
Introduction:
Cancer/testis (CT) genes are normally expressed only in the human germ line and malignant cells [1]. Because of their restricted expression and immunogenicity, CT proteins are being used as targets in several therapeutic vaccination trials [2, 3]. The CT genes located on the X chromosome present the most tissue restricted expression [1] and most of them are encoded by multigene families that are organized in gene clusters. CT gene clusters are present on the telomeric end between Xq24 and Xq28, which includes CT1/MAGEA, CT6/NY-ESO-1, CT7/MAGE-C1, CT10/MAGEC2, and CT14/SAGE, and at a more centromeric position of X chromosome, Xp11.2-11.4, where CT4/GAGE, CT5/SSX and CT12/XAGE1 genes are located [4].
There are relatively few clues regarding function of most of these proteins. Better insights in the function of these genes may uncover links between gametogenesis and tumor growth and could be indicative of their use in additional forms of anti-tumor therapies [1]. In several tumor types, the expression of CT-X genes is associated with advanced disease and poor outcome [5-16] and although these data indicate that CT gene expression might contribute to tumorigenesis, the biological role of these proteins in both germ line tissues and tumors remains poorly understood. Most functional investigations have focused on members of the MAGE proteins on Xq28. Several studies have shown that MAGE proteins are involved in cell survival, can increase tumorigenic properties of cells and may actively contribute to the development of malignancies [17-23]. However, the functional properties of CT-X genes mapping to the short arm of the X-chromosome (CT-Xp) remain poorly investigated. In this study, we used siRNA-mediated knock down in melanoma cell lines to evaluate the potential of CT genes on Xp as therapeutic targets.
Results:
Transfection of 27mers specific to CT-Xp antigens strongly and specifically suppressed gene expression in SK-MEL-37 cells.
We designed and tested siRNAs specific to the CT-Xp genes GAGE/CT4, SSX/CT5 and XAGE/CT12 (Table 1). The GAGE siRNAs were designed to target all members of the GAGE family; those specific to XAGE1 target all isoforms of this gene, while both SSX siRNAs had 100% identity with SSX4 only. These siRNA duplexes targeting the coding regions of the different CT-X and the siRNA specific to HPRT1 were individually introduced into the SK-MEL-37 melanoma cell line and the effect on mRNA level examined by real-time quantitative RT-PCR analysis 24-48 hours post transfection. All siRNA duplexes examined produced a 91–99% reduction in CT-X mRNA compared with the control sample transfected with scrambled siRNA as a negative control (Table 2). In addition, we analyzed the effects of each siRNA duplex on the mRNA level of other CT-Xs, and little to no effect was observed compared with the scrambled control siRNA, suggesting that the effects of the 27mer siRNAs on these genes were sequence-specific. We also examined the kinetics of gene silencing and analyzed the levels of GAGE mRNA at 3, 6, 12, 18, 24 and 48 hours after transfection with GAGE-specific siRNAs (Figure 1A). Around 75-80% mRNA reduction could be observed as early as three hours after transfection at 10nM final duplex concentration.
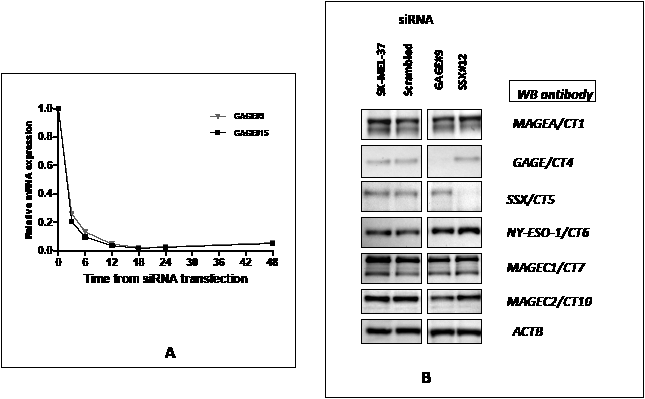
Figure 1: A – Kinetics of siRNA-mediated CT-X knockdown: SK-MEL-37 cells were transfected separately with10 nM of scrambled, GAGE#9 and #15 siRNAs and cells were harvested for real-time PCR 3, 6, 12, 18, 24 and 48 hours after transfection. Relative quantification of gene expression (relative amount of target RNA) was determined using the equation 2−ΔΔCT using the sample transfected with scrambled siRNA as calibrator.
B: Efficiency of siRNA-mediated CT-Xp knockdown:Western blot analysis was used to examine the effect of the specific siRNAs on CT-Xp expression at the protein level. Reduction of protein levels to almost complete depletion was present 72 hours after transfection with all tested siRNAs.
Table 1: Characteristics of the 27mer siRNA duplexes designed for this study
Duplex name |
Sense sequence (5'-3') |
Antisense sequence (5'-3') |
RefSeq |
Sense position |
Antisense position |
Specificity2 |
GAGE #9 |
GUUCAGUGAUGAAGUGGAACCAGCA |
UGCUGGUUCCACUUCAUCACUGAACUG |
NM_ 001468 |
209 |
233 |
GAGE1, 2,8,10,12 |
GAGE #15 |
GAACCAGCAACUCAACGUCAGGATC |
GAUCCUGACGUUGAGUUGCUGGUUCCC |
NM_ 001468 |
249 |
273 |
GAGE1, 2,8,10,12 |
SSX #12 |
CAAGGUCACCCUCCCACCUUUCATG |
CAUGAAAGGUGGGAGGGUGACCUUGAA |
NM_ 005636 |
247 |
271 |
SSX4, SSX4B |
SSX #19 |
CUUGUGUAUCCAUGCACCUACCUCA |
UGAGGUAGGUGCAUGGAUACACAAGCC |
NM_ 005636 |
892 |
916 |
SSX4, SSX4B, SSX6 |
XAGE1 #2 |
GACAGAAGAAGAUCAGGAUACAGCT |
AGCUGUAUCCUGAUCUUCUUCUGUCUG |
NM_ 133430 |
197 |
221 |
XAGE1 |
XAGE1 #9 |
AAGCUGAAACAACGCAAGCUGGUTT |
AAACCAGCUUGCGUUGUUUCAGCUUGU |
NM_ 133430 |
406 |
430 |
XAGE1 |
1 The sense strand has two terminal 3’ nucleotides as DNA, and the remainder bases as RNA for preferential uptake of the antisense strand into RISC (RNA induced silencing) complex.
