
INTRODUCTION
Esophageal cancer is a fast-growing and deadly disease. It presents most often as esophageal squamous cell carcinoma (ESCC), which accounts for 95 % of esophageal cancer worldwide [1–2]. The standard treatment for locally advanced esophageal cancer is concurrent chemoradiotherapy (CCRT) with or without surgery. Patients with esophageal cancer enjoy better survival once they have had a good response to neoadjuvant therapy [3]. Unfortunately, there is no effective targeted therapeutic strategy available for ESCC and the prognosis for esophageal cancer is relatively poor with an average 5-year survival rate of less than 20 % [1, 4–5].
HER2/Neu belongs to the epidermal growth factor receptor (EGFR/ErbB) family. It is an important drug target for cancers, including breast cancer and gastroesophageal (GE) junction adenocarcinoma [6–7]. HER2 inhibition in combination with cisplatin-based chemotherapy has been demonstrated to significantly improve response rate and overall or progression-free survival in patients with adenocarcinoma of the gastro-esophageal junction or the stomach who over-express HER2 [7]. In studies of ESCC, however, the rate of HER2 over-expression has been reported to be less than 10 % [8–10] and the efficacy of HER2 targeted drugs in ESCC has not been demonstrated.
AXL (also call Ark or Ufo) is a receptor tyrosine kinase belonging to the Tyro3/Axl/Mer (TAM) family [11]. Activation of AXL receptors initiates signaling pathways involved in multiple cellular events, including cell survival, anti-apoptosis, proliferation, migration, and cytokine production [12]. AXL is ubiquitously expressed in cells and organs, and its over-expression has been reported in a wide array of human cancers, including breast [13], lung [14], liver [15], colon [16], and esophageal adenocarcinoma [17]. It has also been found to be an important biomarker for prognosis of cancer. Up-regulation of AXL is associated with poor survival of breast cancer [18], lung adenocarcinoma [14], and acute myeloid leukemia [19].
AXL has been reported as an adverse prognostic factor and a therapeutic target in esophageal adenocarcinoma (EAC) [20]. Knockdown of AXL inhibited invasion and migration of EAC cells, and the AXL inhibitor R428 significantly reduced invasion and migration of EAC cells [20]. AXL also mediates TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) and cisplatin resistance in EAC [21–23]. The crucial role of AXL in tumorigenesis and drug resistance in ESCC has been clearly demonstrated only very recently [24–25]. AXL was found to be consistently over-expressed in ESCC cells and human tumor samples [24]. Knockdown of AXL expression was shown to inhibit cell proliferation, survival, migration and invasion both in vitro and in vivo [24]. The tumorigenic function of AXL is mediated by activation of the Akt/NF-κB and Akt/GSK3β pathways [24]. Over-expression of AXL also mediates resistance to treatment with the phosphoinositide -3-kinase-alpha (PI3Kα) inhibitor BYL719 by activating the EGFR/PKC/mTOR axis in ESCC [25]. Resistance to PI3Kα can be reversed by combined treatment with AXL, EGFR, and PKC inhibitors [25].
HER2-targeted agents, including trastuzumab and lapatinib, are a promising targeted therapy, especially in treating breast cancer. Over-expression of AXL has been shown to be a novel mechanism of acquired resistance to HER2-targeted agents in lapatinib-resistant, HER2-positive breast cancer clones [26]. Foretinib (XL880, GSK1363089), an oral multi-kinase inhibitor acting on AXL, c-Met, RON and VEGFR-2, can restore sensitivities to lapatinib and trastuzumab in resistant cells [26]. Synergistic effects of foretinib with HER-targets have been demonstrated in MET and HER1/2 co-activated cells [27]. Meanwhile, the AXL inhibitor BMS777607 and HER2 inhibitor lapatinib exhibit a synergistic cytotoxic effect in breast and ovarian cancer cells [28]. However, the prognostic role of co-expression of AXL and HER2 in cancer cells has hardly been investigated.
Although the molecular function of AXL in ESCC has been demonstrated, clinically there is still a lack of evidence to support the prognostic significance of AXL in ESCC. In our study, we investigated the prognostic relevance of AXL and HER2 expression in operable ESCC patients (116 cases) and the efficacy of the AXL inhibitor, foretinib [29], in wild type and HER2-resistant ESCC cells.
RESULTS
A total of 116 patients who were diagnosed with ESCC and received surgical resection were enrolled in this study. In this cohort, 107 patients (92.2 %) were male and 1 (0.9 %), 25 (21.5%), 54 (46.6%), and 36 (31.0%) were diagnosed with pathologic stage 0, I, II, and III disease, respectively. A total of 75 patients (64.6 %) were treated with CCRT (concurrent chemoradiotherapy) (Table 1). As expected, both pathologic stage and T-stage (tumor stage) were significantly correlated with both survival and recurrence status of patients (P=0.001 for pathologic stage and survival; P<0.001 for pathologic stage and recurrence; P=0.003 for T-stage and survival and P=0.004 for T-stage and recurrence, Table 1). There were also statistically significant differences in the distributions of sex and CCRT treatment by survival and recurrence status (P=0.004 and P=0.023 respectively for survival; P=0.001 and P=0.013 respectively for recurrence, Table 1). A total of 93 patients (80.2 %) exhibited positive expression of AXL in tumor tissue. Significant differences in mortality and disease recurrence status were also observed between AXL-positive patients and AXL-negative patients (Table 1).
