
INTRODUCTION
Acute myeloid leukemia (AML) remains a devastating disease. The overall survival rate for adults is only about 25% [1] and although the overall survival rate for children is around 65% [2], it lags significantly behind that of acute lymphoblastic leukemia, which is about 90% for the pediatric population [3]. A major cause of treatment failure is resistance to cytarabine (ara-C) and anthracycline [e.g., daunorubicin (DNR)]-based chemotherapy [4]. Thus, novel therapies are needed to overcome chemoresistance and improve the overall survival of AML patients.
One strategy to overcome chemoresistance is to target the anti-apoptotic Bcl-2 proteins, as they have been demonstrated to be associated with poor clinical outcome and chemoresistance in leukemic cell line models [5–8]. ABT-199 is the first Bcl-2-selective inhibitor that has demonstrated promising results in multiple cancers, including AML [9–15], though drug resistance remains a concern. We and others have previously demonstrated that Mcl-1 plays a key role in resistance to ABT-199 [12, 16–18]. In our most recent study, we demonstrate that induction of Mcl-1 by ABT-199 represents an intrinsic mechanism of resistance to the agent in AML cells [19]. Thus, rationally designed combinations could prove to be a more effective option for the treatment of AML with ABT-199.
Targeting the DNA damage response (DDR) is another approach to overcome chemotherapy resistance. The DDR involves multiple signaling pathways through which cells maintain genomic integrity following various stresses [20–23]. Checkpoint kinase 1 (CHK1) plays a central role in the DDR and its inhibition can affect replication initiation, replication fork stability, homologous recombination repair, progression of the cell cycle, and the S and G2/M cell cycle checkpoints [20–22, 24]. In addition, CHK1 inhibition has been demonstrated to result in DNA damage [25] and DNA damage has been shown to result in decrease of Mcl-1 expression [26]. Therefore, we hypothesize that inhibition of CHK1 may enhance the cytotoxic effects of ABT-199 by decreasing Mcl-1 expression.
Here, we evaluated the CHK1-selective inhibitor LY2603618 in combination with ABT-199 in AML cell lines and primary patient samples. We demonstrated that LY2603618 treatment resulted in DNA damage and decrease of Mcl-1 expression, which coincided with the initiation of apoptosis. Simultaneous combination of LY2603618 and ABT-199 resulted in synergistic induction of cell death in both AML cell lines and primary patient samples. These findings provide new insights into overcoming intrinsic ABT-199 resistance in AML cells and supports clinical development of the combination of ABT-199 and CHK1 inhibitors.
RESULTS
LY2603618 has antileukemic activity against primary AML patient samples and AML cell lines
To investigate LY2603618 sensitivity in AML patient samples and cell lines, first we determined ex vivo LY2603618 sensitivity in freshly isolated primary AML blast samples obtained either at initial diagnosis (n = 22) or at relapse (n = 4) by MTT (3-[4, 5-dimethyl-thiazol-2-yl]-2, 5-diphenyltetrazoliumbromide) assays. For the majority of the primary patient samples (n = 23), the LY2603618 IC50s were less than 9 μM, which is the maximum clinically achievable concentration of LY2603618 [27]. Interestingly, there was no significant difference between the median LY2603618 IC50 for the AML blasts obtained at initial diagnosis and relapse (p = 0.749, Table 1 and Figure 1A). Next, we determined CHK1 transcript levels in the primary AML patient samples by real-time RT-PCR. CHK1 transcript levels did not correlate with LY2603618 sensitivities in the samples (Figure 1B). Then we tested LY2603618 sensitivities in 11 AML cell lines by MTT assays. The IC50s of LY2603618 in these cell lines ranged from about 0.1–1.6 μM after 72 h treatment (Figure 1C). Consistent with the lack of a correlation between CHK1 transcript levels and LY2603618 IC50s in the primary patient samples, ectopic expression of CHK1 in THP-1 AML cell line had no impact on LY2603618 sensitivity, as assessed by MTT assay (Figure 1D, the western blot confirming overexpression was published previously [28]). However, shRNA knockdown of CHK1 (50% decrease of CHK1 protein compared to NTC shRNA) resulted in a significant increase of LY2603618 sensitivity in THP-1 cells (1.6-fold, p = 0.023, Figure 1E and 1F).
