
INTRODUCTION
Classical Hodgkin lymphoma (cHL) is characterized by the presence of mononucleated Hodgkin cells and multinucleated Reed-Sternberg cells (HRS) surrounded by the overwhelming host cells of “inflammatory infiltrate”. HRS cells are derived from germinal center (GC) B cells which have lost their B cell program in the process of malignant transformation [1]. Multiple pathways that are critical for evading apoptosis and for promotion of cell growth are constitutively activated in HRS cells. They include NF-κB, the Jak-Stat, PI3K-Akt, Erk, AP1, and NOTCH pathways [1]. HRS cells not only have lost their B cell identity, but also acquired expression of markers of other lineages, especially that of T lymphocytes like NOTCH1and GATA3 [1]. Repression of the transcription factors regulating maintenance of B cell program including POU2F2, POU2AF1, SPI1/PU.1, EBF1, and FOXO1 by genetic and epigenetic mechanisms was shown to be responsible for the loss of specific B-cell markers in cHL [1, 2]. Other important B-cell factors are E47 and E12 proteins that are encoded by the TCF3 gene. TCF3 is expressed in HRS cells, but its function is antagonized by MSC/ABF1, ID2, and NOTCH1 [2, 3]. The reprogramming of HRS cells by the abnormal activities of these lineage regulators has been proposed to be of advantage for HRS cell survival [1]. Indeed, aberrant expression of both NOTCH1 and GATA3 was suggested to play important role in cHL tumorigenesis [4, 5]. Moreover, others and we have shown that EBF1, PU.1, and FOXO1 act as tumor suppressors in cHL [6–8]. The role of TCF3 repression in cHL lymphomagenesis has not been addressed so far, but there are several lines of evidences indicating that it may act as a tumor suppressor. Contribution of TCF3 inactivation in oncogenesis have been described in other tumor entities [9–12]. Moreover, recent genome-wide association studies revealed that genetic abnormalities in the TCF3 locus is associated with an increased risk of cHL [13]. In this study we show that reactivation of TCF3 causes cell death and cell cycle arrest in cHL cell lines. The antitumor effect of the TCF3 is accompanied by induction of proapoptotic and antiproliferative genes like BIK, APAF1, FASLG, BOK, and TNFRSF10A/DR4, and p21.
RESULTS
TCF3 negatively regulates proliferation and induces cell death
Since endogenous TCF3 proteins are bona fide inactivated by ID2 and MSC in cHL, we overexpressed E47, a TCF3 transcription variant, in KM-H2 and SUP-HD1 cell lines using doxycycline (DOX)-inducible vector pRTS containing GFP as a fluorescent marker. DOX treatment activated expression of E47 in both cell lines (Figure 1A) and decreased viable cell numbers in time-dependent manner (Figure 1B). In cell lines expressing empty vector, DOX did not influence the viability of cells (Figure 1B). To prove induction of cell death, we used Nicoletti assay. Activation of E47 led to significant nuclear fragmentation in both cell lines (Figure 1C). To assess induction of apoptosis, we used annexin-V staining. Unfortunately, due to the extremely strong GFP fluorescence of the vector that interfered with other channels, we have succeeded only in KM-H2 cells (Supplementary Figure S1A and S1B). E47 activation significantly increased proportion of annexin V positive cells in KM-H2 cells. In addition we investigated the influence of E47 expression on cell cycle progression. In both cell lines, E47 activation led to increasing proportion of cells in G0/G1 phase at the expense of cells in the S phase of the cell cycle (Figure 1D). We tried to extend our experiments on other cHL cell lines. Therefore we cloned E47 into a lentiviral vector harboring fluorescent marker (GFP). L428 and UHO1 cells were infected with lentivirus expressing E47. We validated the expression of E47 by immunoblot (Figure 2A). To assess tumor suppressor effect, we monitored the dynamics of the GFP+ population by flow cytometry (Figure 2B). Compared with cells infected with empty vector, the percentage of cells expressing E47 significantly decreased with time in both lines. Thus, we found that forcible expression of E47 induces cell death and cell cycle arrest in cHL cell lines.