Table 1: Demographic and clinical characteristics of ESCC patients by survival and recurrence status
Survival |
Recurrence |
||||||
|---|---|---|---|---|---|---|---|
Variables |
Total |
Alive |
Dead |
p-value |
no recurrence |
recurrence |
p-value |
26 (22.4) |
90 (77.6) |
21 (18.1) |
95 (81.9) |
||||
Age (years) |
0.299 |
0.104 |
|||||
<40 |
9(7.8) |
4 (44.4) |
5 (55.6) |
4 (44.4) |
5 (55.6) |
||
40-60 |
61 (52.2) |
13 (21.3) |
48 (78.7) |
9 (14.8) |
52 (85.2) |
||
>60 |
46 (39.7) |
9 (19.6) |
37 (80.4) |
8 (17.4) |
38 (82.6) |
||
Sex |
0.004 |
0.001 |
|||||
Male |
107 (92.2) |
20 (18.7) |
87 (81.3) |
15 (14.0) |
92 (86.0) |
||
Female |
9 (7.8) |
6 (66.7) |
3 (33.3) |
6 (66.7) |
3 (33.3) |
||
Stage |
0.001 |
<0.001 |
|||||
0 |
1 (0.9) |
1 (100) |
0 (0) |
1 (100) |
0 (0) |
||
I |
25 (21.5) |
12 (48.0) |
13 (52.0) |
11 (44.0) |
14 (56.0) |
||
II |
54 (46.6) |
10 (18.5) |
44 (81.5) |
7 (13.0) |
47 (87.0) |
||
III |
36 (31.0) |
3 (8.3) |
33 (91.7) |
2 (5.6) |
34 (94.4) |
||
T-stage |
0.003 |
0.004 |
|||||
0 |
1 (0.9) |
1 (100) |
0 (0) |
1 (100) |
0 (0) |
||
1 |
32 (27.6) |
14 (43.8) |
18 (56.3) |
12 (37.5) |
20 (62.5) |
||
2 |
33 (28.4) |
5 (15.2) |
28 (84.8) |
4 (12.1) |
29 (87.9) |
||
3 |
44 (37.9) |
6 (13.6) |
38 (86.4) |
4 (9.1) |
40 (90.9) |
||
4 |
6 (5.2) |
0 (0) |
6 (100.0) |
0 (0) |
6 (100.0) |
||
N-stage |
0.195 |
0.069 |
|||||
0 |
65 (56.0) |
19 (29.2) |
46 (70.8) |
17 (26.2) |
48 (73.8) |
||
1 |
45 (38.8) |
6 (13.3) |
39 (86.7) |
4 (8.9) |
41 (91.1) |
||
2 |
5 (4.3) |
1 (20.0) |
4 (80.0) |
0 (0) |
5 (100) |
||
3 |
1 (0.9) |
0 (0) |
1 (100) |
0 (0) |
1 (100) |
||
Tumor location |
0.409 |
0.424 |
|||||
Upper |
24 (20.7) |
3 (12.5) |
21 (87.5) |
2 (8.3) |
22 (91.7) |
||
Middle |
48 (41.4) |
11 (22.9) |
37 (77.1) |
10 (20.8) |
38 (79.2) |
||
Lower |
44 (37.9) |
12 (27.3) |
32 (72.7) |
9 (20.5) |
35 (79.5) |
||
CCRT |
0.023 |
0.013 |
|||||
No |
38 (32.8) |
14 (36.8) |
24 (63.2) |
12 (31.6) |
26 (68.4) |
||
Yes |
75 (64.6) |
11 (14.7) |
64 (85.3) |
8 (10.7) |
67 (89.3) |
||
CT or RT |
3 (2.6) |
1 (33.3) |
2 (66.7) |
1 (33.3) |
2 (66.7) |
||
AXL expression |
0.002 |
0.020 |
|||||
negative |
23 (19.8) |
11 (47.8) |
12 (52.2) |
8 (34.8) |
15 (65.2) |
||
positive |
93 (80.2) |
15 (16.1) |
78 (83.9) |
13 (14.0) |
80 (86.0) |
||
To analyze the correlation between AXL expression and the prognosis of ESCC patients, we detected AXL expression in cancerous and non-cancerous esophageal tissues by IHC and correlated it with overall survival (OS) and progression-free survival (PFS) by multivariate Cox regression analysis. Expression levels were scored as 0, 1+, 2+, and 3+ (Figure 1A). AXL expression was absent in 19.8 % of the ESCC tissue samples (score=0, 23/116); 48.3 % (56/116) of ESCC tissues showed faint reactivity (score=1+); 24.1 % (28/116) moderate reactivity (score=2+); and 7.8 % (9/116) diffuse and strong reactivity (score=3) (Figure 1A and Table 2). Notably, the expression levels of AXL were significantly elevated in cancerous tissue compared to non-cancerous tissue (P<0.001, one-way ANOVA, Scheffe’s test, Figure 1C). Advanced tumor tissue (stage II and III) also showed increased expression of AXL compared to levels in early-stage tumor tissues, but the increase did not reach statistical significance (P=0.102, one-way ANOVA, Scheffe’s test, Figure 1C). Expression of AXL was significantly associated with increased risk of death (HR [95 % CI]=2.09 [1.09-4.04], P=0.028, Table 2) and borderline significantly correlated with disease progression (P=0.062, Table 2). Patients with strong AXL expression (score=3+) exhibited about a 3-fold higher risk of mortality and disease progression (HR [95 % CI]=2.98[1.17-7.58], P=0.022 for OS; HR [95 % CI]=2.79 [1.14-6.84], P=0.025 for PFS, Table 2). Faint expression of AXL in non-cancerous (normal, 1+, 5.6 % [5/89]) esophageal tissue also significantly correlated with increased risks of death and disease recurrence (HR [95 % CI]=3.63 [1.29-10.29], P=0.015 for OS; HR [95 % CI]=2.72 [1.01-7.33], P=0.048 for PFS, Table 2).
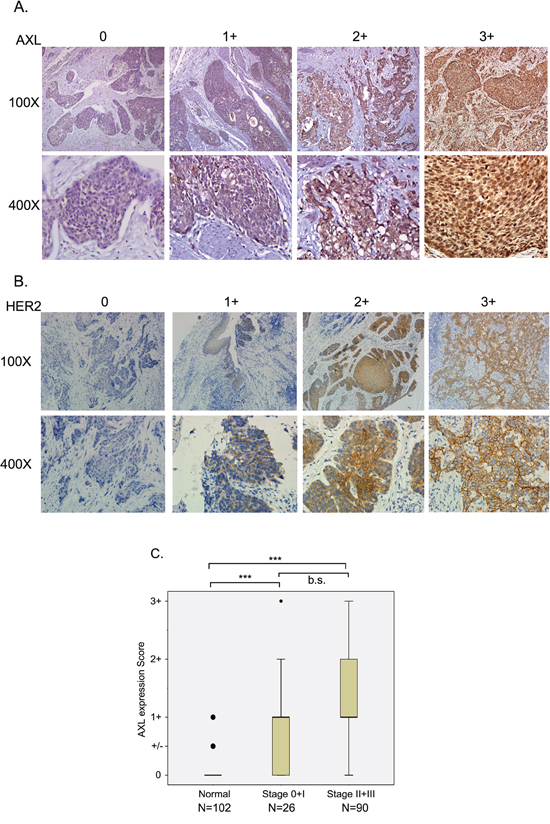
Figure 1: Expression of AXL and HER2 in ESCC tissue specimens. Immuno-histochemical analysis of AXL A. or HER2 B. expression in ESCC tissues is shown by expression level. A. Score 0, no reactivity in any tumor cell; Score 1+, tumor cells cluster with a faint reactivity irrespective of tumor cells stained; Score 2+, tumor cells cluster with a moderate reactivity irrespective of tumor cells stained; Score 3+, tumor cells cluster with a diffuse and strong reactivity. B. Score 0, no reactivity or no membranous reactivity in any tumor cell; Score 1+, tumor cells with a faint or barely perceptible membranous reactivity irrespective of percentage of tumor cells stained; Score 2+, tumor cells cluster with a weak-to-moderate complete membranous reactivity irrespective of percentage of tumor cells stained; Score 3+, tumor cells cluster with a strong complete membranous reactivity irrespective percentage of tumor cells stained. C. AXL expression level was analyzed in adjacent non-cancerous (normal), stage 0 and 1, and stage II and III ESCC tissues. ***, P<0.001; b.s., borderline significance.
