 mAb was chosen, as it had the best combination of high CD22 affinity combined with a high rate of internalization. The mAb is joined to its toxin via the acid hydrolysable 4-(4’-acteylphenoxy)butanoic acid (AcBut) linker. This linker was proven to be more effective than a more stable amide linker in both in vitro cytotoxicity and in vivo anti-tumor assays.[80] The toxin is N-acetyl-γ-calicheamicin dimethyl hydrazide (CalichDMH), which is derived from the γ-calicheamicin antitumor antibiotic naturally produced by the bacterium micromonospora echinospora. This extremely potent product mediates its cytotoxicity by binding DNA in its minor groove, then undergoing thiol-dependent structural changes in its enediyne moiety to generate a di-radical, which abstracts hydrogens from the phosphodiester backbone of DNA, leading to double stranded breaks in the DNA, and ultimately cell death. There is an average of 5-7 moles of toxin per mole of mAb. CMC-544 is seen as more of a targeted chemotherapy as opposed to an immunotherapy, due to the weakness of its mAb, which has nearly no efficacy on its own, owing to its inability to fix complement or initiate ADCC. However, CMC-544 was even more cytotoxic against CD22+ B-lymphoma cell lines than unconjugated CalichDMH, and had strong dose-dependent anti-tumor activity against small and large B-cell lymphoma (BCL) xenografts.[81]
Further studies showed that CMC-544 was also active in both early and late stage disseminated BCL murine models, while Rituximab, a chimeric anti-CD20 mAb, was only effective, and to a lesser degree, at the early stage.[82] Preclinical research showed that CMC-544 could have an additive or synergistic effect when combined with Rituximab. This could be due to their different modes of action, as Rituximab relies on ADCC/fixing complement for its anti-tumor activity, which CMC-544 is unable to perform. Importantly, CMC-544 caused significant tumor regression in a Rituximab-refractory established tumor model.[83] CMC-544 was also found to have strong cytotoxicity against ALL cell lines, along with potent dose dependent anti-tumor activity in both subcutaneous xenograft and disseminated ALL murine models.[84] Finally, CMC-544 demonstrated greater efficacy than either of the frontline combination NHL therapies, CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) or CVP (CHOP without doxorubicin) in both in vitro and in vivo assays. Importantly, CMC-544 retained activity in CHOP/CVP-refractory tumor models, while also showing increased potency when given in combination with CVP (but not CHOP, which was too toxic).[85]
Further in vitro studies showed that CMC-544 efficacy was inversely correlated with P-glycoprotein (P-gp) expression, an efflux pump responsible for multi-drug resistance (MDR) in CLL and NHL cell lines, as well as in patient samples. However, this could be overcome by combining CMC-544 with the MDR modifiers PSC833 or MS209.[86] Additionally, it was discovered that CD22 and CD55 expression decreased after CMC-544 treatment in both BCL cell lines and patient samples, while CD20 and CD59 levels remained the same. This correlated with an increased efficacy in the complement dependent cytotoxicity (CDC) of Rituximab that was seen only when given after CMC-544 treatment, and not when given simultaneously.[87] Research with pre-B ALL patient samples showed that the large variation in sensitivity to CMC-544 was directly correlated with sensitivity of the cells to free calicheamicin and how quickly CMC-544 could be internalized into the cells, but was not dependent on extracellular CD22 levels or the ability of the cells to renew their CD22 expression.[88]
The first Phase I trial of CMC-544 involved 79 patients with refractory NHL, with the majority (35 each) having either follicular lymphoma (FL) or diffuse large B-cell lymphoma (DLBCL). The MTD was established at 1.8 mg/m2 given once every 3-4 weeks, with the DLT being thrombocytopenia. While the ORR was only 39%, the ORR at MTD for FL was 68%, with 32% CRs and a median PFS of 317 days, while for DLBCL it was a lower 15%, with 7.7% CRs and a median PFS of 49 days. Serious side effects at ≥grade 3 included thrombocytopenia, neutropenia, leukopenia, asthenia, arthralgia, and fever. The most common AEs were thrombocytopenia, asthenia, nausea, neutropenia, fever, elevated AST levels, abdominal pain, and anorexia. While thrombocytopenia was a major issue, without a known obvious mechanism for its occurrence, there were no major hemorrhages reported.[89] Another smaller Phase I trial, involving 13 Japanese patients with refractory FL who had previously been treated with Rituximab, had similar findings. The ORR was 85%, with 54% CRs. Serious side effects ≥grade 3 included thrombocytopenia, lymphopenia, neutropenia, leukopenia, hyperbilirubinemia, and hypokalemia. The most common AEs were thrombocytopenia, leukopenia, neutropenia, elevated AST levels, anorexia, and nausea.[90]