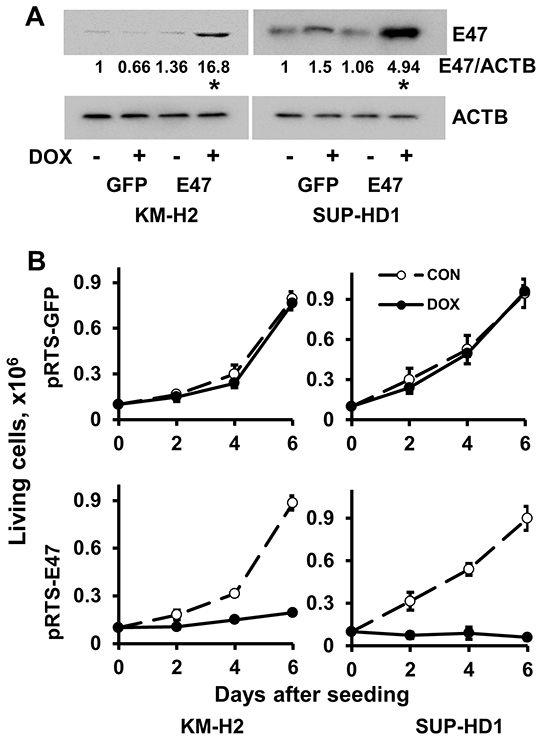
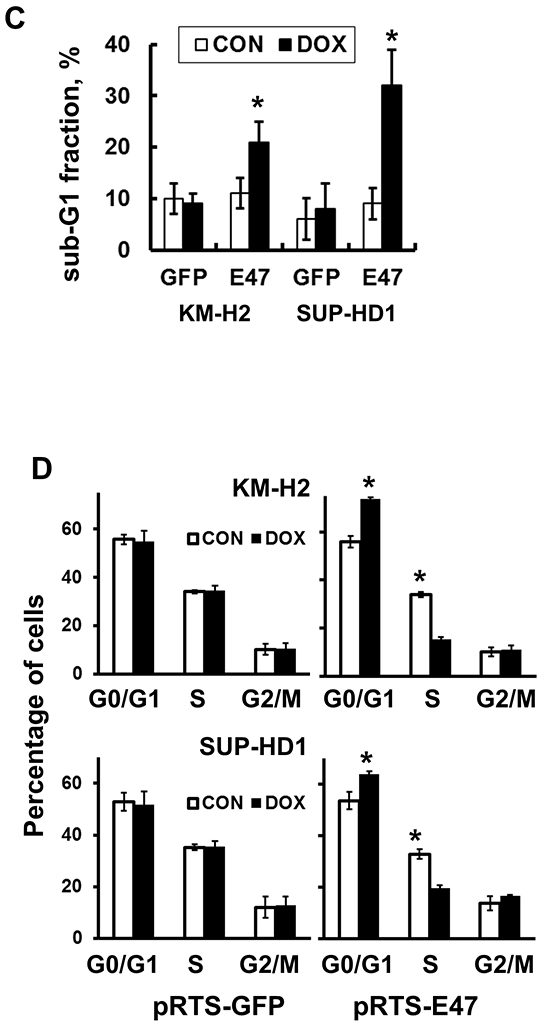
Figure 1: E47 induces cell death in cHL cell lines. A. KM-H2 and SUP-HD1 cell lines conditionally expressing E47 were established using the pRTS vector system. After transfection, cells were selected with hygromycin for two weeks (KM-H2) and one week (SUP-HD1), more than 95% cells were GFP positive upon DOX treatment. Expression of E47 protein was measured by immunobloting using anti-TCF3 antibody. Anti-ACTB antibody was used as a loading control. * p<0.05 (expression levels upon DOX treatment compared with that of samples without DOX treatment) as it was assessed by double sided T-test. B. Cells transfected with empty pRTS vector (pRTS-GFP) or pRTS-E47 were seeded at low density (0.1 × 106 cells per well in 3 ml of complete medium). DOX was added on day 0 and day 3. On day 2, day 4 and day 6, the cells were counted by flow cytometer and viable cells were discriminated using side/forward scatter parameters. C. The nuclear fragmentation was measured by Nicoletti method after 4 days of incubation with DOX. * p<0.05 as it was assessed by double sided T-test. D. The influence of E47 on cell cycle progression. Cells were seeded at density of 0.5×106 per 10 ml of complete medium and treated with 0.5 μg/ml DOX. After 2 days, cell cycle distribution was measured by PI staining as described in the Materials and Methods section. Experiments were repeated three times. Results are presented as mean ± SD. * p<0.05.
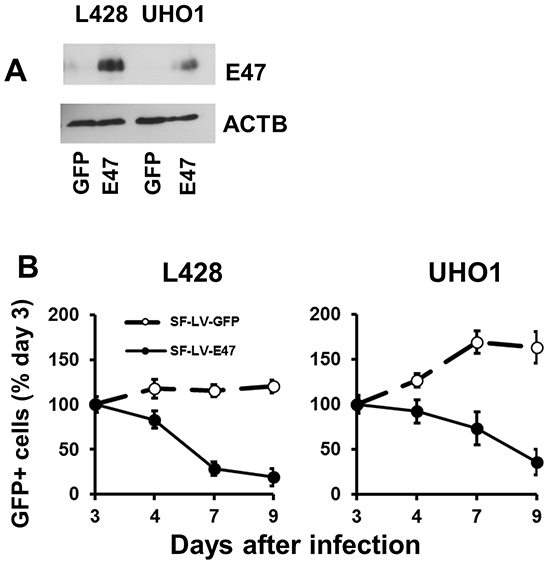
Figure 2: TCF3 negatively regulates proliferation. E47 was over-expressed in L428 and UHO1 cells using the lentiviral SF-LV-cDNA-EGFP vector. A. E47 protein levels were analyzed by immunoblot. The percentage of GFP+ cells determined by flow cytometry was about 40%. Anti-ACTB antibody was used as a loading control. B. Percentage of GFP+ cells expressing E47 or empty vector (SF-LV) was measured at the indicated time points by flow cytometry. Percentage on the third day of infection (day 3) was defined as 100%. The data shown are representative of three independent experiments.