Table 2: Association of AXL and HER2 expression with overall and progression-free survival of ESCC patients under multivariate analysis
Variables |
N |
Overall survival |
*P-value |
Progression-free survival |
*P-value |
|---|---|---|---|---|---|
Tumor_AXL expression |
|||||
0 |
23 (19.8) |
1 |
1 |
||
1+ |
56 (48.3) |
2.24 (1.14 (4.42) |
0.020 |
1.82 (0.99-3.35) |
0.056 |
2+ |
28 (24.1) |
1.60 (0.75-3.39) |
0.224 |
1.42 (0.72-2.81) |
0.318 |
3+ |
9 (7.8) |
2.98 (1.17-7.58) |
0.022 |
2.79 (1.14-6.84) |
0.025 |
Negative |
23 (19.8) |
1 |
1 |
||
Positive |
93 (80.2) |
2.09 (1.09-4.04) |
0.028 |
1.75 (0.97-3.16) |
0.062 |
Normal_AXL expression |
|||||
0 |
73 (82.0) |
1 |
1 |
||
+/- |
11 (12.4) |
1.34 (0.64-2.80) |
0.444 |
1.45 (0.71-2.95) |
0.306 |
1+ |
5 (5.6) |
3.63 (1.29-10.29) |
0.015 |
2.72 (1.01-7.33) |
0.048 |
Tumor_HER2 expression |
|||||
0 |
90 (81.8) |
1 |
1 |
||
1 |
17 (15.5) |
1.47 (0.79-2.76) |
0.228 |
1.54 (0.83-2.86) |
0.173 |
2 |
2 (1.8) |
3.00 (0.69-13.12) |
0.144 |
3.19 (0.73-13.90) |
0.123 |
3 |
1 (0.9) |
2.20 (0.27-17.73) |
0.458 |
1.42 (0.18-11.23) |
0.740 |
Negative |
90 (81.8) |
1 |
1 |
||
Positive |
20 (18.2) |
1.59 (0.89-2.86) |
0.118 |
1.62 (0.91-2.89) |
0.101 |
Tumor_AXL_HER2_expression |
|||||
AXL (-) and HER2 (-) |
17 (15.5) |
1 |
1 |
||
AXL (+) or HER2 (+) |
77 (70.0) |
1.87 (0.91-3.88) |
0.091 |
1.54 (0.79-3.00) |
0.203 |
AXL (+) and HER2 (+) |
16 (14.5) |
3.43 (1.40-8.42) |
0.007 |
3.19 (1.37-7.45) |
0.007 |
*adjusted for stage, age, sex and CCRT
The functional interplay between AXL and HER2 (ErbB2/Neu) has been suggested in previous studies [20, 26]. We also analyzed the prognostic relevance of HER2 in our ESCC subjects (N=110) by IHC. Expression levels of HER2 in ESCC were scored as 0, 1, 2, and 3 (Figure 1B). Only 18.2 % (20/110) of ESCC tissues had positive IHC staining and expression of HER2 did not correlate with either risk of death or of recurrence (Table 2). Cumulative analysis of the effects of expression of AXL and HER2 on prognosis revealed that co-expression of AXL and HER2 notably increased the hazards of both death and recurrence by more than 3-fold (HR [95 % CI]=3.43 [1.40-8.42], P=0.007 for OS; HR [95 % CI]=3.19[1.37-7.45], P=0.007 for PFS, Table 2).
The prognostic relevance of AXL and HER2 in ESCC were also analyzed with Kaplan-Meier estimates. Both OS and PFS differed significantly between patients with different levels of AXL expression (median survival time [MST] = 47.8 vs. 13.4 vs. 12.3 vs. 7.7 months in tissues expressing levels 0, 1+, 2+, 3+ of AXL, log-rank P=0.008 for OS, Figure 2A; MST= 33.0 vs. 8.6 vs. 7.5 vs. 7.7 months in tissues expressing levels 0, 1+, 2+, 3+ of AXL, log-rank P=0.040 for PFS, Figure 2B). Patients with both AXL and HER2 expression exhibited significantly shorter OS and PFS (MST= 47.8 vs. 13.4 vs. 11.6 months in the AXL (-) and HER2 (-), AXL (+) or HER2 (+), and HER2 (+) and AXL (+) subgroups, log-rank P=0.002 for OS, Figure 2C; MST= 26.5 vs. 8.6 vs. 5.3 months in the AXL (-) and HER2 (-), AXL (+) or HER2 (+), and HER2 (+) and AXL (+) subgroups, log-rank P=0.005 for PFS, Figure 2D).
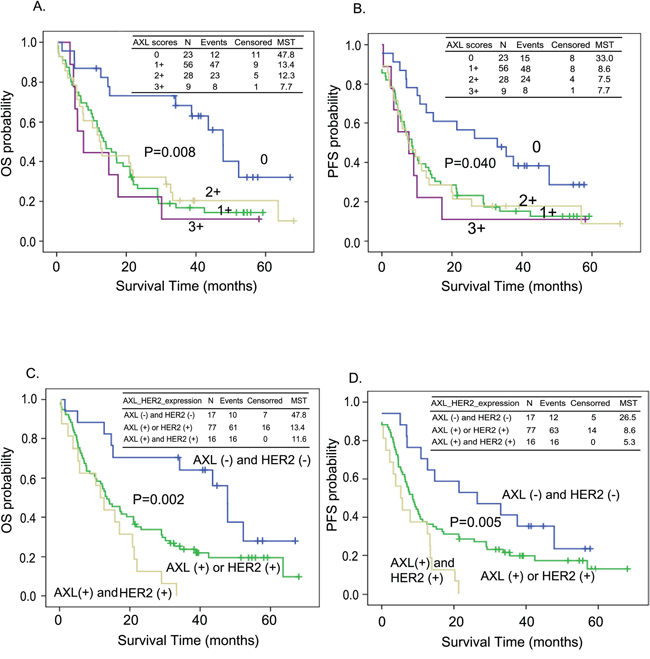
Figure 2: Kaplan-Meier estimates of overall survival (OS, A and C) or progression-free survival (PFS, B and D) by the expression level of AXL only A. and B. or both AXL and HER2 C. and D. MST: median survival time (months).
Patients with expression of AXL in their tumor tissue showed significantly increased risk of distant metastasis, a 3.96 fold increase, compared to patients without AXL in their tumor tissue (OR [95 %CI]=3.96 (1.16-13.60), P=0.029, Table 3). Co-expression of AXL and HER2 also increased the risk of recurrence compared to patients with neither AXL nor HER2 expression, however, without reaching statistical significance (OR [95 % CI]=4.11(0.59-28.46), P=0.152, Table 3). Interestingly, the recurrence pattern of patients differed significantly according to expression of AXL in tumor tissue. Patients testing positive for AXL staining exhibited a significantly increased rate of distant metastasis (the proportion with no recurrence, local recurrence, and distant metastasis were 44.4 % vs. 16.7 % vs. 38.9 % in AXL-negative patients and 18.1 % vs. 8.3 % vs. 73.6 % in AXL-positive patients, respectively, P=0.017, Table 4). HER2 expression, however, was not associated with a different recurrence pattern (P=0.247, Table 4). Co-expression of AXL and HER2 were also associated with an increased incidence of distant metastasis compared to patients with neither AXL nor HER expression (the proportion with no recurrence, local recurrence, and distant metastasis were 1.7 % vs. 16.7 % vs. 41.7% in the AXL (-) and HER2 (-) group and 0 % vs. 25 % vs. 75 % in the AXL (+) and HER2 (+) group, respectively, P=0.012, Table 4). That is, both AXL expression alone and also AXL co-expression with HER2 were associated with increased risk of death and tumor recurrence in patients with ESCC.