A more recent Phase I trial, also done in Japan, which combined CMC-544 treatment one day following Rituximab every 4 weeks in 10 refractory NHL patients (6 of whom had FL) also showed promising results. The ORR was 80%, with 70% CRs, while the CR rate in FL patients was 83%. Serious side effects ≥grade 3 included thrombocytopenia, neutropenia, lymphopenia, leucopenia, hypophosphatemia, and elevated AST levels. The most common AEs were thrombocytopenia, elevated transaminase levels, leukopenia, nausea, neutropenia, and lymphopenia. Importantly, Rituximab did not induce any major changes in the pharmacokinetic or safety profile of CMC-544 than when CMC-544 was used as a monotherapy.[91] Most recently, Fayad and colleagues reported a combined dose-escalation Phase I /II trial of R-INO (rituximab and INO) in patients with relapsed FL and DLBCL, and refractory aggressive NHL (n=118). At the MTD (Rituximab 375mg/m2 on day 1, INO 1.8mg/m2 on day 2, given every 4 weeks for up to 8 cycles), the ORR was 87%, 74%, and 20% respectively for FL (n=39), DLBCL (n=42) and refractory NHL (n=30), with a 2-year progression-free survival (PFS) of 68%, and 42% for FL and DLBCL, respectfully. Thirty percent of patients treated experienced serious AEs, which included pneumonia, sepsis, thrombocytopenia, nausea and/or vomiting, peripheral edema, chest pain, dizziness, infection and nodular regenerative hyperplasia. Treatment at MTD was discontinued in 43% secondary to toxicities, most commonly secondary to thrombocytopenia or hyperbilirubinemia.[92]
The addition of INO to chemotherapy has also been explored in another Japanese Phase 1 study (Ogura M et al. ASH 2011), with the expansion cohort at the MTD having been recently updated. Ogura and colleagues identified the MTD as full dose R-CVP combined with INO at 0.8mg/m2 given on day 2 of a 3-weekly cycle. In the expansion cohort, 32 patients with relapsed or refractory B-cell NHL [FL (n = 15), MCL (n = 1), and DLBCL (n = 16)] were treated at MTD. The ORR was 100% (53% CR) and 605 (7% CR) for patients with FL and DLBCL respectively. The most commonly described AE of any grade were thrombocytopenia, leukopenia, neutropenia, fatigue, constipation, and nausea; the most common grade ≥3 AEs included cytopenias and transaminitis with 1 death secondary to pneumonia in a neutropenic patient (Ogura M et al. ASH 2012).
Another trial looked at Rituximab plus INO (R-INO) salvage therapy followed by autologous SCT (ASCT) for 61 patients with relapsed/refractory DLBCL. Common adverse events during R-INO treatment were again cytopenias, transaminitis, fatigue, pyrexia, and vomiting (24%). Two patients had veno-occlusive disease of the liver (VOD) after ASCT. Fifty-four evaluable patients received a median of 3 cycles of R-INO. The ORR after R-INO was 35% (24% CR, 11% PR), 13% had SD, and 50% progressive lymphoma. Prior response to the most recent therapy was predictive of response to R-INO. Stem cell mobilization and stem cell collection after R-INO appeared acceptable. Six- and 12-month PFS rates for all treated patients were 31% and 13%, respectively (median PFS 2.6 months), and the median OS was 10 months. For patients who underwent ASCT, the 6- and 12-month PFS rates were 79% and 35%, respectively (median PFS 10 months) and the median OS was not reached (Wagner-Johnson N et al. ASH 2011). Multiple other trials with INO in NHL, in combination with Rituximab or chemotherapy, in the upfront and relapsed setting, are planned or ongoing.
In addition to the treatment of NHL, INO shows promise also for ALL. A Phase II trial involving 49 adult and pediatric patients with refractory ALL showed encouraging results. Eighteen percent of patients achieved a CR, and 39% had a marrow CR (<5% lymphoblasts), resulting in an ORR of 57%. The median OS was 5.1 months for all patients, and 7.9 months for responders. Serious side effects ≥grade 3 included thrombocytopenia, neutropenia, fever, and hyperbilirubinemia, which was reversible in most cases. The most common adverse events were thrombocytopenia, neutropenia, elevated ALT/AST levels, fever, hypotension, and hyperbilirubinemia.[93] Two trials explored a weekly schedule of INO for patients with relapsed refractory B-cell ALL (DeAngelo D et al. ASH 2012; Jabbour E et al. ASCO 2012). Responses were consistent in both trials and observed across all INO doses. The ORR ranged around 82% with a CR and CRi (incomplete CR) rate ranging between 45-50%. At the same time the toxicities appeared similar in nature (hematological, liver and gastrointestinal), but possibly less severe and/or frequent.
Given these encouraging early results in both B-cell NHL and ALL, INO is currently being tested in multiple Phase I, II, and III trials alone, in combination with Rituximab, and with other chemotherapeutic regimens (e.g. GemOx [gemcitabine and oxaliplatin], GDP [gemcitabine, dexamethasone, and cisplatin], temsirolimus, and CVP).