Table 1: Patient characteristics and LY2603618 sensitivity in primary AML patient samples (n = 26)
Patient |
Gender |
Age (year) |
Disease status |
FAB subtype |
Cytogenetics |
Blast purity (%) |
LY2603618 IC50 (μM) |
Relative CHK1 transcripts |
|---|---|---|---|---|---|---|---|---|
AML#8 |
Male |
55 |
Newly diagnosed |
M2 |
46, XY |
31.5 |
5.6 |
0.04 |
AML#9 |
Female |
35 |
Relapsed |
M5 |
47, XX,+10,t(16;21)(p11;q22),add(11p) |
87 |
2.6 |
0.04 |
AML#10 |
Male |
23 |
Newly diagnosed |
M2 |
46, XY, del(9q) |
52.5 |
1.2 |
0.20 |
AML#11 |
Female |
43 |
Newly diagnosed |
M2 |
46, XX |
91 |
1.8 |
0.03 |
AML#12 |
Male |
6 |
Newly diagnosed |
M3 |
46, XY, t (15;17) (q22;q21) |
85 |
3.8 |
0.14 |
AML#15 |
Female |
63 |
Newly diagnosed |
M2 |
46, XX |
23 |
1.6 |
0.04 |
AML#16 |
Male |
38 |
Newly diagnosed |
M3 |
46, XY, t (15;17) (q22;q21) |
90 |
1.3 |
0.02 |
AML#17 |
Female |
45 |
Newly diagnosed |
M4 |
46, XX |
35.5 |
18.2 |
0.21 |
AML#18 |
Female |
19 |
Relapsed |
M2 |
46, XX, t (8;21) (q22;q22)/46,XX |
54 |
2.3 |
0.20 |
AML#19 |
Male |
4 |
Newly diagnosed |
M4 |
46, XY |
40.5 |
8.3 |
0.40 |
AML#21 |
Male |
42 |
Newly diagnosed |
M4 |
46, XY/44, XY, -17, -19, (11q-?) |
51 |
3.2 |
0.44 |
AML#23 |
Male |
60 |
Newly diagnosed |
M5 |
48, XY,+2,+8, i (12) (q10)/46, XY |
81 |
3.9 |
1.65 |
AML#24 |
male |
59 |
Newly diagnosed |
M2 |
46, XY |
42 |
19.3 |
0.03 |
AML#25 |
Female |
12 |
Newly diagnosed |
M3 |
46, XX, t (15;17) (q22;q21)/46, XX |
88 |
1.1 |
0.16 |
AML#27 |
female |
41 |
Relapsed |
M4 |
47, XX, del (5q), +8, t (15;18) (q12,q23) |
24 |
19.6 |
0.15 |
AML#29 |
male |
9 |
Newly diagnosed |
M4 |
46, XY, t (6; 9) (p22;q34)/46, XY |
31 |
1.5 |
0.08 |
AML#31 |
Male |
17 |
Newly diagnosed |
M2 |
46, XY |
68.5 |
0.7 |
1.17 |
AML#33 |
Female |
76 |
Newly diagnosed |
M5 |
46, XX |
84.5 |
3.5 |
1.55 |
AML#35 |
Male |
65 |
Newly diagnosed |
M5 |
47, XY, add (7q), -16, -17, +marx3 |
76 |
17.8 |
0.89 |
AML#36 |
Male |
43 |
Newly diagnosed |
M2 |
46, XY, t (8;21) (q22;q22) |
48 |
2.7 |
0.50 |
AML#39 |
Male |
50 |
Newly diagnosed |
M2 |
45, X, -Y, t (8;21) (q22;q22), del (11q) |