Table 3: Association of AXL and HER2 expression with risk of distant metastasis of ESCC patients under multivariate analysis
Variables |
N |
Distant metastasis |
*P-value |
|---|---|---|---|
Tumor_AXL expression |
|||
Negative |
18 |
1 |
|
Positive |
72 |
3.96 (1.16-13.60) |
0.029 |
Tumor_AXL_HER2_expression |
|||
AXL (-) and HER2 (-) |
12 |
1 |
|
AXL (+) or HER2 (+) |
60 |
3.27 (0.80-13.34) |
0.099 |
AXL (+) and HER2 (+) |
12 |
4.11 (0.59-28.46) |
0.152 |
*adjusted for stage and CCRT
In the cell model, we used human ESCC cells cultured from the CE48T cell line to analyze the efficacy of lapatinib, a HER2 inhibitor, and of foretinib (GSK1363089), a multikinase inhibitor of AXL, c-Met and VEGFR-2 [29]. Figure 3A shows the relative cell viabilities of the ESCC cells in response to the indicated concentrations of inhibitors. ESCC cells were more sensitive to foretinib or foretinb plus lapatinib than to lapatinib alone. The IC50 values were 1.891 μM, 0.443 μM and 0.296 μM of lapatinib, foretinib, and lapatinib plus foretinib, respectively (Figure 3A). Synergistic effects of lapatinib and foretinib were demonstrated. The cell viabilities were decreased to 91% and 37% in cells treated with 1 μM of lapatinib and 1 μM of foretinib respectively. The viability was further inhibited to 27 % by combining 1 μM of lapatinib and 1 μM foretinib (Figure 3B). The efficacies of combined targeted therapies in ESCC cells were also examined in two other HER2-targeted drugs, afatinib [38] and AC480 (BMS599626) [39] (Figure 3C and 3D). Cytotoxicity of foretinib was also increased by treatment with either afatinib (Figure 3C) or AC480 (Figure 3D).
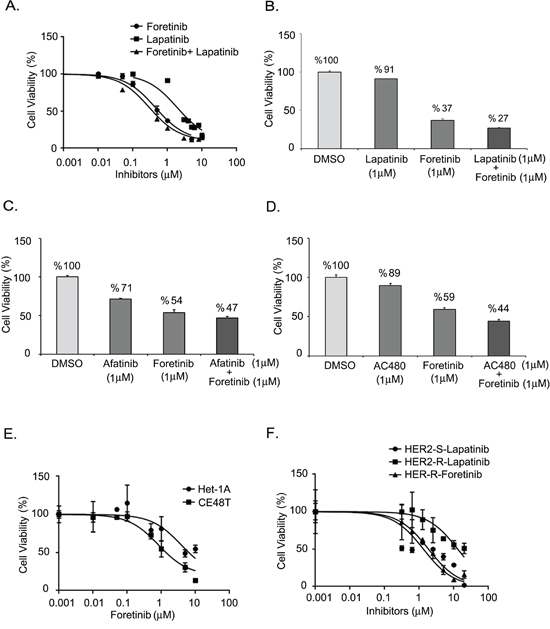
Figure 3: Effect of AXL and HER2 inhibitors on ESCC cells. A. Dose-inhibition curves of CE48T ESCC cells in response to indicated concentration of lapatinib, foretinib, or foretinib plus lapatinib. The respective IC50 values were 1.891 μM, 0.443 μM and 0.296 μM. B-D. Synergistic effects of foretinib and HER2 inhibitors on the cytotoxicities of ESCC. Cell viability of ESCC cells treated by 1 μM of foretinib, or 1 μM of HER2 inhibitors, or foretinib plus HER2 inhibitor. The HER2 inhibitors included 1 μM of lapatinib B. or 1 μM of afatinib C. or 1 μM of AC480 D. E. Relative cell viabilities of Het-1A and CE48T cells treated with indicated concentration of foretinib. The IC50 values were 3.836 μM vs. 0.823 μM for Het-1A and CE48T cells respectively. F. Dose-response curves for HER2-sensitive (HER2-S) and HER2-resistant (HER2-R) ESCC cells in response to increased concentrations of lapatinib or foretinib.
We also evaluated the sensitivity to foretinib in a non-cancerous esophageal cell line, Het-1A (Figure 3E). The IC50 of foretinib was 3.836 μM in Het-1A cells, which was more than 4-fold higher than the value in ESCC cells (0.823 μM). Lapatinib-resistant (HER2 resistant) sub-cells were selected from CE48T cells (Figure 3F) and the IC50 value of lapatinib for these cells was measured to be 10 times greater than for parental cells (IC50= 12.980 μM vs. 1.321 μM in HER2 resistant cells and in parental HER2 sensitive cells respectively). Interestingly, the HER2 resistant ESCC cells displayed sensitivity to foretinib (IC50= 1.856 μM). Notably, AXL expression was hardly detected in Het-1A normal esophageal cells and was increased in HER2-resistant cells compared to the expression level in parental ESCC cells (Figure 4A). The activation profiles of ERK (extracellular signal-regulated protein kinase) and AKT were also analyzed. The levels of phospho-ERK(pERK) and phospho-AKT (pAKT) were similar in parental and HER2-resistant CE48T cells (Figure 4A).
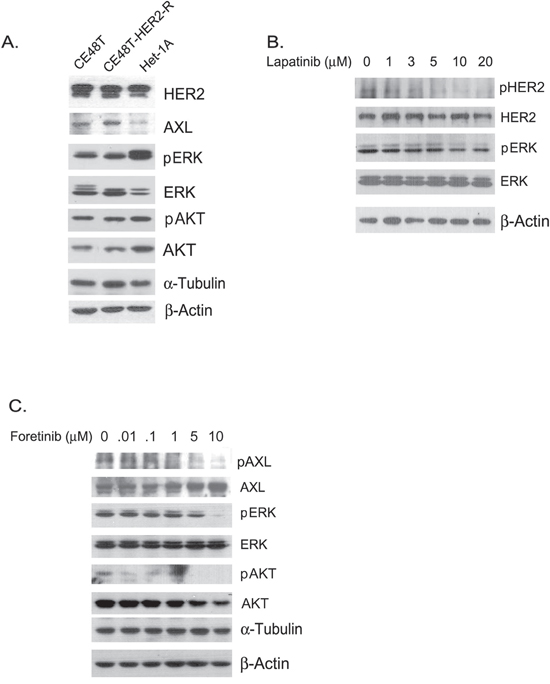
Figure 4: A. Protein expression profiles of CE48T, HER2-resistant CE48T cells (CE48T-HER2-R) and Het-1A cells. α-Tubulin served as a loading control for phospho-ERK (pERK) and ERK whereas β-Actin was used as a loading control for AXL, HER2, phospho-AKT (pAKT), and AKT. B. Phospho-HER2 (pHER2), HER2, pERK, and ERK expression in ESCC cells treated with indicated amounts of lapatinib for 24 hours. β-Actin served as a loading control. C. Phospho-AXL (pAXL), AXL, pERK, ERK, pAKT, and AKT expression in ESCC cells treated with the indicated amounts of foretinib for 24 hours. α-Tubulin served as a loading control for pERK and ERK; β-Actin served as a loading control for pAXL, AXL, pAKT, and AKT.