CONCLUSIONS
The five types of ADCs described here in detail illustrate some of the many different strategies research groups are taking to bring ADCs from bench to bedside (Table 2). The two most recently FDA approved ADCs, Brentuximab vedotin and Trastuzumab emtansine, were welcome by clinicians with great anticipation. These ADCs were developed using novel peptide and thioether linkers, respectively, giving them an advantage over older ADCs using linkers with less stability, and therefore causing more collateral damage secondary to systemic off-target effects by free toxin. Following the biotechnological progress in ADC development there has to be a similar progress in finding the best dosing strategies and combination with other drugs to maximize their efficacy. The inherent specificity of ADCs may serve as the perfect complement to the broad based killing effects of standard cytotoxic chemotherapies, as ADCs are able to destroy slower growing and even quiescent cancer cells that are more resistant to chemotherapy and most likely responsible for relapse and refractoriness. Determining the optimal dosing schedule as part of combination therapy might help increase efficacy, while reducing the side effects of both the ADC and chemotherapy. Certain classes of chemotherapies may also work in a synergistic manner with specific ADC toxins and drugs that increase antigen expression might counter resistance to targeted agents. Addition of ADCs to the armamentarium of oncologists has offered novel and more targeted treatment strategies that might enable clinicians to safely extend the lifespan of patients with various cancer diagnoses.
References
1. Strebhardt K and Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008; 8(6):473-480.
2. Mathe G, LO TB and Bernard J. Effect on mouse leukemia 1210 of a combination by diazo-reaction of amethopterin and gamma-globulins from hamsters inoculated with leukemia by heterografts. C R Hebd Seances Acad Sci. 1958; 246(10):1626-1628.
3. Piascik PP. FDA approves fusion protein for treatment of lymphoma. J Am Pharm Assoc (Wash). 1999; 39(4):571-572.
4. Leget GA and Czuczman MS. Use of rituximab, the new FDA-approved antibody. Current Opinion in Oncology. 1998; 10(6):548-551.
5. Hussain N. Ligand-mediated tissue specific drug delivery. Advanced Drug Delivery Reviews. 2000; 43(2–3):95-100.
6. Goodman LS, Wintrobe MM, Damehek W, Goodman MJ, Gilman A, McLennan MT. Nitrogen mustard therapy: Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. JAMA. 1984; 251(17):2255-2261.
7. Polson AG, Calemine-Fenaux J, Chan P, Chang W, Christensen E, Clark S, de Sauvage FJ, Eaton D, Elkins K, Elliott JM, Frantz G, Fuji RN, Gray A, Harden K, Ingle GS, Kljavin NM, et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: target and linker-drug selection. Cancer Res. 2009; 69(6):2358-2364.
8. Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, Wilhelm SD and Singh R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2010; 21(1):84-92.
9. Alley SC, Benjamin DR, Jeffrey SC, Okeley NM, Meyer DL, Sanderson RJ and Senter PD. Contribution of Linker Stability to the Activities of Anticancer Immunoconjugates. Bioconjugate Chemistry. 2008; 19(3):759-765.
10. Crocker PR, McMillan SJ and Richards HE. CD33-related siglecs as potential modulators of inflammatory responses. Annals of the New York Academy of Sciences. 2012; 1253(1):102-111.
11. Tedder TF and Isaacs CM. Isolation of cDNAs encoding the CD19 antigen of human and mouse B lymphocytes. A new member of the immunoglobulin superfamily. The Journal of Immunology. 1989; 143(2):712-717.
12. Wilson GL, Fox CH, Fauci AS and Kehrl JH. cDNA cloning of the B cell membrane protein CD22: a mediator of B-B cell interactions. The Journal of Experimental Medicine. 1991; 173(1):137-146.
13. Hynes NE and Stern DF. The biology of erbB-2/nue/HER-2 and its role in cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1994; 1198(2–3):165-184.
14. Soler-Rodriguez AM, Uhr JW, Richardson J and Vitetta ES. The toxicity of chemically deglycosylated ricin A-chain in mice. International Journal of Immunopharmacology. 1992; 14(2):281-291.
15. Kato J, O’Donnell RT, Abuhay M and Tuscano JM. Efficacy and toxicity of a CD22-targeted antibody-saporin conjugate in a xenograft model of non-Hodgkin’s lymphoma. OncoImmunology. 2012; 1(9):1469-1475.
16. Morris JC and Waldmann TA. Antibody-based therapy of leukaemia. Expert Reviews in Molecular Medicine. 2009; 11:null-null.
17. Du X, Beers R, FitzGerald DJ and Pastan I. Differential Cellular Internalization of Anti-CD19 and -CD22 Immunotoxins Results in Different Cytotoxic Activity. Cancer Research. 2008; 68(15):6300-6305.
18. Ghetie M, Tucker K, Richardson J, Uhr J and Vitetta E. The antitumor activity of an anti-CD22 immunotoxin in SCID mice with disseminated Daudi lymphoma is enhanced by either an anti-CD19 antibody or an anti-CD19 immunotoxin. Blood. 1992; 80(9):2315-2320.