TCF3 activates proapoptotic and antiproliferative genes in cHL cell lines
To elucidate mechanisms responsible for E47 induced cell death, we used the Human Apoptosis Primer Library comprising 88 genes of the apoptosis pathway. Fifteen genes were up-regulated by more than 2-fold after E47 activation in both KM-H2 and SUP-HD1 cells (Table 1). Interestingly, no gene was downregulated by more than 2-fold in both cell lines. This is in agreement with previous observations that E47 induces far more genes than it repressed [9]. Among the 15 upregulated genes, only BIRC1 and CD40LG were anti-apoptotic and the rest 13 genes might be considered as pro-apoptotic. Eight of them belong to the class of TNF receptors and their ligands, which have not been reported to play a major role in apoptosis in cHL. The same is true for caspases 5 and 10 as well as for the BCL2-related ovarian killer BOK. The APAF1 is an essential component of the apoptosome, but it is ubiquitously expressed and its upregulation hardly can initiate apoptosis by itself. So, by exclusion we identified BCL2-interacting killer (BIK) as the most interesting candidate for further analysis. Noteworthy, BIK is at the top of the list of genes down-regulated in primary HRS cells over B-NHLs [14], and it was shown to be involved in B cell apoptosis [15]. BIK has also been reported to be upregulated upon over-expression of E47 in T cell lymphoma [9] and in a murine TCF3/E2A-deficient hematopoietic progenitor cell line [16].
Table 1: Genes modulated by TCF3/E2A in cHL cell lines
Official symbol |
Full name |
Fold of induction |
|
|---|---|---|---|
Sup-HD1 |
KM-H2 |
||
APAF1 |
apoptotic peptidase activating factor 1 |
16.5±22.2 |
2.83±0.81 |
BIK |
BCL2-interacting killer (apoptosis-inducing) |
10.3±2.6 |
4.77±1.93 |
BIRC1 |
baculoviral IAP repeat-containing 1 |
2.31±0.06 |
2.21±0.09 |
BOK |
BCL2-related ovarian killer |
2.66±0.44 |
4.18±1.55 |
CASP10 |
Caspase 10 |
4.26±0.62 |
5.21±0.54 |
CASP5 |
Caspase 5 |
20.5±21.5 |
4.54±0.45 |
TNFRSF10A |
tumor necrosis factor receptor superfamily, member 10A |
2.55±0.34 |
3.62±0.4 |
TNFRSF11B |
tumor necrosis factor receptor superfamily, member 11b |
3.15±1.05 |
2.87±1.32 |
TNFRSF17 |
tumor necrosis factor receptor superfamily, member 17 |
5.04±1.5 |
13.7±7.76 |
TNFSF11 |
tumor necrosis factor (ligand) superfamily, member 11 |
5.95±1.41 |
2.73±1.98 |
TNFSF13 |
tumor necrosis factor (ligand) superfamily, member 13 |
3.8±0.63 |
2.14±0.14 |
CD40LG |
CD40 ligand |
7.74±1.21 |
5.37±2.24 |
FASLG |
Fas ligand (TNF superfamily, member 6) |
4.42±1.55 |
2.32±1.76 |
TNFSF8 |
tumor necrosis factor (ligand) superfamily, member 8 |
6.23±0.51 |
2.18±1.58 |
TRAF4 |
TNF receptor-associated factor 4 |
5.86±3.06 |
8.53±0.78 |
A total of 2 × 106 KM-H2-E47 and SUP-HD1-E47 cells were seeded in 10 mL of complete culture medium and treated with or without DOX for 2 days. Then, cells were harvested, and expression of genes involved in apoptosis was analyzed using the Human Apoptosis Primer Library (Real Time Primers) Genes whose expression was changed more than 2-fold in both cell lines are shown. The data are shown as fold modulation ± SD of 3 independent experiments.
Consequently, we validated the upregulation of BIK by E47 in KM-H2 and SUP-HD1 cell lines using Q-RT-PCR. BIK was found upregulated in both cell lines, more pronounced in SUP-HD1 than in KM-H2 cells (over 9 and 3 folds in SUP-HD1 and KM-H2 cells, respectively) (Figure 3A). The upregulation of BIK was also detected on protein level using immunoblot (Figure 3B). Since E47 induced BIK in our cHL cell lines, we asked whether there is a correlation between expression levels of the two proteins in other tumor entities. Using immunoblot, we found that expression of BIK in most NHL cell lines was higher than in normal CD19+ B cells. cHL cell lines showed a virtual absence of BIK expression (Figure 4A), which is in agreement with a previous study [14]. Interestingly, MedB1 and Karpas1106, two cell lines derived from primary mediastinal B cell lymphoma (PMBL), a subtype of DLBCL that share morphological and molecular similarities with cHL, also did not express BIK (Figure 4A). Furthermore, we re-analyzed published gene expression profiling (GEP) data of microdissected tumor cells of 12 cHL cases, 30 B cell lymphoma cases, normal B cell subtypes (http://www.ncbi.nlm.nih.gov/geo/; GSE12453, 17.06.2013) [17]. We found that BIK and TCF3 were highly expressed in GC B cells, plasma cells, B-NHLs and nodular lymphocyte predominant Hodgkin’s lymphoma (NLPHL). HRS cells of cHL demonstrated lowest expression levels of BIK and TCF3 (Figure 4B).