2 Only genes presenting 100% identity with the siRNA, as assessed with the BLAST tool at NCBI are listed.
Western blot analysis was used to examine the effect of GAGE and SSX-specific siRNAs on CT-X expression at the protein level (Figure 1B). Reduction of protein levels to almost complete depletion was present 72 hours after transfection with all siRNAs tested. Similarly to the RT-PCR results, we found that the GAGE and SSX siRNAs do not alter the expression of the other CT-X proteins tested. No commercially available anti-XAGE1 antibody was found to be adequate for Western blotting analyses and our own attempts to produce anti-XAGE1 monoclonal or polyclonal antibodies failed. However, we assume that since in all other cases tested, the 27-mer induced gene knock down was very efficient at the protein level that it was for XAGE1 as well.
Effects on of GAGE, XAGE1 and SSX4 knockdown on SK-MEL-37 proliferation and clonogenic survival.
To investigate the biological result of depletion of CT-Xp by RNAi, we examined growth phenotypes of the melanoma cell line SK-MEL-37, which expresses high levels of the CT genes studied. First, the effect of CT-Xp knockdown on cell proliferation was determined by the MTT assay. The knockdown of the genes tested did not exert effects on cell proliferation, as determined by MTT assay performed with cells up to 120 hours after transfection (Figure 2A).
We next analyzed the ability of the siRNA-treated cells to form colonies between 10 and 14 days after transfection. The clonogenic assay relies on the ability of cells to form viable colonies derived from a single cell. In this colony formation assay, only 5-10% of control cells gave rise to colonies (plating efficiency). Depletion of SSX4 and XAGE1 significantly reduced the colony-forming ability of SK-MEL-37 cells to 50% or less of control levels (Figure 2B) (p<0.05). 27mer siRNA mediated silencing of SSX4 and XAGE1 using different duplexes (Table 1) that had similar specificity and efficiency of gene knock down (Table 2), equally reduced clonogenic survival and migration of SK-MEL-37 melanoma cell-line, reducing the possibility of off-target effects. Depletion of GAGE genes did not alter cell colony formation in SK-MEL-37.
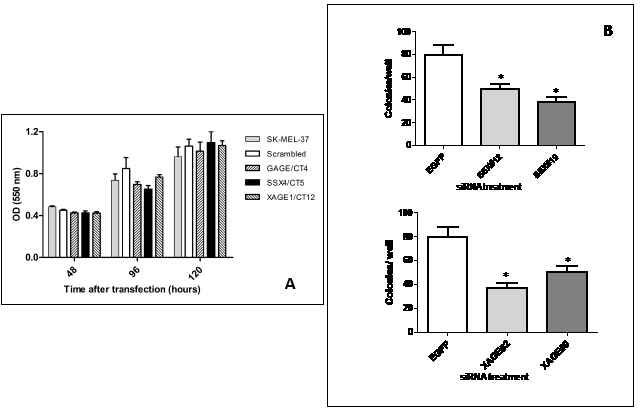
Figure 2: A: Effect of CT-X knockdown cell proliferation as determined by the MTT assay. Results were representative of two experiments with each siRNA. The knock down levels in these experiments were confirmed by real-time PCR. Data are means ± SD.
B: siRNA duplexes specific to SSX and XAGE1 inhibit colony formation of SK-MEL-37 cell line. Significantly reduced colony numbers transfected with SSX#12 and SSX#19 and XAGE1#2 and XAGE1#9 were observed as compared to cells transfected with non-targeting siRNA. All experiments were repeated at least three times and representative data are presented. The knock down levels of SSX4 and XAGE1 in these experiments were confirmed by real-time PCR. Bars, SD. *, P < 0.05 relative to non-targeting siRNA.
Table 2: Degree and specificity of gene knock down
PCR Probe 27mer duplex |
MAGEA3 |
GAGE |
SSX4 |
NY-ESO-1 |
MAGEC1 |
XAGE1 |
Scrambled |
1 |
1 |
1 |
1 |
1 |
1 |
GAGE#15 |
0.823 |
0.056 |
1.112 |
0.556 |
0.814 |
0.821 |
GAGE#9 |
0.974 |
0.050 |
0.569 |
0.339 |
0.749 |
0.538 |
SSX#12 |
1.029 |
0.924 |
0.054 |
0.659 |
0.688 |
0.911 |
SSX#19 |
0.458 |
0.638 |
0.088 |
0.458 |
0.350 |
0.405 |
XAGE1#2 |
0.427 |
0.814 |
1.142 |
0.520 |
0.699 |
0.086 |
XAGE1#9 |
0.323 |
0.638 |
0.373 |
0.723 |
0.498 |
0.030 |
Effects on of CT-X knockdown on SK-MEL-37 migration and invasion.
To determine the possible role of CT-X in the migration of melanoma cells we used a transwell migration assay. siRNAs specific to GAGE and XAGE1 significantly inhibited migration of melanoma cells (Figure 3A and B), while SSX4 siRNA had no effect on cell migration. The influence of GAGE and XAGE1 expression on cell invasion was also assessed using a modified Boyden chamber assay. A consistent and significant decrease in invasion of SK-MEL-37 with decreased GAGE and XAGE1 levels was observed (Figure 3) (p<0.05). 27mer siRNA mediated silencing of GAGE and XAGE1 using a different duplex equally reduced invasion of SK-MEL-37 (Figure 3). GAGE and XAGE1 knockdown also significantly decreased transwell migration and invasion in another melanoma cell line (SK-MEL-119) (Figure 4), suggesting that these genes may have a positive effect on melanoma cell migration and invasion. However, the siRNAs specific to XAGE1 had no effect on migration of a XAGE1-negative melanoma cell line, SK-MEL-124 (data not shown).