19. Messmann RA, Vitetta ES, Headlee D, Senderowicz AM, Figg WD, Schindler J, Michiel DF, Creekmore S, Steinberg SM, Kohler D, Jaffe ES, Stetler-Stevenson M, Chen H, Ghetie V and Sausville EA. A Phase I Study of Combination Therapy with Immunotoxins IgG-HD37-Deglycosylated Ricin A Chain (dgA) and IgG-RFB4-dgA (Combotox) in Patients with Refractory CD19(+), CD22(+) B Cell Lymphoma. Clinical Cancer Research. 2000; 6(4):1302-1313.
20. Herrera L, Farah RA, Pellegrini VA, Aquino DB, Sandler ES, Buchanan GR and Vitetta ES. Immunotoxins against CD19 and CD22 are effective in killing precursor-B acute lymphoblastic leukemia cells in vitro. Leukemia. 2000; 14(5):853-858.
21. Herrera L, Yarbrough S, Ghetie V, Aquino DB and Vitetta ES. Treatment of SCID//human B cell precursor ALL with anti-CD19 and anti-CD22 immunotoxins. Leukemia. 2003; 17(2):334-338.
22. Herrera LMD, Bostrom BMD, Gore LMD, Sandler EMD, Lew GMD, Schlegel PGMD, Aquino VMD, Ghetie VP, Vitetta ESP and Schindler JP. A Phase 1 Study of Combotox in Pediatric Patients With Refractory B-lineage Acute Lymphoblastic Leukemia. Journal of Pediatric Hematology/Oncology. 2009; 31(12):936-941.
23. Schindler J, Gajavelli S, Ravandi F, Shen Y, Parekh S, Braunchweig I, Barta S, Ghetie V, Vitetta E and Verma A. A phase I study of a combination of anti-CD19 and anti-CD22 immunotoxins (Combotox) in adult patients with refractory B-lineage acute lymphoblastic leukaemia. British Journal of Haematology. 2011; 154(4):471-476.
24. Barta SK, Zou Y, Schindler J, Shenoy N, Bhagat TD, Steidl U and Verma A. Synergy of sequential administration of a deglycosylated ricin A chain-containing combined anti-CD19 and anti-CD22 immunotoxin (Combotox) and cytarabine in a murine model of advanced acute lymphoblastic leukemia. Leukemia & Lymphoma. 2012; 53(10):1999-2003.
25. Younes A, Yasothan U and Kirkpatrick P. Brentuximab vedotin. Nat Rev Drug Discov. 2012; 11(1):19-20.
26. Deutsch YE, Tadmor T, Podack ER and Rosenblatt JD. CD30: an important new target in hematologic malignancies. Leukemia & Lymphoma. 2011; 52(9):1641-1654.
27. Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki BE, Law C-L, Doronina SO, Siegall CB, Senter PD and Wahl AF. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003; 102(4):1458-1465.
28. Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL and Francisco JA. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clinical Cancer Research. 2004; 10(20):7063-7070.
29. Sanderson RJ, Hering MA, James SF, Sun MMC, Doronina SO, Siadak AW, Senter PD and Wahl AF. In vivo Drug-Linker Stability of an Anti-CD30 Dipeptide-Linked Auristatin Immunoconjugate. Clinical Cancer Research. 2005; 11(2):843-852.
30. Sutherland MSK, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, Lewis TS, Meyer DL, Zabinski RF, Doronina SO, Senter PD, Law C-L and Wahl AF. Lysosomal Trafficking and Cysteine Protease Metabolism Confer Target-specific Cytotoxicity by Peptide-linked Anti-CD30-Auristatin Conjugates. Journal of Biological Chemistry. 2006; 281(15):10540-10547.
31. Oflazoglu E, Kissler KM, Sievers EL, Grewal IS and Gerber H-P. Combination of the anti-CD30-auristatin-E antibody-drug conjugate (SGN-35) with chemotherapy improves antitumour activity in Hodgkin lymphoma. British Journal of Haematology. 2008; 142(1):69-73.
32. Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL and Forero-Torres A. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. New England Journal of Medicine. 2010; 363(19):1812-1821.
33. Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ, Ramchandren R, Bartlett NL, Cheson BD, de Vos S, Forero-Torres A, Moskowitz CH, Connors JM, Engert A, Larsen EK, Kennedy DA, et al. Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients With Relapsed or Refractory Hodgkin’s Lymphoma. Journal of Clinical Oncology. 2012; 30(18):2183-2189.
34. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, Matous J, Ramchandren R, Fanale M, Connors JM, Yang Y, Sievers EL, Kennedy DA and Shustov A. Brentuximab Vedotin (SGN-35) in Patients With Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. Journal of Clinical Oncology. 2012; 30(18):2190-2196.
35. Fanale MA, Forero-Torres A, Rosenblatt JD, Advani RH, Franklin AR, Kennedy DA, Han TH, Sievers EL and Bartlett NL. A Phase I Weekly Dosing Study of Brentuximab Vedotin in Patients with Relapsed/Refractory CD30-Positive Hematologic Malignancies. Clinical Cancer Research. 2012; 18(1):248-255.