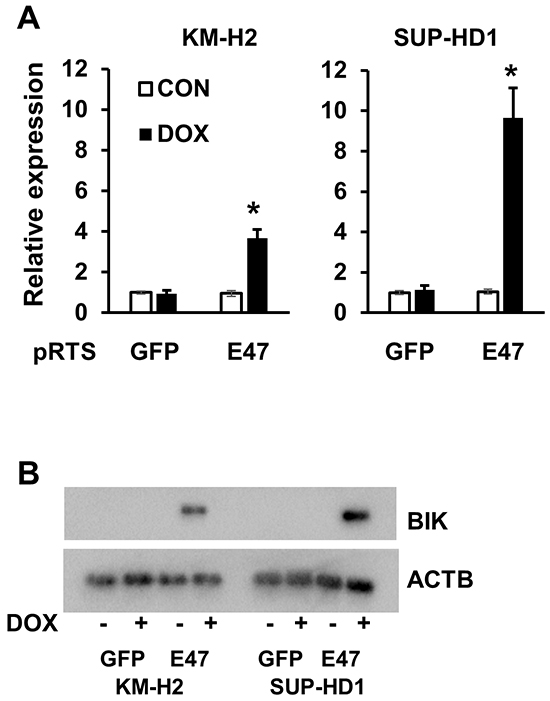
Figure 3: TCF3/E2A induced expression of BIK. BIK is an E47 target gene in cHL cell lines. Expression of BIK in cHL cells expressing pRTS-GFP or pRTS-E47. Cells were treated with DOX for 2 days. RNA and protein were prepared as described in the Materials and methods section. A. BIK mRNA expression was measured by qRT-PCR. * p<0.05. B. BIK protein in E47-expressing cells was detected by immunobloting, in comparison with empty vector-expressing cells. The data are representative of three independent experiments with similar results.
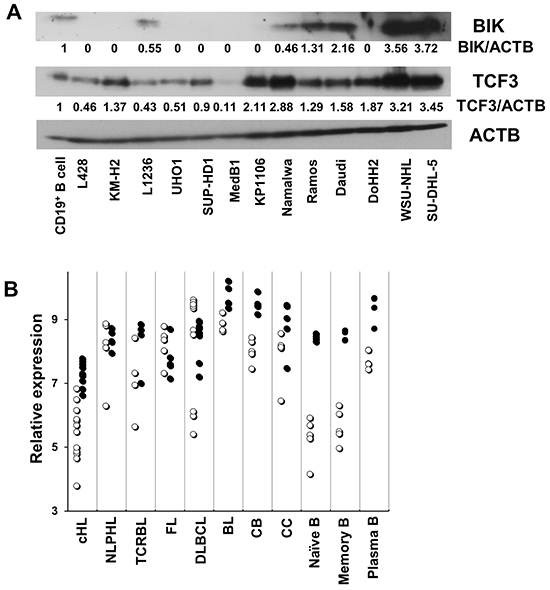
Figure 4: The expression of BIK correlates with that of TCF3/E2A. A. The expression of BIK and TCF3 in various B cell lymphoma cell lines and CD19+ B cells was measured by immunobloting. Anti-ACTB antibody was used as a loading control. The immunoblots in (A) were quantified using ImageJ software. B. Published gene expression data of microdissected tumor cells including 12 cHL cases, 5 nodular lymphocyte predominant Hodgkin’s lymphoma (NLPHL) cases, 4 T cell - rich B cell lymphoma (TCRBL) cases, 5 follicular lymphoma (FL) cases, 11 diffuse large B cell lymphoma (DLBCL) cases, 5 BL cases, and normal B cell subtypes (5 samples each) including centroblast (CB), centrocyte (CC), Naïve B cell (N), memory B cell (M), plasma cell (PC), were re-analyzed with help of the Genesifter software (Perkin Elmer, Seattle, WA). The data are shown as log2 of fluorescence intensity. The hollow circle (○) stands for BIK, the solid circle (•) stands for TCF3.
Finally we investigated whether E47 is able to reactivate known TCF3 targets like FOXO1, ABF1/MSC, POU2F2, CD19, CD79, EBF1, MYC and CDK6. We found that E47 significantly upregulated expression of NOTCH1, ID2, AICDA, and cell cycle regulator CDKN1A (Figure 5). The other genes (FOXO1, ABF1/MSC, POU2F2, CD19, CD79, EBF1, MYC, and CDK6) were not significantly modulated (data not shown). We conclude that TCF3 inactivation contributes to oncogenic program of cHL by repression of pro-apoptotic and anti-proliferative genes.