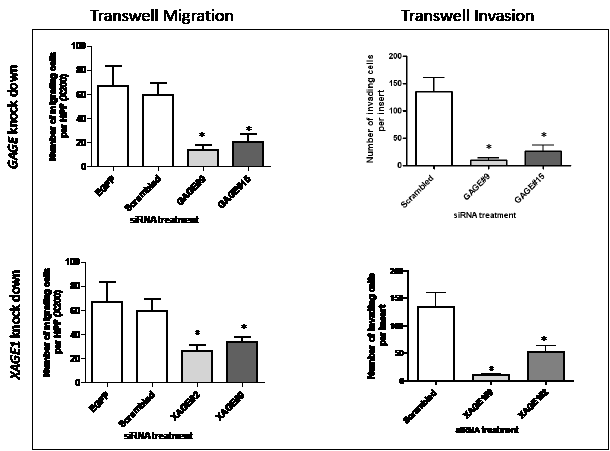
Figure 3: Depletion of GAGE and XAGE1 in the melanoma cell line SK-MEL-37 results in reduced migration and invasion. Transwell migration and invasion assays with SK-MEL-37 cells treated with nontargeting siRNA, GAGE-specific or XAGE1-specific siRNAs. All experiments were repeated at least three times and representative data are presented. The knock down levels of GAGE and XAGE1 in these experiments were confirmed by real-time PCR. Bars, SD. *, P < 0.05 relative to non-targeting siRNA.
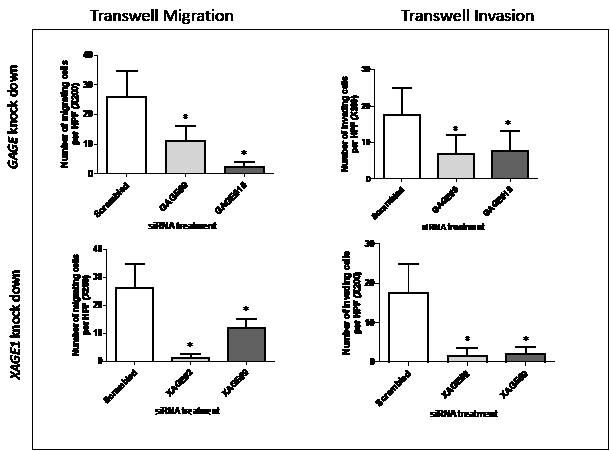
Figure 4: Depletion of GAGE and XAGE1 in the melanoma cell line SK-MEL-119 results in reduced migration and invasion. Transwell migration and invasion assays with SK-MEL-119 cells treated with nontargeting siRNA, GAGE-specific or XAGE1-specific siRNAs. All experiments were repeated at least three times and representative data are presented. The knock down levels of GAGE and XAGE1 in these experiments were confirmed by real-time PCR. Bars, SD. *, P < 0.05 relative to non-targeting siRNA.
In vitro effects of GAGE shRNA induction
To confirm the function of GAGE in the tumor migration and invasion processes, we used a melanoma cell system with inducible expression of GAGE shRNA (double-stable Tet-On/GAGE shRNAmir). Addition of doxycycline (DOX) induces GAGE shRNA in SK-MEL-37 and almost complete depletion of GAGE protein can be observed one week after exposure to DOX (Figure 5). Using this system, the role of GAGE in the migratory properties of tumor cells was examined in the transwell assay. We observed that cells exposed to DOX migrated significantly less than untreated cells (Figure 5B), while no effect was observed in the parental cells treated with DOX (data not shown). The inducible GAGE shRNA system was also tested in proliferation assays. Consistent with the results obtained with the transient siRNA transfections, no effect was observed on the rate of cell proliferation (data not shown).
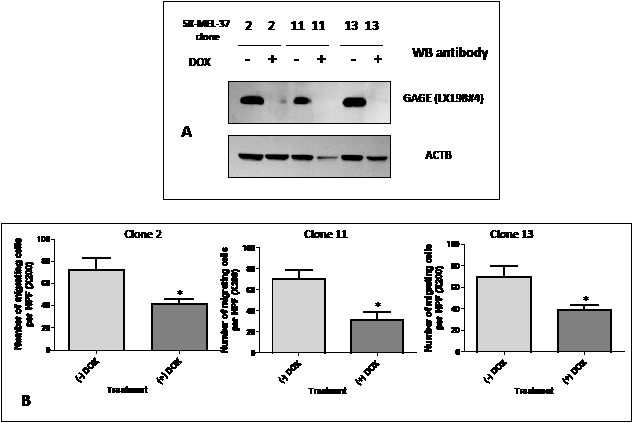
Figure 5: A: Western blot analysis of GAGE expression in double-stable Tet-On/GAGE shRNAmir clones, untreated (- DOX) or treated with doxycycline (+ DOX). Reduction of GAGE protein levels to almost complete depletion was present seven days after exposure to DOX.
B: Analysis of the migratory properties using a modified Boyden chamber assay of three double-stable Tet-On/GAGE shRNAmir clones, untreated (-DOX) or treated with doxycycline (+DOX). All experiments were repeated at least two times and representative data are presented. The knock down levels of GAGE in these experiments were confirmed by western blot. Bars, SD. *, P < 0.05 relative to non-DOX treated cell line.