36. Wagner-Johnston ND, Bartlett NL, Cashen A and Berger JR. Progressive multifocal leukoencephalopathy in a patient with Hodgkin lymphoma treated with brentuximab vedotin. Leukemia & Lymphoma. 2012; 53(11):2283-2286.
37. Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blattler WA, Lambert JM, Chari RVJ, Lutz RJ, Wong WLT, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody-Cytotoxic Drug Conjugate. Cancer Research. 2008; 68(22):9280-9290.
38. Erickson HK, Lewis Phillips GD, Leipold DD, Provenzano CA, Mai E, Johnson HA, Gunter B, Audette CA, Gupta M, Pinkas J and Tibbitts J. The Effect of Different Linkers on Target Cell Catabolism and Pharmacokinetics/Pharmacodynamics of Trastuzumab Maytansinoid Conjugates. Molecular Cancer Therapeutics. 2012; 11(5):1133-1142.
39. Chen X, Nguyen M, Jacobson F and Ouyang J. Charge-based analysis of antibodies with engineered cysteines: From multiple peaks to a single main peak. mAbs. 2009; 1(6):563-571.
40. Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotech. 2008; 26(8):925-932.
41. Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, Mai E, Lewis Phillips GD, Hiraragi H, Fuji RN, Tibbitts J, Vandlen R, et al. Engineered Thio-Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. Clinical Cancer Research. 2010; 16(19):4769-4778.
42. Junttila TT, Li G, Parsons K, Phillips GL and Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Research and Treatment. 2011; 128(2):347-356.
43. Barok M, Tanner M, Koninki K and Isola J. Trastuzumab-DM1 causes tumour growth inhibition by mitotic catastrophe in trastuzumab-resistant breast cancer cells in vivo. Breast Cancer Research. 2011; 13(2):R46.
44. Cufí S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, Martin-Castillo B and Menendez JA. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012; 3(4):395-398.
45. Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, Girish S, Tibbitts J, Yi J-H, Sliwkowski MX, Jacobson F, Lutzker SG and Burris HA. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, Given Every 3 Weeks to Patients With HER2-Positive Metastatic Breast Cancer. Journal of Clinical Oncology. 2010; 28(16):2698-2704.
46. Burris HA, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, Tan-Chiu E, Krop IE, Michaelson RA, Girish S, Amler L, Zheng M, Chu Y-W, Klencke B and O’Shaughnessy JA. Phase II Study of the Antibody Drug Conjugate Trastuzumab-DM1 for the Treatment of Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Breast Cancer After Prior HER2-Directed Therapy. Journal of Clinical Oncology. 2011; 29(4):398-405.
47. Krop IE, LoRusso P, Miller KD, Modi S, Yardley D, Rodriguez G, Guardino E, Lu M, Zheng M, Girish S, Amler L, Winer EP and Rugo HS. A Phase II Study of Trastuzumab Emtansine in Patients With Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer Who Were Previously Treated With Trastuzumab, Lapatinib, an Anthracycline, a Taxane, and Capecitabine. Journal of Clinical Oncology. 2012; 30(26):3234-3241.
48. Beeram M, Krop IE, Burris HA, Girish SR, Yu W, Lu MW, Holden SN and Modi S. A phase 1 study of weekly dosing of trastuzumab emtansine (T-DM1) in patients with advanced human epidermal growth factor 2–positive breast cancer. Cancer. 2012:118(23):5733-5740.
49. Olson EM, Lin NU, DiPiro PJ, Najita JS, Krop IE, Winer EP and Burstein HJ. Responses to subsequent anti-HER2 therapy after treatment with trastuzumab-DM1 in women with HER2-positive metastatic breast cancer. Annals of Oncology. 2012; 23(1):93-97.
50. Thon JN, Devine MT, Jurak Begonja A, Tibbitts J and Italiano JE. High-content live-cell imaging assay used to establish mechanism of trastuzumab emtansine (T-DM1)-mediated inhibition of platelet production. Blood. 2012; 120(10):1975-1984.
51. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh D-Y, Dieras Vr, Guardino E, Fang L, Lu MW, Olsen S and Blackwell K. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. New England Journal of Medicine. 2012; 367(19):1783-1791.
52. Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, Guardino E, Song C, Tong B, Ng V, Chu Y-W and Perez EA. Phase II Randomized Study of Trastuzumab Emtansine Versus Trastuzumab Plus Docetaxel in Patients With Human Epidermal Growth Factor Receptor 2–Positive Metastatic Breast Cancer. Journal of Clinical Oncology. 2013.
53. Mansfield E, Pastan I and FitzGerald DJ. Characterization of RFB4-Pseudomonas Exotoxin A Immunotoxins Targeted to CD22 on B-Cell Malignancies. Bioconjugate Chemistry. 1996; 7(5):557-563.