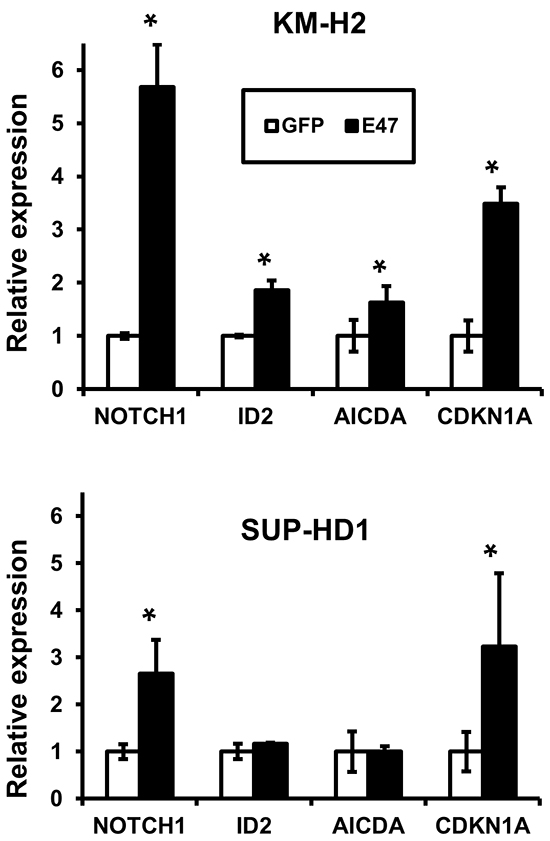
Figure 5: TCF3 induces expression of NOTCH1 and CDKN1A in cHL cell lines. Expression of multiple genes in cHL cells expressing pRTS-GFP or pRTS-E47. Cells were treated with DOX for 2 days. RNA was prepared as described in the Materials and methods section. mRNA expression was measured by qRT-PCR. The data represent mean ± SD of three experiments. * p<0.05.
DISCUSSION
In this study, we have shown that forced expression of TCF3 in cHL cell lines causes apoptosis and cell cycle arrest that is associated with expression of antiproliferative and pro-apoptotic target genes. cHL is characterized by loss of the B-cell phenotype caused by repression of critical regulators of B-cell differentiation. Recently, we and other authors have shown that repression of these transcription factors including FOXO1 [6], EBF1 [8] and SPI1/PU.1 [7] has a critical impact on oncogenic program of cHL. Here we show that inactivation TCF3 also contributes to the oncogenic program of cHL. These transcription factors function in concert to regulate B cell differentiation and tumorigenesis, forming an interconnected regulatory network [18–20]. It is conceivable that deregulation of one of the regulatory units would lead to malfunction of the whole system. Particularly, given that TCF3 contributes to FOXO1 induction [20], inactivation of TCF3 might explain partially repression of FOXO1 in cHL. Our previous study showed that KLF4 repression contributes to MSC/ABF-1 activation [21]. Therefore, KLF4 inhibition might be also responsible for TCF3 inactivation.
Given that TCF3 is not only an essential regulator in B- and T- lymphocyte development, but also a tumor suppressor in T cell tumors [9, 22–25], our finding is not surprising. Similarly to cHL, in T cell lymphomas, ID proteins including ID2, block lymphocyte differentiation and promote T-cell lymphomagenesis by antagonizing TCF3 [24, 25]. In T cell lymphomas TCF3 exerts its tumor suppressing function by repression of proto-oncogenes MYC, CDK6 and by up-regulation of cell cycling inhibitor CDKN1A/p21, thereby leading to cell cycle arrest [9, 26].
In regard to molecular mechanisms of TCF3 antitumor effects, in Sézary syndrome (a subtype of T cell lymphoma) derived cells, no significant cell death induction was observed after E47 overexpression, whereas, up-regulation of proapoptotic genes including BCL2L11 and BIK have been seen [9, 26]. In our study, E47 induced cell death of cHL cells, which was associated with activation of multiple proapoptotic genes including BIK, APAF1, FASLG, BOK, and TNFRSF10A/DR4. Given that BIK is strongly expressed in GC B cells [27] and specifically repressed in HRS cells [14], this pro-apoptotic factor has attracted special attention. We have shown that the BIK expression significantly correlated with TCF3 in normal B cell subtypes and B cell lymphomas. Another interesting finding was the absence of BIK expression in PMBL cell lines. As a distinct subtype of DLBCL, PMBL shares morphological and molecular similarities with cHL. Nevertheless, we were not able to prove contribution of BIK in the E47-induced cell death due to technical reasons. Given that TCF3 activates numerous proapoptotic genes, it is conceivable that tumor-suppressor effect does not depend on a single factor but develops as a “death by a thousand cuts” as it was supposed for IRF4 depletion in multiple myeloma [28].
BIK expression is regulated by multiple mechanisms. A recent study demonstrated that BIK expression might be a consequence of G1 cell cycle arrest in solid tumor cell lines [29]. BIK was also shown to be a p53 target gene [30] and silenced by DNA hypermethylation in some tumor entities [31, 32]. Previous studies and the present study found that BIK was upregulated by TCF3 [9, 16]. Therefore, the regulation of BIK by TCF3 might be conserved in multiple cell types. Whether this regulation is direct or indirect deserves further investigation.
We conclude that inactivation of TCF3 contributes to oncogenic program of cHL and might be critical for tumor maintenance.
MATERIALS AND METHODS
Cell culture and primary B cells
cHL cell lines (KM-H2, SUP-HD1, L428, UHO1, and L1236), Burkitt’s lymphoma (BL) cell lines (Ramos, Namalwa, Daudi) and other B cell lymphoma cell lines (MedB1, Karpas 1106, DOHH2, WSU-NHL and SU-DHL-5) were cultured in complete RPMI 1640 medium as described earlier [21, 33]. Primary CD19+ B cells were isolated using microbeads (Miltenyi Biotec) as described before [21].