Table 3: Effect of GAGE and XAGE1 knock down on migration of cell lines from different origins
Cell line |
Origin |
GAGE siRNA (effect on migration) 1 |
GAGE status2 |
XAGE1 siRNA1 (effect on migration) |
XAGE1 status |
SK-MEL-37 |
Melanoma |
Decreased |
+++ |
Decreased |
+++ |
SK-MEL-119 |
Melanoma |
Decreased |
+++ |
Decreased |
+++ |
LM-MEL-34 |
Melanoma |
No effect |
+++ |
NT |
+++ |
SK-MEL-128 |
Melanoma |
NT |
+++ |
Decreased |
+++ |
SK-MEL-124 |
Melanoma |
NT |
++ |
No effect |
Negative |
SK-MEL-131 |
Melanoma |
NT |
+++ |
Decreased |
+++ |
U343MG |
Glioma |
Decreased |
++ |
NT |
Negative |
A172 |
Glioma |
No effect |
+++ |
NT |
+ |
SK-LC-19 |
Lung |
Decreased |
+++ |
Decreased |
+++ |
SK-LC-5 |
Lung |
NT |
+ |
Decreased |
+++ |
DU145 |
Prostate |
No effect |
Negative |
Decreased |
+++ |
22RV1 |
Prostate |
No effect |
++ |
Decreased |
+++ |
NT: not tested
1 As determined by transwell assays
2GAGE and XAGE1 status were determined by semiquantitative RT-PCR and evaluation of the intensity of the band in ethidium bromide stained agarose gels.
Effects on of GAGE and XAGE1 knockdown on migration of cell lines from different origins.
To evaluate the effects of GAGE and XAGE1 knockdown on the migration of additional GAGE and XAGE1-overexpressing cell lines (Table 3). Cell lines were tested for the expression of GAGE and XAGE1 by RT-PCR, for their ability to migrate through transwell membranes and lastly for the efficiency of the transfection using the GAGE and XAGE1 siRNAs and Lipofectamine 2000. For XAGE1, in addition to the two melanoma cell lines shown previously (Figures 3 and 4), transwell migration was also significantly reduced in two other melanoma cell lines, two lung cancer cell lines and two prostate cancer cell lines. Neither of the two XAGE1-specific siRNAs had effect on migration of SK-MEL-124, which does not express XAGE1. While treatment with XAGE1 siRNA decreased migration in all XAGE1 expressing cell lines tested, the treatment with GAGE-specific siRNA failed to decrease migration in three cell lines tested (LM-MEL-34, A172 and 22RV1) that expressed high levels of GAGE, although gene knock down, assessed by real-time PCR, was achieved.
Discussion
Overall, our results suggest that inhibition on SSX4, XAGE1 and GAGE expression in cancer cell lines interferes with tumor cell migration and/or reduce cell viability. We demonstrate that the observed RNAi-induced phenotype is probably a result of the suppression of CT-antigen expression and not an off-target effect. The finding that multiple siRNAs targeting different regions of the same gene have the same phenotypic effect indicates that these effects are indeed dependent on gene depletion.
Effects of members of the MAGEA family on survival of cancer cell lines have been shown before, using a similar approach used in our study [20-22, 24, 25], but the effects of depletion of GAGE, XAGE1 and SSX4 have not been previously investigated.
SSX proteins are thought to act as transcriptional corepressors. They were identified as fusion partners of the SS18 gene in synovial sarcomas carrying t(X;18) translocations [26], which typically, result in fusion of the 78 most C-terminal amino acids of SSX genes to SS18, replacing its eight most C-terminal amino acids. SSX presents two transcriptional repressor domains, a Kruppel-associated box (KRAB) and an SSX repressor domain (SSXRD), both retained in the SS18-SSX fusion proteins [27]. Both SS18 and SSX gene products, together with the fusion proteins, are localized in the nucleus but lack obvious DNA binding motifs. SS18-SSX was shown to block the tumor-suppressive function of p53 in the absence of inactivating p53 mutations by increasing its degradation, therefore promoting cell survival [28]. We speculate that the effects of SSX4 inhibition seen in this study may be analogous to the effects of inhibition of the SS18-SSX fusion proteins.
The XAGE family of genes was first identified in a search for expressed sequence tags (ESTs) homologous to another CT gene, CT16/PAGE-4. This approach led to the identification of three novel PAGE-GAGE-related genes termed XAGE-1, -2 and -3 [29]. The GAGEs, PAGEs, and XAGEs form one large supercluster of related genes, which are expressed in various reproductive tissues as well as in different tumors [29]. One transcript variant (XAGE-1b), was identified as a dominant antigen recognized by sera from lung adenocarcinoma patients [30]. XAGE-1b mRNA expression was also observed in 26% of prostate cancer specimens [31].
GAGE-1 was the first gene of the GAGE family to be identified from the melanoma cell line MZ2-Mel. Subsequent screening of the MZ2-MEL cDNA library with a GAGE-1 probe identified five cDNAs, designated GAGE-2, -3, -4, -5, and -6 sharing nucleotide identities with the GAGE-1 sequence. GAGE-1 differs from these other GAGE genes by the presence of a 143-bp insertion. The GAGE genes are located on chromosome Xp11 Protein products of GAGE genes have more than 95% of homology. GAGE gene transcripts have been found in numerous types of cancers, most frequently in melanomas and lung adenocarcinomas [32, 33], in which up to 54% of specimens were found to express GAGE, as well as in gastric cancers [34] and hepatocellular carcinomas [35]. Using a panel of mAbs, GAGE protein expression was identified in specimens of malignant melanoma, breast carcinoma, bladder carcinoma, lung carcinoma, liver carcinoma, thyroid carcinoma, mesothelioma and germinal cell cancers [36]. GAGE expression in melanoma cell lines ranged from 41% to 58% and in melanoma tissues from 22% to 53%. Immunohistochemical analysis of melanoma tumors revealed a rather heterogeneous expression of GAGE resulting in individual positive cells or foci of stained cells. Furthermore, autoantibodies against GAGE family proteins were detectable in 6% of melanoma patients [33]. GAGE has been correlated with poor prognosis in stomach cancer, esophageal carcinoma and neuroblastoma [34, 37, 38]. The function of GAGE proteins remains largely unknown, although antiapoptotic properties of GAGE-7 have been reported [39, 40].