54. Mansfield E, Amlot P, Pastan I and FitzGerald DJ. Recombinant RFB4 Immunotoxins Exhibit Potent Cytotoxic Activity for CD22-Bearing Cells and Tumors. Blood. 1997; 90(5):2020-2026.
55. Kreitman RJ, Wang Q-C, FitzGerald DJP and Pastan I. Complete regression of human B-cell lymphoma xenografts in mice treated with recombinant anti-CD22 immunotoxin RFB4(dsFv)-PE38 at doses tolerated by cynomolgus monkeys. International Journal of Cancer. 1999; 81(1):148-155.
56. Kreitman RJ, Margulies I, Stetler-Stevenson M, Wang Q-C, FitzGerald DJP and Pastan I. Cytotoxic Activity of Disulfide-stabilized Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) toward Fresh Malignant Cells from Patients with B-Cell Leukemias. Clinical Cancer Research. 2000; 6(4):1476-1487.
57. Kreitman RJ, Wilson WH, Bergeron K, Raggio M, Stetler-Stevenson M, FitzGerald DJ and Pastan I. Efficacy of the Anti-CD22 Recombinant Immunotoxin BL22 in Chemotherapy-Resistant Hairy-Cell Leukemia. New England Journal of Medicine. 2001; 345(4):241-247.
58. Salvatore G, Beers R, Margulies I, Kreitman RJ and Pastan I. Improved Cytotoxic Activity toward Cell Lines and Fresh Leukemia Cells of a Mutant Anti-CD22 Immunotoxin Obtained by Antibody Phage Display. Clinical Cancer Research. 2002; 8(4):995-1002.
59. Bang S, Nagata S, Onda M, Kreitman RJ and Pastan I. HA22 (R490A) Is a Recombinant Immunotoxin with Increased Antitumor Activity without an Increase in Animal Toxicity. Clinical Cancer Research. 2005; 11(4):1545-1550.
60. Ho M, Kreitman RJ, Onda M and Pastan I. In Vitro Antibody Evolution Targeting Germline Hot Spots to Increase Activity of an Anti-CD22 Immunotoxin. Journal of Biological Chemistry. 2005; 280(1):607-617.
61. Onda M, Beers R, Xiang L, Nagata S, Wang Q-c and Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proceedings of the National Academy of Sciences. 2008; 105(32):11311-11316.
62. Weldon JE, Xiang L, Chertov O, Margulies I, Kreitman RJ, FitzGerald DJ and Pastan I. A protease-resistant immunotoxin against CD22 with greatly increased activity against CLL and diminished animal toxicity. Blood. 2009; 113(16):3792-3800.
63. Onda M, Beers R, Xiang L, Lee B, Weldon JE, Kreitman RJ and Pastan I. Recombinant immunotoxin against B-cell malignancies with no immunogenicity in mice by removal of B-cell epitopes. Proceedings of the National Academy of Sciences. 2011; 108(14):5742-5747.
64. Liu W, Onda M, Lee B, Kreitman RJ, Hassan R, Xiang L and Pastan I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proceedings of the National Academy of Sciences. 2012; 109(29):11782-11787.
65. Keppler-Hafkemeyer A, Kreitman RJ and Pastan I. Apoptosis induced by immunotoxins used in the treatment of hematologic malignancies. International Journal of Cancer. 2000; 87(1):86-94.
66. Decker T, Oelsner M, Kreitman RJ, Salvatore G, Wang Q-c, Pastan I, Peschel C and Licht T. Induction of caspase-dependent programmed cell death in B-cell chronic lymphocytic leukemia by anti-CD22 immunotoxins. Blood. 2004; 103(7):2718-2726.
67. Bogner C, Dechow T, Ringshausen I, Wagner M, Oelsner M, Lutzny G, Licht T, Peschel C, Pastan I, Kreitman RJ and Decker T. Immunotoxin BL22 induces apoptosis in mantle cell lymphoma (MCL) cells dependent on Bcl-2 expression. British Journal of Haematology. 2010; 148(1):99-109.
68. Kreitman RJ, Squires DR, Stetler-Stevenson M, Noel P, FitzGerald DJP, Wilson WH and Pastan I. Phase I Trial of Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) in Patients With B-Cell Malignancies. Journal of Clinical Oncology. 2005; 23(27):6719-6729.
69. Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, FitzGerald DJP, Wilson WH and Pastan I. Phase II Trial of Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) in Patients With Hairy Cell Leukemia. Journal of Clinical Oncology. 2009; 27(18):2983-2990.
70. Wayne AS, Kreitman RJ, Findley HW, Lew G, Delbrook C, Steinberg SM, Stetler-Stevenson M, FitzGerald DJ and Pastan I. Anti-CD22 Immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-Positive Hematologic Malignancies of Childhood: Preclinical Studies and Phase I Clinical Trial. Clinical Cancer Research. 2010; 16(6):1894-1903.