46 |
4.0 |
0.24 |
AML#40 |
Male |
12 |
Newly diagnosed |
M3 |
46, XY, t (15;17) (q22;q21) |
92.5 |
2.6 |
0.19 |
AML#41 |
Male |
74 |
Newly diagnosed |
M5 |
47, XY,+8 |
95 |
6.5 |
0.23 |
AML#43 |
Male |
19 |
Newly diagnosed |
M2 |
45, X, -Y, t (8;21) (q22;q22), del (9q) |
47 |
3.4 |
0.85 |
AML#44 |
Male |
25 |
Newly diagnosed |
M3 |
46, XY, t (15;17) (q22;q21) |
94 |
3.2 |
0.50 |
AML#45 |
Male |
48 |
Relapsed |
M2 |
46, XY, t (7;11) (p15;p15) |
39.5 |
3.6 |
1.04 |
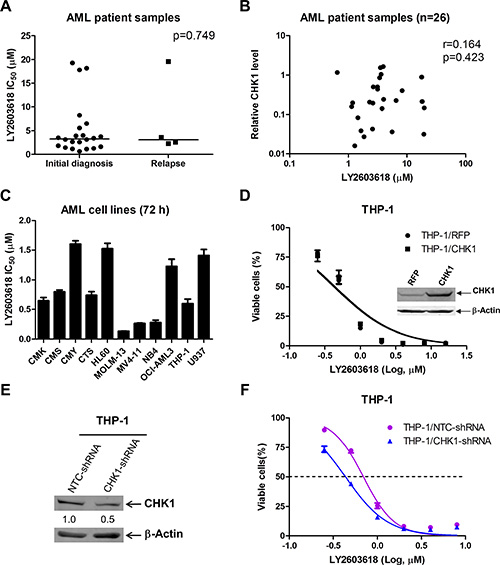
Figure 1: AML cells are sensitive to LY2603618. (A) Primary AML patient samples were treated with variable concentrations of LY2603618 in 96-well plates for 72 h and viable cells were determined using MTT reagent. IC50 values were calculated as drug concentration necessary to inhibit 50% OD590 compared to vehicle control treated cells. The IC50 values are means of duplicates from one experiment due to limited sample. The horizontal lines indicate the median. (B) Total RNAs were isolated from primary AML patient samples and gene transcript levels were determined by Real-time RT-PCR. Transcript levels were normalized to GAPDH and relative expression levels were calculated using the comparative Ct method. The relationship between the CHK1 expression levels and LY2603618 sensitivities was determined by the nonparametric Spearman rank correlation coefficient. (C) LY2603618 sensitivities for AML cell lines were determined using MTT assays. (D) THP-1 cells were infected with Precision LentiORF CHK1 (THP-1/CHK1) or red fluorescent protein control (THP-1/RFP) lentivirus. LY2603618 sensitivities were determined using MTT assays. Whole cell lysates were subjected to Western blotting (Western blot from a previous publication [28]). (E and F) THP-1 cells were transiently infected with CHK1- or non-target control (NTC)-shRNA lentivirus particles overnight, the cells were washed and resuspended in fresh complete media and cultured for 48 h. Then Western blotting and MTT assays were perform in the cells to determine CHK1 knockdown (E) and sensitivity to LY2603618 (F).