Vectors and transfections
pSFI and pRTS vectors were kindly provided by D. Eick and G. W. Bornkamm [34] and were described before [18]. E47 2/5-pCS2 vector [35] was obtained from Addgene (www.addgene.org). Human E47 coding sequence was first cut out from pCS2-E47, subsequently the fragment was blunt inserted to EcoRV sites of the pSFI vector and then transferred into the pRTS-1 vector to construct the pRTS-E47 plasmid. Cells were transfected with an Amaxa Nucleofector device (Lonza, Cologne, Germany) using nucleofection buffer “V”, program Q-07 for SUP-HD1 and U-01 for KM-H2 (Lonza, Cologne, Germany). Twenty-four hours later, transfected cells were selected with 150 μg/mL Hygromycin B (InvivoGen, Toulouse, France). Efficiency of selection was monitored by FACS analysis of green fluorescent protein (GFP) expression and immunoblotting of the transgene expression 24 h after treatment with 0.5 μg/mL of Doxycycline (DOX).
To construct SF-LV-E47 vector, E47 was amplified from pCDNA3-E47 using primers: 5′-TTAACTCGAGTTAACGCCACCATGAACCAGCCGCAGAGGATG-3 and 5′-GGAGGGAGAGGGGGCTCACATGTGCCCGGCGGG-3; with restriction sites for NotI. Subsequently, E47 was introduced into the NotI restriction site of the lentiviral vector SF-LV-cDNA-EGFP (a gift of K. L. Rudolph, Leibniz Institute for Age Research, Germany [36]). Lentiviral particles were generated and cell lines were transduced as we described [37].
Quantitative real-time PCR (qRT-PCR)
The total RNA was isolated using High Pure RNA Isolation Kit (Roche, Mannheim, Germany), first-strand cDNA with MMLV reverse transcriptase (Promega, Madison, WI). Templates amplification was done using QuantiTect SYBR Green PCR Kit (Qiagen, Hilden, Germany) on a LightCycler 480 (Roche). Primers were designed with Genscript on-line software (www.genscript.com) and synthesized by biomers.net (Ulm, Germany), sequences 5′ to 3′, sense and antisense, annealing temperature 60°C:
BIK: AGCTCCTGGAACCCCCGACC and CGC AGGGCCAATGCGTCACT [9];RPL13A: CGGACCGT GCGAGGTAT and CACCA TCCGCTTTTTCTTGTC;POU2F2: ATGGAGAAGGAAGTGATCCG and TTGA TGCGTTTCTCCTTCTG;CD19: GCAACCTGACCAT GTCATTC and TCA CAGCTGAGACCTTCCAG; CD79A: TCCTCCTCTTCCTGCTGTCT and ATC AATGATGCTGGGACCTT;EBF1: TGCTGGTCTGGAGTGA GTTG and ATGA ATCTGCCTGGTGTTCC;AICDA: CAT GGTCACCTTCAAGCTA and TTGC GTTTCCAGAAG ATTTG;CDK6: CTAGCAACCATCCCTCCATT and GGA AAGGAGCAAGAGCATTC;CDKN1A: GCAGACCAG CATGACAGATTT and GGATTAGGGCTTCCTCTTG GA;MYC: TCGGATTCTCTGCTCTCCTC and TGTT CCTCCTCAGAGTCGCT;NOTCH1: AAGATGCTCCA GCAACACAG and GGCTCTGGCAAGTCTCCTAC. Reference genes including ACTB, HPRT1, HMBS, and RPL13A were evaluated by geNorm software (http://medgen.ugent.be/~jvdesomp/genorm/). RPL13A was selected for calculations because it was found to be the most reliable. Human apoptosis primer library (Real Time Primers, LCC, Elkins Park, PA) was used to identify TCF3/E2A target genes as described before [21].
Immunoblot
Immunoblot was done as described before [21]. Rabbit anti-Actin antibody (A5060, 1:15000 dilution) was from Sigma-Aldrich (Saint Louis, Missouri, USA). Rabbit antibody against human BIK protein (#4592) and TCF3/E2A (#4865) was from Cell Signaling (1:1000 dilution). Secondary antibodies included goat anti-rabbit IgG-horseradish peroxidase (HRP; sc-2004), donkey anti-goat IgG-horseradish peroxidase (HRP; sc-2020; both Santa Cruz Biotechnology). Immunoblots were quantified using ImageJ software. Anti-ACTB antibody was used as a loading control. The relative expression levels of target proteins were normalized to those of ACTB proteins.
Cell proliferation assay and cell cycle analysis
Cell proliferation was analyzed by cell counting using a Vi-cell XR cell viability analyzer based on the trypan blue dye exclusion method (Beckman Coulter, Krefeld, Germany). Viable and dead cells were differentiated and quantified by the cell viability analyzer. Apoptosis was measured by Propidium Iodide (PI) staining and flow cytometry as described [38]. Cell cycle distribution was determined as described before [33]. Briefly, 1×106 cells were fixed and stained with 70% cold ethanol and PI. DNA contents were measured by flow cytometry with a FACSCalibur flow cytometer (BD Biosciences). Data were analyzed using ModFit cell cycle analysis software (Verity Software, Topsham ME, USA).