The activation profiles of downstream factors in response to lapatinib and foretinib in ESCC were analyzed. As expected, lapatinib dose-dependently and time-dependently inhibited tyrosine phosphorylation of ERK in ESCC cells (Figure 4B and Figure S1). Phosphorylation of HER2 also decreased with increased doses of lapatinib, which revealed the specific inhibition of HER2 activation in ESCC cells (Figure 4B). Oncogenic function of AXL has been demonstrated to function through activating Akt/NF-κB and Akt/GSK3β pathways [24]. In agreement with a previous report, we demonstrated that higher doses of foretinib inhibited phosphorylation of Akt in ESCC cells (Figure 4C). However, total AKT also decreased with increased amounts of foretinib (Figure 4C). Meanwhile, phosphorylation levels of AXL and ERK were also reduced with elevated amounts of foretinib (Figure 4C). Thus, foretinib might induce cytotoxicity of ESCC cells through specific inhibition of AXL activation and of the signaling cascades of PI3K/AKT and Ras/ERK.
DISCUSSION
ESCC is a deadly cancer with poor prognosis and no clinically approved targeted therapy. The EGFR (epidermal growth factor receptor)-targeted drugs, gefitinib (Iressa) and erlotinib (Tarceva) were evaluated in esophageal cancer in phase II and phase III studies [42–45]. Other targeting agents have been studied for treatment of ESCC, though most have been restricted to pre-clinical tests or phase I studies [46]. A growing body of studies has demonstrated the promise of AXL as a novel target for cancer targeted therapy, including in esophageal cancer. In our current study, we provide further clinical evidence of the oncogenic role of AXL in ESCC. We found that AXL expression was significantly elevated in tumor tissues and associated with pathological stage (Figure 1). Over-expression of AXL increased risk of death (Table 2 and Figure 2), and distant metastasis of ESCC (Table 3 and 4). Patients positive for AXL have about a 2-fold greater hazard of death and about a 4-fold increased risk of disease recurrence. The median survival time was also drastically decreased from 47 months to less than 14 months.
Table 4: Recurrence patterns of patients with different AXL and HER2 expression profiles
Variables |
N |
Recurrence pattern |
p-value |
||
|---|---|---|---|---|---|
No recurrence |
Local recurrence |
Distant metastasis |
|||
21 (23.3) |
9 (10.0) |
60 (66.7) |
|||
Tumor_AXL |
0.017 |
||||
Negative |
18 (20) |
8 (44.4) |
3 (16.7) |
7 (38.9) |
|
Positive |
72 (80) |
13 (18.1) |
6 (8.3) |
53 (73.6) |
|
Tumor_HER2 |
0.247 |
||||
Negative |
68 (81.0) |
17 (25.0) |
5 (7.4) |
46 (67.6) |
|
Positive |
18 (19.0) |
2 (12.5) |
3 (18.8) |
11 (68.8) |
|
Tumor_AXL and HER2 |
0.012 |
||||
AXL (-) and HER2 (-) |
12 (14.3) |
5 (41.7) |
2 (16.7) |
5 (41.7) |
|
AXL (+) or HER2 (+) |
60 (71.4) |
14 (23.3) |
3 (5.0) |
43 (71.7) |
|
AXL (+) and HER2 (+) |
12 (14.3) |
0 (0) |
3 (25.0) |
9 (75.0) |
|
In our operable ESCC patients, the rate of positive AXL expression in ESCC tissue was about 80 %, which was markedly higher than in adjacent normal esophageal tissue (5.6 %). AXL expression has been found to be induced by chemotherapy drugs in U937 acute myeloid leukemia cells [47]. About 65 % of our patients were treated with operative neoadjuvant CCRT, which makes reasonable the speculation that AXL expression may have been induced by CCRT. However, the rate of AXL-positive samples were not significantly different between patients who received CCRT and those who did not (positive and negative rates were 84.2 % vs. 15.8 % and 78.2 % vs. 21.8 % in the CCRT [-] and CCRT [+] groups respectively, P=0.446, Table S1). CCRT treatment did not correlate with the expression of HER2 in ESCC either (P=1.000, Table S1). Thus, CCRT was not a major factor in inducing the expression of AXL or HER2 in ESCC tissues.
AXL expression has been found to be induced by epithelial-mesenchymal transition (EMT) and to correlate with induction of EMT in breast cancer [18, 48] and in ESCC [49]. EMT is a critical factor in promoting metastasis. Our results demonstrated that AXL expression significantly correlated with incidence of distant metastasis, providing strong clinical support for the crucial role of AXL in ESCC metastasis.
AXL has also been suggested as a downstream effector of transforming growth factor-beta 1 (TGF-β1) during langerhans cell differentiation [50]. In hepatocellular carcinoma (HCC), the molecular collaboration of AXL/14-3-3ζ and TGF-β/Smad signaling on cancer progression has been demonstrated [51]. TGF-β1 is an upstream factor of EMT [52] and has been shown to be induced by environmental carcinogens, such as tobacco smoking [53], alcohol drinking [54] and betel nut chewing [55–56]. Incidence of ESCC is well-known to highly correlate with these environmental carcinogens. Among our study population, over 90 % of patients have at least one of these unfavorable habits. Interestingly, in our patients, 90.5 % of betel nut chewers were positive for AXL in tumor tissue, which was significantly higher than in non-chewers (P=0.029, data not shown). Such results suggest that these environmental carcinogens might increase the level of TGF-β1 and thus contribute to the over-expression of AXL in esophageal tissue.
Expression of HER2 is highly correlated with the prognosis of esophageal adenocarcinom [6], while the prognostic role in ESCC has hardly been investigated. We found that about 20 % of our patients were positive for HER2. Even though expression of HER2 alone did not associate with the clinical outcome of ESCC, cumulative expression of AXL and HER2 showed significant clinical impact on ESCC. AXL expression was increased in HER2-resistant ESCC cells compared to expression in HER2-sensitive ESCC cells revealing the interplay of AXL and HER2 in ESCC.
The activity of foretinib was demonstrated in a phase II trial in patients with advanced papillary renal cell carcinoma (PRCC), especially in those with germline MET mutations [57]. Because c-Met is also an adverse prognostic factor for ESCC [58], we suggest foretinib has great potential for ESCC targeted therapy in patients over-expressing AXL or c-Met. The synergistic cytotoxicity of foretinib with HER2 inhibitors, including lapatinib, afatinib, and AC480 have also been demonstrated in ESCC cells. Combination therapy of AXL and HER2 inhibitors is, therefore, a possible direction in ESCC patients co-overexpressing AXL and HER2.
Collectively, our results provide clinical evidence that AXL is a strong adverse prognostic factor, which is significantly correlated with pathological stage, overall survival, and distant metastasis of operable ESCC. Therapeutic agents targeting AXL, therefore, have great potential to improve the clinical outcome of operable ESCC.