We observed inhibition of cell migration and invasion in this study following the knock down of XAGE1 and GAGE genes, both members of the GAGE family indicating that targeting of these genes may be useful for cancer treatment. The effects of XAGE1 inhibition were very consistent, not only in melanoma cell lines, but also in cell lines from other tumors, including lung adenocarcinomas and prostate cancer that were shown to present frequent expression of XAGE1 [30, 31]. Although the mechanism of the impairment of migration following XAGE1 or GAGE knock down was not yet elucidated, these results warrant further investigation of CT-X as therapeutic targets for melanoma and other malignancies.
Material and methods
Cell lines and tumor tissues
The cell lines SK-MEL-37, SK-MEL-119, SK-MEL-124, SK-MEL-128 and SK-MEL-131 were obtained from the cell culture bank of the New York Branch of the Ludwig Institute for Cancer Research. They were maintained in RPMI medium containing 10% fetal bovine serum (FBS) and non-essential amino acids. These cell lines were selected for study because they express high levels of the CT genes analyzed.
27mer siRNA oligonucleotide design – Dicer substrate RNAs
Dicer-Substrate RNAs are chemically synthesized 27-mer RNA duplexes that are optimized for Dicer processing and show increased potency when compared with 21-mer duplexes [41, 42]. The duplexes were chosen by a rational design algorithm that integrates both traditional 21-mer siRNA design rules as well as new 27-mer design criteria available at http://www.idtdna.com/Scitools/Applications/RNAi/RNAi.aspx. The approximately 20 options identified by the algorithm in each case were optimized at several levels. We first level aimed to exclude off-target complementarity. This was undertaken with the BLAST tool at NCBI adjusted for analyzing short sequences (http://www.ncbi.nlm.nih.gov/BLAST/). Sequences were excluded if total or partial complementarity with other genes was noted. Further selection was based on published criteria for selection of active siRNA [43, 44]. siRNA sequences were designed to target all isoforms and possible members of the gene families. The selected siRNA sequences are shown in Table 1.
Gene downregulation by 27-mer siRNAs
siRNAs were purchased from IDT (Integrated DNA Technologies, Coralville, IA). The RNAs were resuspended in the RNase-free Duplex Buffer (IDT, Coralville, IA) to 20 µM final concentration; vortexed thoroughly, microfuged and heated to 94°C for 2 minutes, and allowed to cool to room temperature to ensure that the formation of duplexes. Once hydrated, duplexes were stored at -80oC in aliquots. A scrambled universal negative control RNA duplex (DS Scrambled Neg) and a siRNA specific to EGFP, both absent in human, mouse, and rat genomes, and a positive control Dicer-Substrate RNA duplex (HPRT-S1 DS Positive Control), targeting HPRT (hypoxanthine guanine phosphoribosyltransferase 1) and prevalidated to give >90% knockdown of HPRT when transfected at 10 nM concentration, were also purchased from IDT (IDT, Coralville, IA) and used as negative and positive controls, respectively. siRNA duplexes were used to transfect the melanoma cell lines cells using LipofectamineTM 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s recommended protocols to a final concentration of 10nM. Briefly, 1 × 105 cells were seeded into 60mm dishes containing antibiotic-free medium and incubated overnight to reach a density of 50-70%. For each dish, 5 µL of 10µM siRNA solution was mixed with 500 µl of OPTI-MEM I. The mixture was then combined with a solution of 10 µL lipofectamine 2000 in 500 µL OPTI-MEM I and, after 20 minutes at RT, the mixture was applied to the cells, and the final concentration in the dish was 10 nmol/l for each siRNA. After incubation for 24 h at 37℃, the medium was changed to RPMI-1640 supplemented with serum and cells were then cultured for an additional 24-48 h at 37℃ before analysis.
RNA extraction and reverse transcription
Total RNA from the cell pellets was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA). RNA amounts were estimated by spectrophotometric analysis (Nanophotometer, Implen, Germany). One µg of RNA was reverse transcribed into cDNA by using an Omniscript RT kit according to the manufacturer’s protocol using oligo (dT)18 primers.
Semi-quantitative reverse transcription -PCR
RT-PCR was undertaken with Jump-Start master mix (Sigma) plus 10 pmol of each of the following primers (5’-3’): GAGE F: GACCAAGACGCTACGTAG, GAGE R: CCATCAGGACCATCTTCA, XAGE1F: TCCCAGGAGCCCAGTAATGGAGA, XAGE1R: CAGCTTGTCTTCATTTAAACTTGTGGTTGC, ACTBF: AAATCTGGCACCACACCTTC, ACTBR: CACTGTGTTGCCGTACAGGT. The amplification involved three stages in which the annealing temperature was higher (60°C) in the first ten cycles and reduced in two degrees in the following stage (ten cycles) and other two degrees in the last 15 cycles and involved an initial denaturation at 94°C for 5min. Each cycle consisted of a denaturation step at 94°C for 30s, followed by 30 s at the annealing temperature and extension at 72°C for 30 s followed by a final 7-min extension. Controls without DNA and using cDNA of testis as a positive control were carried out for each set of reaction. PCR products were loaded onto 2% agarose gels, stained with ethidium bromide and visualized by UV illumination. The predicted sizes of the PCR products were 243 bp and 257 bp for GAGE and XAGE1, respectively. A 644 fragment from ACTB was amplified as an endogenous control.