Statistical analysis
All experiments were independently performed three times. Data were expressed as mean ± standard deviation (SD). Student’s t-test was used to determine the significance of difference between two groups, one-way ANOVA was used for multiple comparisons. All statistical analyses were carried out with SPSS13.0 software (SPSS, Chicago, IL, USA), p < 0.05 was considered statistically significant. The correlation analysis was calculated using the CORREL function in Microsoft Office Excel (Microsoft Corporation).
ACKNOWLEDGMENTS
We thank Anita Kick for excellent technical assistance. We thank D. Eick and G. W. Bornkamm for the inducible gene expression vector system, and Stephen Tapscott for the E47 2/5-pCS2 plasmid. We are grateful to Bernd Baumann for his critical reading of the manuscript and valuable suggestions.
CONFLICTS OF INTEREST
The authors indicated no potential conflict of interest.
FUNDING
This study was supported in part by grant 107547 from Deutsche Krebshilfe e.V. to T.W. and A.U, National Natural Science Foundation of China (No. 81101788 and 81572857 to HG, and No. 81201867 to LX), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars by the State Education Ministry to HG.
Author contributions
HG and LX performed the research; AU and TW designed the study; LX, AU and TW wrote the paper.
REFERENCES
1. Küppers R. The biology of Hodgkin’s lymphoma. Nat Rev Cancer. 2009; 9:15-27.
2. Mathas S, Janz M, Hummel F, Hummel M, Wollert-Wulf B, Lusatis S, Anagnostopoulos I, Lietz A, Sigvardsson M, Jundt F, Johrens K, Bommert K, Stein H and Dorken B. Intrinsic inhibition of transcription factor E2A by HLH proteins ABF-1 and Id2 mediates reprogramming of neoplastic B cells in Hodgkin lymphoma. Nat Immunol. 2006; 7:207-215.
3. Jundt F, Acikgoz O, Kwon SH, Schwarzer R, Anagnostopoulos I, Wiesner B, Mathas S, Hummel M, Stein H, Reichardt HM and Dorken B. Aberrant expression of Notch1 interferes with the B-lymphoid phenotype of neoplastic B cells in classical Hodgkin lymphoma. Leukemia. 2008; 22:1587-1594.
4. Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H and Dorken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002; 99:3398-3403.
5. Stanelle J, Doring C, Hansmann ML and Kuppers R. Mechanisms of aberrant GATA3 expression in classical Hodgkin lymphoma and its consequences for the cytokine profile of Hodgkin and Reed/Sternberg cells. Blood. 2010; 116:4202-4211.
6. Xie L, Ushmorov A, Leithäuser F, Guan H, Steidl C, Färbinger J, Pelzer C, Vogel MJ, Maier HJ, Gascoyne RD, Möller P and Wirth T. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012; 119:3503-3511.
7. Yuki H, Ueno S, Tatetsu H, Niiro H, Iino T, Endo S, Kawano Y, Komohara Y, Takeya M, Hata H, Okada S, Watanabe T, Akashi K, Mitsuya H and Okuno Y. PU.1 is a potent tumor suppressor in classical Hodgkin lymphoma cells. Blood. 2013; 121:962-970.
8. Bohle V, Doring C, Hansmann ML and Kuppers R. Role of early B-cell factor 1 (EBF1) in Hodgkin lymphoma. Leukemia. 2013; 27:671-679.
9. Steininger A, Mobs M, Ullmann R, Kochert K, Kreher S, Lamprecht B, Anagnostopoulos I, Hummel M, Richter J, Beyer M, Janz M, Klemke CD, Stein H, Dorken B, Sterry W, Schrock E, et al. Genomic loss of the putative tumor suppressor gene E2A in human lymphoma. The Journal of experimental medicine. 2011; 208:1585-1593.
10. Lasorella A, Noseda M, Beyna M, Yokota Y and Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000; 407:592-598.
11. Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, Cao TC, Carano RA and Dixit VM. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell. 2011; 146:918-930.
12. Iavarone A, Garg P, Lasorella A, Hsu J and Israel MA. The helix-loop-helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes & development. 1994; 8:1270-1284.
13. Cozen W, Timofeeva MN, Li D, Diepstra A, Hazelett D, Delahaye-Sourdeix M, Edlund CK, Franke L, Rostgaard K, Van Den Berg DJ, Cortessis VK, Smedby KE, Glaser SL, Westra HJ, Robison LL, Mack TM, et al. A meta-analysis of Hodgkin lymphoma reveals 19p13.3 TCF3 as a novel susceptibility locus. Nature communications. 2014; 5:3856.
14. Tiacci E, Doring C, Brune V, van Noesel CJ, Klapper W, Mechtersheimer G, Falini B, Kuppers R and Hansmann ML. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood. 2012; 120:4609-4620.
15. Jiang A and Clark EA. Involvement of Bik, a proapoptotic member of the Bcl-2 family, in surface IgM-mediated B cell apoptosis. Journal of immunology. 2001; 166:6025-6033.
16. Ikawa T, Kawamoto H, Goldrath AW and Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. The Journal of experimental medicine. 2006; 203:1329-1342.
17. Brune V, Tiacci E, Pfeil I, Doring C, Eckerle S, van Noesel CJ, Klapper W, Falini B, von Heydebreck A, Metzler D, Brauninger A, Hansmann ML and Kuppers R. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. The Journal of experimental medicine. 2008; 205:2251-2268.