MATERIALS AND METHODS
Study population
Our study subjects were collected in the pathological and surgical department of National Taiwan University Hospital from 2005 to 2013. The consent procedure of the clinical study was approved by the Research Ethics Committee of National Taiwan University Hospital (201402056RINA). The inclusion criteria were patients aged above 20 years who were histologically confirmed with primary ESCC and received surgical resection. The exclusion criteria were pregnant women, those unwilling to give informed consent, and those without complete clinical records. Surgical resection of esophagectomy with two or three field lymph node dissection and esophageal reconstruction were performed in all of the recruited patients.
CCRT was administered to patients with advanced TNM stages (IIb or more advanced) diagnosed by endoscopic ultrasound or computed tomography before surgery. The CCRT procedures were described in a previous study [30–31]. Information regarding the demographics, disease characteristics (tumor location, TNM stage), histology, clinical treatments, recurrence status, and survival were obtained from the Tumor Registry of the National Taiwan University Hospital and/or medical chart-review. The TNM staging of the patients receiving CCRT was re-staged according to the pathological reports after surgery [32]. Overall survival duration (OS) was defined as the interval between esophagectomy and either death from disease or last follow-up. Progression-free survival (PFS) was defined as the interval between esophagectomy (complete resection) and detection of local recurrence or distant metastasis of the tumor.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded (FFPE) blocks with ESCC and adjacent non-tumor tissue samples were obtained from the Department of Pathology in our hospital. Briefly, FFPE tissue sections (4 μm) were dewaxed and rehydrated. After antigen retrieval at 37°C for 2 hours in a Ventana BenchMark XT processor (Ventana, Tucson, AZ), tissue sections on the slides were incubated with primary antibodies. After reincubation with polymer-horseradish peroxidase reagent, the sections were visualized with Ventana ultraview DAB (diaminobenzidine 3,3′ tetrahydrochloride) detection kit according to the manufacturer’s protocol. The primary antibodies used were a rabbit polyclonal antibody against AXL (1:100, ab72069, abcam) and HER2 (ready- to-use, DAKO). The tissue slides were then counterstained with hematoxylin.
Cell culture
CE48T/VGH (CE48T, BCRC 60165), a human ESCC cell line derived from a 57 year-old Taiwanese man [33–34], was cultured in RPMI complete medium. Het-1A, a non-tumorigenic primary esophageal squamous cell line, was transformed by SV40 T antigen [35] and was cultured on CellBind dishes (Corning) in BEGM medium (Lonza). All the cells were cultured at 37°C in an incubator containing 5% CO2.
Isolation of HER2-resistant ESCC cells
HER2-resistant ESCC sub-cells were selected by repeated treatment with the HER2 inhibitor lapatinib (Tykerb, synthesized by Selleckchem, S2111) [36] of different concentrations (5 to 20 μM/L). Briefly, the 50 % and 90 % inhibition concentrations (IC50 and IC90) of lapatinib for a 72 hour treatment period were determined. The parental cells were then treated with the IC90 dose for 24 hours. After removing the medium, the cells were then maintained and regrown in medium containing the IC50 dose of lapatinib. When cells covered 60-70 % of the dish surface, the cells were re-incubated with an increased concentration (1.5 fold) of the drug. Cells resistant to 20 μM/L of lapatinib were harvested and were subjected to cell viability assay.
Cell viability assay
The CE48T/VGH cells, lapatinib-resistant sub-cells and Het-1A cells were plated on 96-well plates (8000 cells/well) and treated with the indicated amounts of AXL inhibitor, foretinib [37] (GSK1363089, provided by Santa Cruz Biotechnology, Inc. SC-364492) or HER2 inhibitors. The HER2 inhibitors included lapatinib, afatinib [38] (Tovok, provided by Boehringer Ingelheim, Taiwan), and AC480 [39] (BMS599626, synthesized by Selleckchem, S1056). After incubation for 72 hours, cell survival was determined by MTT assay as described previously [40–41].
Protein extraction and western blotting
To detect protein expression in the esophageal cells, total protein was extracted from the cells by RIPA buffer (150 mM NaCl, 50 mM Tris–HCl [pH 7.5], 1 % Igepal-CA630, 0.5 % sodium deoxycholate, 0.1 % SDS, 50 mM NaF, 1 mM Na3VaO4, and complete protease inhibitor cocktail). Protein expression was analyzed by SDS-PAGE and western blotting with specific antibodies as described previously [40–41]. The primary antibodies for protein detection were anti-AXL rabbit polyclonal antibody (pAb) (ab72069, Abcam), anti-phospho-AXL (Tyr702, D12B2) rabbit monoclonal antibody (mAb) (#5724, Cell Signaling Technology), anti-HER2 rabbit antibody (#2242, Cell signaling), anti-phospho -HER2/ErbB2 (Tyr1221/1222, 6B12) pAb (#2243, Cell Signaling Technology), anti-Actin mAb (clone C4, Millipore), anti-ERK 1 (K-23) (Santa Cruz), anti-phospho-p44/42 Erk1/2 (Thr202/Tyr204) mAb (#4370, Cell Signaling Technology), anti-Akt antibody (#9272, Cell Signaling Technology), anti-phospho-Akt (Ser473) (#9271, Cell Signaling Technology), and anti-α-Tubulin (DM1A, Abcam).
Statistical analysis
The distribution of demographic variables, clinical characteristics, and indicated protein expression levels among the subgroups with different prognostic results were compared using a Pearson χ2 test or Fisher exact test (N≤ 5). Protein expression levels of esophageal tissues among non-tumor and tumor tissue of different stages were analyzed by one-way ANOVA and box-plot. Multivariate Cox regression analysis was used to evaluate the hazard ratios (HRs) of death and disease progression adjusted for potential significant clinical covariates and indicated protein expression. The odds ratios (ORs) obtained from logistic regression were used to describe association of distant metastasis and protein expression profiles. Data were expressed as mean value and 95% confidence interval (CI). Crude correlations between genotypes and OS or PFS were assessed using the Kaplan-Meier survival function and compared using the log-rank test. All statistical analyses were conducted with SPSS 17.0 (SPSS Institute, Chicago, IL). A p-value ≤ 0.05 was considered statistically significant. Sigmoidal dose-response curve and the corresponding IC50 and IC90 values were generated by Graph-Pad Prism software (Graph-Pad soft ware, Inc.).
ACKNOWLEDGMENTS
This study was supported by the Ministry of Science and Technology (MOST 103-2320-B-002-020; MOST 104-2314-B-002-182-MY3; MOST 104-2811-B-002-090), National Taiwan University Hospital (NTUH.104-S2650), Excellent Translational Medicine Research Projects of National Taiwan University College of Medicine and National Taiwan University Hospital, and Taiwan Health Foundation of the Republic of China. We thank the staff of the Second Core Lab, Department of Medical Research, NTUH for technical support.
CONFLICTS OF INTEREST
There is no conflict of interest for any author regarding the publication of this manuscript.
REFERENCES
1. Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010; 19:1893-1907.
2. Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006; 24:2137-2150.
3. Miyashita M, Tajiri T, Sasajima K, Makino H, Maruyama H, Nomura T, Futami R, Hagiwara N, Tsuchiya Y, Yamashita K. Response to preoperative chemotherapy affects prognosis in esophageal cancer. Dis Esophagus. 2003; 16:99-101.