71. Biberacher V, Decker T, Oelsner M, Wagner M, Bogner C, Schmidt B, Kreitman RJ, Peschel C, Pastan I, Meyer zum Buschenfelde C and Ringshausen I. The cytotoxicity of anti-CD22 immunotoxin is enhanced by bryostatin 1 in B-cell lymphomas through CD22 upregulation and PKC-βII depletion. Haematologica. 2012; 97(5):771-779.
72. Li Z, Mahesh SP, Shen DF, Liu B, Siu WO, Hwang FS, Wang Q-C, Chan C-C, Pastan I and Nussenblatt RB. Eradication of Tumor Colonization and Invasion by a B Cell-Specific Immunotoxin in a Murine Model for Human Primary Intraocular Lymphoma. Cancer Research. 2006; 66(21):10586-10593.
73. Alderson RF, Kreitman RJ, Chen T, Yeung P, Herbst R, Fox JA and Pastan I. CAT-8015: A Second-Generation Pseudomonas Exotoxin A-Based Immunotherapy Targeting CD22-Expressing Hematologic Malignancies. Clinical Cancer Research. 2009; 15(3):832-839.
74. Mussai F, Campana D, Bhojwani D, Stetler-Stevenson M, Steinberg SM, Wayne AS and Pastan I. Cytotoxicity of the anti-CD22 immunotoxin HA22 (CAT-8015) against paediatric acute lymphoblastic leukaemia. British Journal of Haematology. 2010; 150(3):352-358.
75. Wei H, Xiang L, Wayne AS, Chertov O, FitzGerald DJ, Bera TK and Pastan I. Immunotoxin resistance via reversible methylation of the DPH4 promoter is a unique survival strategy. Proceedings of the National Academy of Sciences. 2012; 109(18):6898-6903.
76. Kreitman RJ, Tallman MS, Robak T, Coutre S, Wilson WH, Stetler-Stevenson M, FitzGerald DJ, Lechleider R and Pastan I. Phase I Trial of Anti-CD22 Recombinant Immunotoxin Moxetumomab Pasudotox (CAT-8015 or HA22) in Patients With Hairy Cell Leukemia. Journal of Clinical Oncology. 2012; 30(15):1822-1828.
77. Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, Roy S, Sridhara R, Rahman A, Williams G and Pazdur R. Approval Summary. Clinical Cancer Research. 2001; 7(6):1490-1496.
78. Estey E. Treatment of AML: resurrection for gemtuzumab ozogamicin? Lancet. 2012; 379(9825):1468-1469.
79. Castaigne S, Pautas C, Terre C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, de Revel T, Gastaud L, de Gunzburg N, Contentin N, Henry E, Marolleau JP, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012; 379(9825):1508-1516.
80. Burnett AK, Russell NH, Hills RK, Kell J, Freeman S, Kjeldsen L, Hunter AE, Yin J, Craddock CF, Dufva IH, Wheatley K and Milligan D. Addition of Gemtuzumab Ozogamicin to Induction Chemotherapy Improves Survival in Older Patients With Acute Myeloid Leukemia. Journal of Clinical Oncology. 2012; 30(32):3924-3931.
81. DiJoseph JF, Popplewell A, Tickle S, Ladyman H, Lawson A, Kunz A, Khandke K, Armellino DC, Boghaert ER, Hamann PR, Zinkewich-Peotti K, Stephens S, Weir N and Damle NK. Antibody-targeted chemotherapy of B-cell lymphoma using calicheamicin conjugated to murine or humanized antibody against CD22. Cancer Immunology, Immunotherapy. 2005; 54(1):11-24.
82. DiJoseph JF, Armellino DC, Boghaert ER, Khandke K, Dougher MM, Sridharan L, Kunz A, Hamann PR, Gorovits B, Udata C, Moran JK, Popplewell AG, Stephens S, Frost P and Damle NK. Antibody-targeted chemotherapy with CMC-544: a CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood. 2004; 103(5):1807-1814.
83. DiJoseph JF, Goad ME, Dougher MM, Boghaert ER, Kunz A, Hamann PR and Damle NK. Potent and Specific Antitumor Efficacy of CMC-544, a CD22-Targeted Immunoconjugate of Calicheamicin, against Systemically Disseminated B-Cell Lymphoma. Clinical Cancer Research. 2004; 10(24):8620-8629.
84. DiJoseph JF, Dougher MM, Kalyandrug LB, Armellino DC, Boghaert ER, Hamann PR, Moran JK and Damle NK. Antitumor Efficacy of a Combination of CMC-544 (Inotuzumab Ozogamicin), a CD22-Targeted Cytotoxic Immunoconjugate of Calicheamicin, and Rituximab against Non-Hodgkin’s B-Cell Lymphoma. Clinical Cancer Research. 2006; 12(1):242-249.
85. DiJoseph JF, Dougher MM, Armellino DC, Evans DY and Damle NK. Therapeutic potential of CD22-specific antibody-targeted chemotherapy using inotuzumab ozogamicin (CMC-544) for the treatment of acute lymphoblastic leukemia. Leukemia. 2007; 21(11):2240-2245.