18. Grosschedl R. Establishment and maintenance of B cell identity. Cold Spring Harbor symposia on quantitative biology. 2013; 78:23-30.
19. Mansson R, Welinder E, Ahsberg J, Lin YC, Benner C, Glass CK, Lucas JS, Sigvardsson M and Murre C. Positive intergenic feedback circuitry, involving EBF1 and FOXO1, orchestrates B-cell fate. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:21028-21033.
20. Welinder E, Mansson R, Mercer EM, Bryder D, Sigvardsson M and Murre C. The transcription factors E2A and HEB act in concert to induce the expression of FOXO1 in the common lymphoid progenitor. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:17402-17407.
21. Guan H, Xie L, Leithauser F, Flossbach L, Moller P, Wirth T and Ushmorov A. KLF4 is a tumor suppressor in B-cell non-Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood. 2010; 116:1469-1478.
22. Park ST, Nolan GP and Sun XH. Growth inhibition and apoptosis due to restoration of E2A activity in T cell acute lymphoblastic leukemia cells. The Journal of experimental medicine. 1999; 189:501-508.
23. Bain G, Engel I, Robanus Maandag EC, te Riele HP, Voland JR, Sharp LL, Chun J, Huey B, Pinkel D and Murre C. E2A deficiency leads to abnormalities in alphabeta T-cell development and to rapid development of T-cell lymphomas. Molecular and cellular biology. 1997; 17:4782-4791.
24. Morrow MA, Mayer EW, Perez CA, Adlam M and Siu G. Overexpression of the Helix-Loop-Helix protein Id2 blocks T cell development at multiple stages. Molecular immunology. 1999; 36:491-503.
25. Yan W, Young AZ, Soares VC, Kelley R, Benezra R and Zhuang Y. High incidence of T-cell tumors in E2A-null mice and E2A/Id1 double-knockout mice. Molecular and cellular biology. 1997; 17:7317-7327.
26. Prabhu S, Ignatova A, Park ST and Sun XH. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Molecular and cellular biology. 1997; 17:5888-5896.
27. Shaffer AL, Rosenwald A, Hurt EM, Giltnane JM, Lam LT, Pickeral OK and Staudt LM. Signatures of the immune response. Immunity. 2001; 15:375-385.
28. Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, Zeng Y, Chen B, Epstein J and Staudt LM. IRF4 addiction in multiple myeloma. Nature. 2008; 454:226-231.
29. Sale MJ and Cook SJ. The increase in BIK expression following ERK1/2 pathway inhibition is a consequence of G(1) cell-cycle arrest and not a direct effect on BIK protein stability. The Biochemical journal. 2014; 459:513-524.
30. Hur J, Bell DW, Dean KL, Coser KR, Hilario PC, Okimoto RA, Tobey EM, Smith SL, Isselbacher KJ and Shioda T. Regulation of expression of BIK proapoptotic protein in human breast cancer cells: p53-dependent induction of BIK mRNA by fulvestrant and proteasomal degradation of BIK protein. Cancer research. 2006; 66:10153-10161.
31. Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, Jung K, Loening S, Dorken B and Daniel PT. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell death and differentiation. 2006; 13:619-627.
32. Hatzimichael E, Dasoula A, Kounnis V, Benetatos L, Lo Nigro C, Lattanzio L, Papoudou-Bai A, Dranitsaris G, Briasoulis E and Crook T. Bcl2-interacting killer CpG methylation in multiple myeloma: a potential predictor of relapsed/refractory disease with therapeutic implications. Leukemia & lymphoma. 2012; 53:1709-1713.
33. Guan H, Xie L, Klapproth K, Weitzer CD, Wirth T and Ushmorov A. Decitabine represses translocated MYC oncogene in Burkitt lymphoma. J Pathol. 2013; 229:775-783.
34. Bornkamm GW, Berens C, Kuklik-Roos C, Bechet JM, Laux G, Bachl J, Korndoerfer M, Schlee M, Holzel M, Malamoussi A, Chapman RD, Nimmerjahn F, Mautner J, Hillen W, Bujard H and Feuillard J. Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic acids research. 2005; 33:e137.
35. Yang Z, MacQuarrie KL, Analau E, Tyler AE, Dilworth FJ, Cao Y, Diede SJ and Tapscott SJ. MyoD and E-protein heterodimers switch rhabdomyosarcoma cells from an arrested myoblast phase to a differentiated state. Genes & development. 2009; 23:694-707.
36. Wang J, Sun Q, Morita Y, Jiang H, Gross A, Lechel A, Hildner K, Guachalla LM, Gompf A, Hartmann D, Schambach A, Wuestefeld T, Dauch D, Schrezenmeier H, Hofmann WK, Nakauchi H, et al. A differentiation checkpoint limits hematopoietic stem cell self-renewal in response to DNA damage. Cell. 2012; 148:1001-1014.
37. Vogel MJ, Xie L, Guan H, Tooze RM, Maier T, Kostezka U, Maier HJ, Holzmann K, Chan FC, Steidl C, Reichel JB, Weitzer CD, Gehringer F, Kick AB, Cesarman E, Roshal M, et al. FOXO1 repression contributes to block of plasma cell differentiation in classical Hodgkin lymphoma. Blood. 2014; 124:3118-3129.
38. Riccardi C and Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nature protocols. 2006; 1:1458-1461.