4. Allum WH, Stenning SP, Bancewicz J, Clark PI, Langley RE. Long-term results of a randomized trial of surgery with or without preoperative chemotherapy in esophageal cancer. J Clin Oncol. 2009; 27:5062-5067.
5. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003; 349:2241-2252.
6. Almhanna K, Meredith KL, Hoffe SE, Shridhar R, Coppola D. Targeting the human epidermal growth factor receptor 2 in esophageal cancer. Cancer Control. 2013; 20:111-116.
7. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Ruschoff J, Kang YK. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010; 376:687-697.
8. Wei Q, Chen L, Sheng L, Nordgren H, Wester K, Carlsson J. EGFR, HER2 and HER3 expression in esophageal primary tumours and corresponding metastases. Int J Oncol. 2007; 31:493-499.
9. Huang JX, Zhao K, Lin M, Wang Q, Xiao W, Lin MS, Yu H, Chen P, Qian RY. HER2 gene amplification in esophageal squamous cell carcinoma is less than in gastroesophageal junction and gastric adenocarcinoma. Oncol Lett. 2013; 6:13-18.
10. Kato H, Arao T, Matsumoto K, Fujita Y, Kimura H, Hayashi H, Nishiki K, Iwama M, Shiraishi O, Yasuda A, Shinkai M, Imano M, Imamoto H, Yasuda T, Okuno K, Shiozaki H, et al. Gene amplification of EGFR, HER2, FGFR2 and MET in esophageal squamous cell carcinoma. Int J Oncol. 2013; 42:1151-1158.
11. O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, Espinosa R, 3rd, Le Beau MM, Earp HS, Liu ET. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991; 11:5016-5031.
12. Linger RMA, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008; 100:35-83.
13. Meric F, Lee WP, Sahin A, Zhang H, Kung HJ, Hung MC. Expression profile of tyrosine kinases in breast cancer. Clin Cancer Res. 2002; 8:361-367.
14. Shieh YS, Lai CY, Kao YR, Shiah SG, Chu YW, Lee HS, Wu CW. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005; 7:1058-1064.
15. Tsou AP, Wu KM, Tsen TY, Chi CW, Chiu JH, Lui WY, Hu CP, Chang C, Chou CK, Tsai SF. Parallel hybridization analysis of multiple protein kinase genes: identification of gene expression patterns characteristic of human hepatocellular carcinoma. Genomics. 1998; 50:331-340.
16. Craven RJ, Xu LH, Weiner TM, Fridell YW, Dent GA, Srivastava S, Varnum B, Liu ET, Cance WG. Receptor tyrosine kinases expressed in metastatic colon cancer. Int J Cancer. 1995; 60:791-797.
17. Nemoto T, Ohashi K, Akashi T, Johnson JD, Hirokawa K. Overexpression of protein tyrosine kinases in human esophageal cancer. Pathobiology. 1997; 65:195-203.
18. Gjerdrum C, Tiron C, Hoiby T, Stefansson I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT, Micklem DR, Akslen LA, Glackin C, Lorens JB. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. P Natl Acad Sci USA. 2010; 107:1124-1129.
19. Rochlitz C, Lohri A, Bacchi M, Schmidt M, Nagel S, Fopp M, Fey MF, Herrmann R, Neubauer A. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia. 1999; 13:1352-1358.
20. Alvarez H, Montgomery EA, Karikari C, Canto M, Dunbar KB, Wang JS, Feldmann G, Hong SM, Haffner MC, Meeker AK, Holland SJ, Yu JX, Heckrodt TJ, Zhang J, Ding PY, Goff D, et al. The Axl receptor tyrosine kinase is an adverse prognostic factor and a therapeutic target in esophageal adenocarcinoma. Cancer Biology & Therapy. 2010; 10:1009-1018.
21. Hong J, Peng DF, El-Rifai W, Belkhiri A. Regulation of Death-Inducing Signaling Complex by Axl Mediates TRAIL Resistance in Esophageal Adenocarcinoma. Gastroenterology. 2013; 144:S144-S144.
22. Hong J, Belkhiri A. AXL Mediates TRAIL Resistance in Esophageal Adenocarcinoma. Neoplasia. 2013; 15:296-304.
23. Hong J, Peng DF, Chen Z, Sehdev V, Belkhiri A. ABL Regulation by AXL Promotes Cisplatin Resistance in Esophageal Cancer. Cancer Research. 2013; 73:331-340.
24. Paccez JD, Duncan K, Vava A, Correa RG, Libermann TA, Parker MI, Zerbini LF. Inactivation of GSK3beta and activation of NF-kappaB pathway via Axl represents an important mediator of tumorigenesis in esophageal squamous cell carcinoma. Mol Biol Cell. 2015; 26:821-831.
25. Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, Benzaken AO, Rodon J, Morse N, Yan JJ, Liu M, Das R, Chen Y, Tam A, Wang H, Liang J, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015; 27:533-546.
26. Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin AM, Gilmer TM. Novel Mechanism of Lapatinib Resistance in HER2-Positive Breast Tumor Cells: Activation of AXL. Cancer Res. 2009; 69:6871-6878.
27. Liu L, Shi H, Liu Y, Anderson A, Peterson J, Greger J, Martin AM, Gilmer TM. Synergistic effects of foretinib with HER-targeted agents in MET and HER1- or HER2-coactivated tumor cells. Mol Cancer Ther. 2011; 10:518-530.
28. Torka R, Penzes K, Gusenbauer S, Baumann C, Szabadkai I, Orfi L, Keri G, Ullrich A. Activation of HER3 interferes with antitumor effects of Axl receptor tyrosine kinase inhibitors: suggestion of combination therapy. Neoplasia. 2014; 16:301-318.
29. Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, Lougheed J, Yakes FM, Bentzien F, Xu W, Zaks T, Wooster R, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009; 69:8009-8016.
30. Hsu FM, Lee YC, Lee JM, Hsu CH, Lin CC, Tsai YC, Wu JK, Cheng JC. Association of clinical and dosimetric factors with postoperative pulmonary complications in esophageal cancer patients receiving intensity-modulated radiation therapy and concurrent chemotherapy followed by thoracic esophagectomy. Ann Surg Oncol. 2009; 16:1669-1677.
31. Hsu FM, Lee JM, Huang PM, Lin CC, Hsu CH, Tsai YC, Lee YC, Chia-Hsien Cheng J. Retrospective analysis of outcome differences in preoperative concurrent chemoradiation with or without elective nodal irradiation for esophageal squamous cell carcinoma. Int J Radiat Oncol Biol Phys. 2011; 81:e593-599.
32. Korst RJ, Kansler AL, Port JL, Lee PC, Kerem Y, Altorki NK. Downstaging of T or N predicts long-term survival after preoperative chemotherapy and radical resection for esophageal carcinoma. Ann Thorac Surg. 2006; 82:480-484; discussion 484-485.
33. Hu CP, Hsieh HG, Chien KY, Wang PY, Wang CI, Chen CY, Lo SJ, Wuu KD, Chang CM. Biologic properties of three newly established human esophageal carcinoma cell lines. J Natl Cancer Inst. 1984; 72:577-583.