86. DiJoseph J, Dougher M, Evans D, Zhou B-B and Damle N. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemotherapy and Pharmacology. 2011; 67(4):741-749.
87. Takeshita A, Shinjo K, Yamakage N, Ono T, Hirano I, Matsui H, Shigeno K, Nakamura S, Tobita T, Maekawa M, Ohnishi K, Sugimoto Y, Kiyoi H, Naoe T and Ohno R. CMC-544 (inotuzumab ozogamicin) shows less effect on multidrug resistant cells: analyses in cell lines and cells from patients with B-cell chronic lymphocytic leukaemia and lymphoma. British Journal of Haematology. 2009; 146(1):34-43.
88. Takeshita A, Yamakage N, Shinjo K, Ono T, Hirano I, Nakamura S, Shigeno K, Tobita T, Maekawa M, Kiyoi H, Naoe T, Ohnishi K, Sugimoto Y and Ohno R. CMC-544 (inotuzumab ozogamicin), an anti-CD22 immuno-conjugate of calicheamicin, alters the levels of target molecules of malignant B-cells. Leukemia. 2009; 23(7):1329-1336.
89. de Vries JF, Zwaan CM, De Bie M, Voerman JSA, den Boer ML, van Dongen JJM and van der Velden VHJ. The novel calicheamicin-conjugated CD22 antibody inotuzumab ozogamicin (CMC-544) effectively kills primary pediatric acute lymphoblastic leukemia cells. Leukemia. 2012; 26(2):255-264.
90. Advani A, Coiffier B, Czuczman MS, Dreyling M, Foran J, Gine E, Gisselbrecht C, Ketterer N, Nasta S, Rohatiner A, Schmidt-Wolf IGH, Schuler M, Sierra J, Smith MR, Verhoef G, Winter JN, et al. Safety, Pharmacokinetics, and Preliminary Clinical Activity of Inotuzumab Ozogamicin, a Novel Immunoconjugate for the Treatment of B-Cell Non-Hodgkin’s Lymphoma: Results of a Phase I Study. Journal of Clinical Oncology. 2010; 28(12):2085-2093.
91. Ogura M, Tobinai K, Hatake K, Uchida T, Kasai M, Oyama T, Suzuki T, Kobayashi Y, Watanabe T, Azuma T, Mori M, Terui Y, Yokoyama M, Mishima Y, Takahashi S, Ono C, et al. Phase I study of inotuzumab ozogamicin (CMC-544) in Japanese patients with follicular lymphoma pretreated with rituximab-based therapy. Cancer Science. 2010; 101(8):1840-1845.
92. Ogura M, Hatake K, Ando K, Tobinai K, Tokushige K, Ono C, Ishibashi T and Vandendries E. Phase I study of anti-CD22 immunoconjugate inotuzumab ozogamicin plus rituximab in relapsed/refractory B-cell non-Hodgkin lymphoma. Cancer Science. 2012; 103(5):933-938.
93. Fayad L, Offner F, Smith MR, Verhoef G, Johnson P, Kaufman JL, Rohatiner A, Advani A, Foran J, Hess G, Coiffier B, Czuczman M, Gine E, Durrant S, Kneissl M, Luu KT, et al. Safety and Clinical Activity of a Combination Therapy Comprising Two Antibody-Based Targeting Agents for the Treatment of Non-Hodgkin Lymphoma: Results of a Phase I/II Study Evaluating the Immunoconjugate Inotuzumab Ozogamicin With Rituximab. Journal of Clinical Oncology. 2013.
94. Kantarjian H, Thomas D, Jorgensen J, Jabbour E, Kebriaei P, Rytting M, York S, Ravandi F, Kwari M, Faderl S, Rios MB, Cortes J, Fayad L, Tarnai R, Wang SA, Champlin R, et al. Inotuzumab ozogamicin, an anti-CD22?calecheamicin conjugate, for refractory and relapsed acute lymphocytic leukaemia: a phase 2 study. The Lancet Oncology. 2012; 13(4):403-411.
95. von Deimling A, Sahm F and Capper D. Mutation specific antibodies: tool or dinosaur? Oncotarget. 2012; 3(9):907-908.
96. Aurisicchio L, Marra E, Roscilli G, Mancini R and Ciliberto G. The promise of anti-ErbB3 monoclonals as new cancer therapeutics. Oncotarget. 2012; 3(8):744-758.
97. Guo K, Tang JP, Li J, Tan CP, Al-Aidaroos AQO, Hong CW, Varghese L, Feng ZW, Park JE, Zhou JB, Chng WJ and Zeng Q. Engineering the First Chimeric Antibody in Targeting Intracellular PRL-3 Oncoprotein for Cancer Therapy in Mice. Oncotarget 2012; 3(2):158-171.
98. Schlaak M, Schmidt P, Bangard C, Kurschat P, Mauch C and Abken H. Regression of metastatic melanoma in a patient by antibody targeting of cancer stem cells. Oncotarget. 2012; 3(1):22-30.