Quantitative real-time reverse transcription-PCR
cDNA samples were run in duplicate for the genes of interest and for the reference gene within the same experiment using the Applied Biosystem apparatus 7500 Fast Real-Time PCR system and Taqman platform (Applied Biosystems, Foster City, CA). TFRC was amplified as an internal reference gene. The PCR primers and probes for all tested genes (MAGEA3, Hs00366532_m1; GAGE1, Hs00275620_m1; SSX4, Hs00171942_m1; NY-ESO-1, Hs00265824_m1; MAGEC1, Hs00193821_m1; XAGE1, Hs00220764_m1) and TFRC internal control gene (4326323E) were purchased from Applied Biosystems. Primers used for PCR amplification were chosen to encompass intron between exon sequences to avoid amplification of genomic DNA (Applied Biosystems, Foster City, CA). The gene-specific probes were labeled with the reporter dye 6-FAM at the 5’-end. The TFRC probe was labeled with a reporter dye (VIC) to the 5’-end of the probe and all probes had minor groove binder/nonfluorescent quencher at the 3’-end of the probe (Applied Biosystems, Foster City, CA). The PCR conditions were 95°C for 10 minutes followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Duplicate threshold cycles (CT) were averaged for each sample. Relative quantification of gene expression (relative amount of target RNA) was determined using the equation 2−ΔΔCT.
Determination of rate of cell proliferation.
The rate of proliferation was determined using the 3-(4,5-dimethyl thizol-2-yl) 2,5-diphenyl tetrazolium bromide (MTT) assay (Roche, Indianapolis, IN). Cells (5x103cells per well) were incubated in 96-well plates and maintained in complete medium 24 h after transfection. After 48, 96 and 120 hours, 10 µL of sterile MTT dye was added to the cells and incubated for 4 hours at 37°C, and then 100 µL of solubilization buffer was added. Spectrometric absorbance at a wavelength of 550 nm was measured on an enzyme immunoassay analyzer (Molecular Devices) after overnight incubation. Experiments were performed at least three times, with six replicate measurements, and data are presented as the average OD ± SD.
Migration and invasion assays
Cell migration and invasion were assessed in 12-well Boyden Chambers (BD Biosciences, San Diego, CA) according to the protocol of the manufacturer. Invasion assays were carried out in chamber equipped with an 8 µm polycarbonate membrane coated with Matrigel. Briefly, cells were serum-starved for 2 hr, and 500 µl containing 25,000 cells in medium supplemented with 1% FBS were loaded into the upper chamber. The lower chamber contained medium supplemented with 10% FBS as the chemoattractant. Cells were incubated at 37°C overnight, fixed in 10% formalin for 20 min and stained with 0.2% crystal violet (Fisher Scientific, Pittsburgh, PA). Non-invading cells on the top of the membrane were wiped off using cotton swabs, and invading cells affixed to the underside of the membranes on each insert were counted at 100 x magnification in 10 random areas. The migration assay was done in a similar fashion except the 8.0-µm pore size membrane inserts were not coated with Matrigel. Results were expressed as mean ± SD. Experiments were performed at least three times.
Colony formation assay
At 48h after transfection with each siRNA, cells were trypsinized, counted and 1,000 cells were seeded in duplicate in 6-well plates and allowed to form colonies for 2 weeks. The colonies were fixed with 10% formalin and stained with 0.2% crystal violet (Fisher Scientific, Pittsburgh, PA). The number of colonies with 30 cells or larger than 1mm in diameter in each well was counted. Experiments were repeated at least three times.
Western blotting analyses
Cells were harvested and washed with cold phosphate-buffered saline solution, and total proteins were extracted in the extraction buffer (50 mM Tris-Cl pH 7.4, 0.15 M NaCl, 2mM EDTA 1% NP40), containing protease inhibitors (Protease Inhibitor Cocktail, Roche, Indianapolis, IN). Equal amounts of protein (20 µg per lane) were mixed with an equal volume of 2x loading buffer (125 mM Tris-HCl pH 6.8, 4% SDS, 10% glycerol, 0.006% bromophenol blue, 2% β-mercaptoethanol), incubated at 95°C for 3 min, and loaded in 10% SDS Bis-Tris gels (Invitrogen, Carlsbad, CA). After electrophoresis, proteins were transferred to nitrocellulose membranes. The membranes were blocked by incubation in PBST (PBS 0.1% Tween 20) 3% bovine serum albumin (BSA) for 1 h, then incubated with the primary antibody overnight at 4°C in PBST 1% BSA. After washing four times in PBST, the membranes were incubated either with peroxidase-conjugated anti-rabbit or anti-mouse IgG (Jackson Immunoresearch, Bar Harbor, ME) for 1 h at room temperature. Antibody binding was detected using the system Western Lightening Chemiluminescence Reagent Plus (Perkin Elmer, Emeryville, CA). The antibodies used were: monoclonal antibodies anti-NY-ESO-1 (E978, Sigma-Aldrich, St. Louis, MO), anti-MAGEC1 (CT7.33, Sigma-Aldrich, St. Louis, MO), anti-MAGEC2 (CT10#5), anti-GAGE LX198, anti-SSX (LX#1), anti-MAGEA (6C1, Santa Cruz Biotechnology, Santa Cruz, CA) and a rabbit polyclonal anti-actin (20-33, Sigma-Aldrich, St. Louis, MO).
Establishment of the Double-stable Tet-On/GAGE shRNAmir cell lines
The double-stable Tet-On/GAGE shRNAmir SK-MEL-37 cell line was established using the Tet-On Advanced Inducible Gene Expression System (Clontech Laboratories). SK-MEL-37 was initially transfected with 5 μg of the tetracycline-controlled transactivator (pTet-On advanced) encoding the G418-resistant gene. After selection, a clone that presented high levels of expression of the transativator as assessed by the transfection of a reporter plasmid containing tetracycline response elements (TRE) within the promoter was identified. The pTet-On advanced cells were then submitted to retroviral infection with the microRNA-adapted retroviral vector pTMP (Open Biosystems), containing TET responsive promoter and the puromycin-resistant gene. Double-stable cells were then selected and further screened for GAGE protein expression by Western blot using the anti-GAGE LX198 antibody four days after exposure to 1μg/ml of doxycycline. For cloning in pTMP, a standard GAGE 21mer was created from the dicer substrate RNAi (GAACCAGCAACUCAACGUCAGGATC) by removing bases on the 3’ end of the sense strand and on the 5’end of the antisense strand.