34. Wuu KD, Cheng MY, Wang-Wuu S, Hu CP, Chang CM. Chromosome analysis on a cell line (CE48T/VGH) derived from a human esophageal carcinoma. Cancer Genet Cytogenet. 1986; 20:279-285.
35. Stoner GD, Kaighn ME, Reddel RR, Resau JH, Bowman D, Naito Z, Matsukura N, You M, Galati AJ, Harris CC. Establishment and characterization of SV40 T-antigen immortalized human esophageal epithelial cells. Cancer Res. 1991; 51:365-371.
36. Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TF, Jr., Hudson-Curtis BL, Gilmer TM. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007; 40:580-594.
37. Zillhardt M, Park SM, Romero IL, Sawada K, Montag A, Krausz T, Yamada SD, Peter ME, Lengyel E. Foretinib (GSK1363089), an Orally Available Multikinase Inhibitor of c-Met and VEGFR-2, Blocks Proliferation, Induces Anoikis, and Impairs Ovarian Cancer Metastasis. Clin Cancer Res. 2011; 17:4042-4051.
38. Suzawa K, Toyooka S, Sakaguchi M, Morita M, Yamamoto H, Tomida S, Ohtsuka T, Watanabe M, Hashida S, Maki Y, Soh J, Asano H, Tsukuda K, Miyoshi S. Antitumor effect of afatinib, as a human epidermal growth factor receptor 2-targeted therapy, in lung cancers harboring HER2 oncogene alterations. Cancer Sci. 2016; 107:45-52.
39. Wong TW, Lee FY, Yu C, Luo FR, Oppenheimer S, Zhang HJ, Smykla RA, Mastalerz H, Fink BE, Hunt JT, Gavai AV, Vite GD. Preclinical antitumor activity of BMS-599626, a pan-HER kinase inhibitor that inhibits HER1/HER2 homodimer and heterodimer signaling. Clin Cancer Res. 2006; 12:6186-6193.
40. Yang PW, Hung MC, Hsieh CY, Tung EC, Wang YH, Tsai JC, Lee JM. The effects of Photofrin-mediated photodynamic therapy on the modulation of EGFR in esophageal squamous cell carcinoma cells. Lasers Med Sci. 2013; 28:605-614.
41. Yang PW, Chiang TH, Hsieh CY, Huang YC, Wong LF, Hung MC, Tsai JC, Lee JM. The effect of ephrin-A1 on resistance to Photofrin-mediated photodynamic therapy in esophageal squamous cell carcinoma cells. Lasers Med Sci. 2015; 30:2353-2361.
42. Ilson DH, Kelsen D, Shah M, Schwartz G, Levine DA, Boyd J, Capanu M, Miron B, Klimstra D. A Phase 2 Trial of Erlotinib in Patients With Previously Treated Squamous Cell and Adenocarcinoma of the Esophagus. Cancer. 2011; 117:1409-1414.
43. Dutton SJ, Ferry DR, Blazeby JM, Abbas H, Dahle-Smith A, Mansoor W, Thompson J, Harrison M, Chatterjee A, Falk S, Garcia-Alonso A, Fyfe DW, Hubner RA, Gamble T, Peachey L, Davoudianfar M, et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): a phase 3, multicentre, double-blind, placebo-controlled randomised trial. Lancet Oncol. 2014; 15:894-904.
44. Janmaat ML, Gallegos-Ruiz MI, Rodriguez JA, Meijer GA, Vervenne WL, Richel DJ, Van Groeningen C, Giaccone G. Predictive factors for outcome in a phase II study of gefitinib in second-line treatment of advanced esophageal cancer patients. Journal of Clinical Oncology. 2006; 24:1612-1619.
45. Rodriguez CP, Adelstein DJ, Rice TW, Rybicki LA, Videtic GMM, Saxton JP, Murthy SC, Mason DP, Ives DI. A Phase II Study of Perioperative Concurrent Chemotherapy, Gefitinib, and Hyperfractionated Radiation Followed by Maintenance Gefitinib in Locoregionally Advanced Esophagus and Gastroesophageal Junction Cancer. Journal of Thoracic Oncology. 2010; 5:229-235.
46. Digklia A, Voutsadakis IA. Targeted treatments for metastatic esophageal squamous cell cancer. World J Gastrointest Oncol. 2013; 5:88-96.
47. Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, Lai GM, Chuang SE. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 2008; 268:314-324.
48. Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB, Ivaska J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011; 30:1436-1448.
49. Paccez JD, Duncan K, Vava A, Correa RG, Libermann TA, Parker MI, Zerbini LF. Inactivation of GSK3 beta and activation of NF-kappa B pathway via Axl represents an important mediator of tumorigenesis in esophageal squamous cell carcinoma. Mol Biol Cell. 2015; 26:821-831.
50. Bauer T, Zagorska A, Jurkin J, Yasmin N, Koffel R, Richter S, Gesslbauer B, Lemke G, Strobl H. Identification of Axl as a downstream effector of TGF-beta 1 during Langerhans cell differentiation and epidermal homeostasis. J Exp Med. 2012; 209:2033-2047.
51. Reichl P, Dengler M, van Zijl F, Huber H, Fuhrlinger G, Reichel C, Sieghart W, Peck-Radosavljevic M, Grubinger M, Mikulits W. Axl activates autocrine transforming growth factor-beta signaling in hepatocellular carcinoma. Hepatology. 2015; 61:930-941.
52. Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007; 178:437-451.
53. Mak JCW, Chan-Yeung MMW, Ho SP, Chan KS, Choo K, Yee KS, Chau CH, Cheung AHK, Ip MSM, Study HKTSC. Elevated plasma TGF-beta(1) levels in patients with chronic obstructive pulmonary disease. Resp Med. 2009; 103:1083-1089.
54. Ciuclan L, Ehnert S, Ilkavets I, Weng HL, Gaitantzi H, Tsukamoto H, Ueberham E, Meindl-Beinker NM, Singer MV, Breitkopf K, Dooley S. TGF-beta enhances alcohol dependent hepatocyte damage via down-regulation of alcohol dehydrogenase I. J Hepatol. 2010; 52:407-416.
55. Pant I, Kumar N, Khan I, Rao SG, Kondaiah P. Role of Areca Nut Induced TGF-beta and Epithelial-Mesenchymal Interaction in the Pathogenesis of Oral Submucous Fibrosis. Plos One. 2015; 10.
56. Khan I, Kumar N, Pant I, Narra S, Kondaiah P. Activation of TGF-beta Pathway by Areca Nut Constituents: A Possible Cause of Oral Submucous Fibrosis. Plos One. 2012; 7.
57. Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM, Ottesen LH, Laubscher KH, Sherman L, McDermott DF, Haas NB, Flaherty KT, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013; 31:181-186.
58. Ozawa Y, Nakamura Y, Fujishima F, Felizola SJ, Takeda K, Okamoto H, Ito K, Ishida H, Konno T, Kamei T, Miyata G, Ohuchi N, Sasano H. c-Met in esophageal squamous cell carcinoma: an independent prognostic factor and potential therapeutic target. BMC Cancer. 2015; 15:451.