Statistical analyses:
Statistical comparisons were performed using analysis of variance for analysis of significance between different values using GraphPad Prism software (San Diego, CA). Values are expressed as mean with SD from an experiment representative of at least three separate experiments, and differences were considered significant at a p value of less than 0.05.
Acknowledgements:
The authors would like to thank Dr. Lloyd J. Old for his important contribution not only to the current study, but also for his mentoring and support over the years. We are thankful to Erika Ritter, from LICR NY branch, for providing the cell lines used in this study. This work was conducted as part of the Hilton-Ludwig Cancer Metastasis Initiative, funded by the Conrad N. Hilton Foundation and the Ludwig Institute for Cancer Research.
References
1. Simpson AJ, Caballero OL, Jungbluth A, Chen YT and Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nature reviews. 2005; 5(8):615-625.
2. Brichard VG and Lejeune D. Cancer immunotherapy targeting tumour-specific antigens: towards a new therapy for minimal residual disease. Expert Opin Biol Ther. 2008; 8(7):951-968.
3. Tyagi P and Mirakhur B. MAGRIT: the largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clin Lung Cancer. 2009; 10(5):371-374.
4. Caballero OL and Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009; 100(11):2014-2021.
5. Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, Ritter G, Simpson AJ, Chen YT, Old LJ and Altorki NK. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. 2005; 11(22):8055-8062.
6. Kim J, Reber HA, Hines OJ, Kazanjian KK, Tran A, Ye X, Amersi FF, Martinez SR, Dry SM, Bilchik AJ and Hoon DS. The clinical significance of MAGEA3 expression in pancreatic cancer. Int J Cancer. 2006; 118(9):2269-2275.
7. Li M, Yuan YH, Han Y, Liu YX, Yan L, Wang Y and Gu J. Expression profile of cancer-testis genes in 121 human colorectal cancer tissue and adjacent normal tissue. Clin Cancer Res. 2005; 11(5):1809-1814.
8. Andrade VC, Vettore AL, Felix RS, Almeida MS, Carvalho F, Oliveira JS, Chauffaille ML, Andriolo A, Caballero OL, Zago MA and Colleoni GW. Prognostic impact of cancer/testis antigen expression in advanced stage multiple myeloma patients. Cancer Immun. 2008; 8:2.
9. Atanackovic D, Luetkens T, Hildebrandt Y, Arfsten J, Bartels K, Horn C, Stahl T, Cao Y, Zander AR, Bokemeyer C and Kroger N. Longitudinal analysis and prognostic effect of cancer-testis antigen expression in multiple myeloma. Clin Cancer Res. 2009; 15(4):1343-1352.
10. Taylor BJ, Reiman T, Pittman JA, Keats JJ, de Bruijn DR, Mant MJ, Belch AR and Pilarski LM. SSX cancer testis antigens are expressed in most multiple myeloma patients: co-expression of SSX1, 2, 4, and 5 correlates with adverse prognosis and high frequencies of SSX-positive PCs. J Immunother. 2005; 28(6):564-575.
11. Jungbluth AA, Ely S, DiLiberto M, Niesvizky R, Williamson B, Frosina D, Chen YT, Bhardwaj N, Chen-Kiang S, Old LJ and Cho HJ. The cancer-testis antigens CT7 (MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma and correlate with plasma-cell proliferation. Blood. 2005; 106(1):167-174.
12. Condomines M, Hose D, Raynaud P, Hundemer M, De Vos J, Baudard M, Moehler T, Pantesco V, Moos M, Schved JF, Rossi JF, Reme T, Goldschmidt H and Klein B. Cancer/testis genes in multiple myeloma: expression patterns and prognosis value determined by microarray analysis. J Immunol. 2007; 178(5):3307-3315.
13. Yakirevich E, Sabo E, Lavie O, Mazareb S, Spagnoli GC and Resnick MB. Expression of the MAGE-A4 and NY-ESO-1 cancer-testis antigens in serous ovarian neoplasms. Clin Cancer Res. 2003; 9(17):6453-6460.
14. Patard JJ, Brasseur F, Gil-Diez S, Radvanyi F, Marchand M, Francois P, Abi-Aad A, Van Cangh P, Abbou CC, Chopin D and et al. Expression of MAGE genes in transitional-cell carcinomas of the urinary bladder. Int J Cancer. 1995; 64(1):60-64.
15. Kurashige T, Noguchi Y, Saika T, Ono T, Nagata Y, Jungbluth A, Ritter G, Chen YT, Stockert E, Tsushima T, Kumon H, Old LJ and Nakayama E. Ny-ESO-1 expression and immunogenicity associated with transitional cell carcinoma: correlation with tumor grade. Cancer Res. 2001; 61(12):4671-4674.
16. Velazquez EF, Jungbluth AA, Yancovitz M, Gnjatic S, Adams S, O’Neill D, Zavilevich K, Albukh T, Christos P, Mazumdar M, Pavlick A, Polsky D, Shapiro R, Berman R, Spira J, Busam K, et al. Expression of the cancer/testis antigen NY-ESO-1 in primary and metastatic malignant melanoma (MM)--correlation with prognostic factors. Cancer Immun. 2007; 7:11.
17. Bai S, He B and Wilson EM. Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol Cell Biol. 2005; 25(4):1238-1257.
18. Nagao T, Higashitsuji H, Nonoguchi K, Sakurai T, Dawson S, Mayer RJ, Itoh K and Fujita J. MAGE-A4 interacts with the liver oncoprotein gankyrin and suppresses its tumorigenic activity. J Biol Chem. 2003; 278(12):10668-10674.
19. Peikert T, Specks U, Farver C, Erzurum SC and Comhair SA. Melanoma antigen A4 is expressed in non-small cell lung cancers and promotes apoptosis. Cancer Res. 2006; 66(9):4693-4700.
20. Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, Rodolfo M and Schneider C. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103(30):11160-11165.
21. Kondo T, Zhu X, Asa SL and Ezzat S. The cancer/testis antigen melanoma-associated antigen-A3/A6 is a novel target of fibroblast growth factor receptor 2-IIIb through histone H3 modifications in thyroid cancer. Clin Cancer Res. 2007; 13(16):4713-4720.
22. Liu W, Cheng S, Asa SL and Ezzat S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 2008; 68(19):8104-8112.
23. Doyle JM, Gao J, Wang J, Yang M and Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell. 39(6):963-974.
24. Yang B, O’Herrin SM, Wu J, Reagan-Shaw S, Ma Y, Bhat KM, Gravekamp C, Setaluri V, Peters N, Hoffmann FM, Peng H, Ivanov AV, Simpson AJ and Longley BJ. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res. 2007; 67(20):9954-9962.
25. Atanackovic D, Hildebrandt Y, Jadczak A, Cao Y, Luetkens T, Meyer S, Kobold S, Bartels K, Pabst C, Lajmi N, Gordic M, Stahl T, Zander AR, Bokemeyer C and Kroger N. Cancer-testis antigens MAGE-C1/CT7 and MAGE-A3 promote the survival of multiple myeloma cells. Haematologica. 95(5):785-793.
26. Ladanyi M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001; 20(40):5755-5762.
27. de Bruijn DR, van Dijk AH, Willemse MP and van Kessel AG. The C terminus of the synovial sarcoma-associated SSX proteins interacts with the LIM homeobox protein LHX4. Oncogene. 2008; 27(5):653-662.
28. D’Arcy P, Maruwge W, Ryan BA and Brodin B. The oncoprotein SS18-SSX1 promotes p53 ubiquitination and degradation by enhancing HDM2 stability. Mol Cancer Res. 2008; 6(1):127-138.
29. Brinkmann U, Vasmatzis G, Lee B and Pastan I. Novel genes in the PAGE and GAGE family of tumor antigens found by homology walking in the dbEST database. Cancer Res. 1999; 59(7):1445-1448.
30. Nakagawa K, Noguchi Y, Uenaka A, Sato S, Okumura H, Tanaka M, Shimono M, Ali Eldib AM, Ono T, Ohara N, Yoshino T, Yamashita K, Tsunoda T, Aoe M, Shimizu N and Nakayama E. XAGE-1 expression in non-small cell lung cancer and antibody response in patients. Clin Cancer Res. 2005; 11(15):5496-5503.
31. Koizumi F, Noguchi Y, Saika T, Nakagawa K, Sato S, Eldib AM, Nasu Y, Kumon H and Nakayama E. XAGE-1 mRNA expression in prostate cancer and antibody response in patients. Microbiol Immunol. 2005; 49(5):471-476.
32. De Backer O, Arden KC, Boretti M, Vantomme V, De Smet C, Czekay S, Viars CS, De Plaen E, Brasseur F, Chomez P, Van den Eynde B, Boon T and van der Bruggen P. Characterization of the GAGE genes that are expressed in various human cancers and in normal testis. Cancer Res. 1999; 59(13):3157-3165.
33. Bazhin AV, Wiedemann N, Schnolzer M, Schadendorf D and Eichmuller SB. Expression of GAGE family proteins in malignant melanoma. Cancer Lett. 2007; 251(2):258-267.
34. Zambon A, Mandruzzato S, Parenti A, Macino B, Dalerba P, Ruol A, Merigliano S, Zaninotto G and Zanovello P. MAGE, BAGE, and GAGE gene expression in patients with esophageal squamous cell carcinoma and adenocarcinoma of the gastric cardia. Cancer. 2001; 91(10):1882-1888.
35. Kobayashi Y, Higashi T, Nouso K, Nakatsukasa H, Ishizaki M, Kaneyoshi T, Toshikuni N, Kariyama K, Nakayama E and Tsuji T. Expression of MAGE, GAGE and BAGE genes in human liver diseases: utility as molecular markers for hepatocellular carcinoma. J Hepatol. 2000; 32(4):612-617.
36. Gjerstorff MF, Johansen LE, Nielsen O, Kock K and Ditzel HJ. Restriction of GAGE protein expression to subpopulations of cancer cells is independent of genotype and may limit the use of GAGE proteins as targets for cancer immunotherapy. Br J Cancer. 2006; 94(12):1864-1873.
37. Cheung IY, Chi SN and Cheung NK. Prognostic significance of GAGE detection in bone marrows on survival of patients with metastatic neuroblastoma. Med Pediatr Oncol. 2000; 35(6):632-634.
38. Kong U, Koo J, Choi K, Park J and Chang H. The expression of GAGE gene can predict aggressive biologic behavior of intestinal type of stomach cancer. Hepatogastroenterology. 2004; 51(59):1519-1523.
39. Kular RK, Yehiely F, Kotlo KU, Cilensek ZM, Bedi R and Deiss LP. GAGE, an antiapoptotic protein binds and modulates the expression of nucleophosmin/B23 and interferon regulatory factor 1. J Interferon Cytokine Res. 2009; 29(10):645-655.
40. Cilensek ZM, Yehiely F, Kular RK and Deiss LP. A member of the GAGE family of tumor antigens is an anti-apoptotic gene that confers resistance to Fas/CD95/APO-1, Interferon-gamma, taxol and gamma-irradiation. Cancer Biol Ther. 2002; 1(4):380-387.
41. Kim DH and Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007; 8(3):173-184.
42. Amarzguioui M, Lundberg P, Cantin E, Hagstrom J, Behlke MA and Rossi JJ. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat Protoc. 2006; 1(2):508-517.
43. Kurreck J. siRNA Efficiency: Structure or Sequence-That Is the Question. J Biomed Biotechnol. 2006; 2006(4):83757.
44. Patzel V. In silico selection of active siRNA. Drug Discov Today. 2007; 12(3-4):139-148.