LY2603618 induces bak-dependent apoptosis in AML cell lines
To assess the effect of LY2603618 treatment on cell death, first we treated five AML cell lines (CTS, MOLM-13, MV4-11, THP-1, and U937) and one primary AML patient sample (AML #31) with variable concentrations of LY2603618 for 24 h and then subjected them to Annexin V/propidium iodide (PI) staining, and flow cytometry analyses. LY2603618 treatment resulted in concentration-dependent increase in Annexin V positive cells (Figure 2A and 2B). The U937 cells treated with LY2603618 for 24 h were mostly dead, as indicated by AnnexinV+/PI+, so the treatment was performed with a shorter incubation to determine if the cells underwent apoptosis. After 8 h treatment, U937 cells showed concentration-dependent increase in AnnexinV+PI- cells, indicating that the cells underwent apoptosis. LY2603618-induced cell death, in all cell lines tested, was accompanied by cleavage of caspase 3 and PARP (poly ADP ribose polymerase, Figure 2C), demonstrating that the cells underwent apoptosis. To determine if the cells did indeed undergo intrinsic apoptosis, we performed shRNA knockdown of Bax and Bak, at least one of which is required for intrinsic apoptosis [29]. The LY2603618-induced increase of Annexin V positive cells was diminished by shRNA knockdown of Bak, but not Bax, demonstrating that LY2603618 induces Bak-dependent intrinsic apoptosis in AML cells (Figure 2D and 2E).
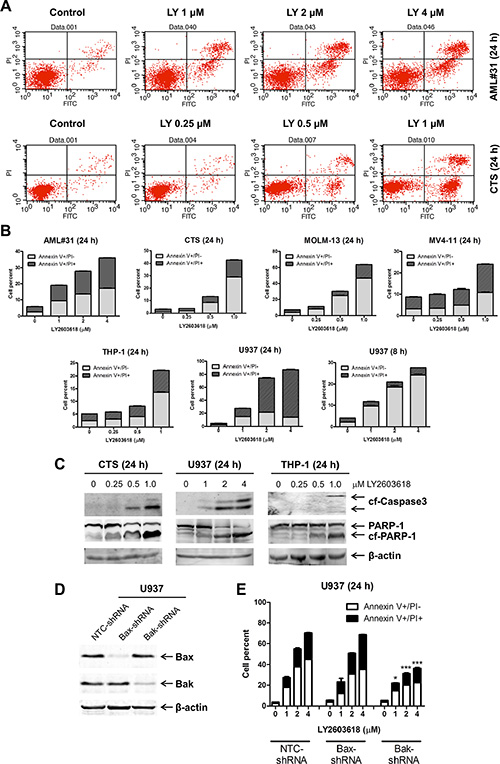
Figure 2: LY2603618 induces apoptosis in AML cell lines and a primary patient sample. (A) AML cells were treated with LY2603618 (LY) for 24 h and then subjected to Annexin V-FITC/PI staining and flow cytometry analyses. Representative dot plots are shown for primary patient AML#31 (top) and CTS cells (bottom). (B) Mean percent Annexin V+ cells ± s.e.m are shown. (C) Whole cell lysates were subjected to Western blotting. (D) U937 cells were infected with NTC-, Bax- or Bak-shRNA lentivirus particles overnight, then washed and incubated for 48 h prior to adding puromycin to the culture medium. Whole cell lysates were subjected to Western blotting. (E) U937 NTC-, Bax-, and Bak-shRNA cells were treated with LY for 24 h and then subjected to Annexin V/PI staining and flow cytometry analysis. *indicates p < 0.05, ***indicates p < 0.0005.
LY2603618 treatment decreases the G2/M cell population and increases DNA damage in AML cell lines
To investigate the effects of LY2603618 treatment on cell cycle progression, we treated CTS, THP-1, and U937 cells with LY2603618 for 24 h. PI staining and flow cytometry analyses revealed a concentration-dependent decrease of the G2/M population accompanied by concentration-dependent increase of sub-G1 population (Figures 3A, S1 and S2). Decreased protein levels for CHK1 were detected after LY2603618 treatment of the U937 cells. Although treatment with high concentrations of LY2603618 resulted in decreased levels of p-CDC25C and p-CDK1 (phosphorylated cyclin-dependent kinase 1), it did not alter the levels of phosphorylated histone H3 (p-H3, a marker for mitosis) (Figure 3B). Increased levels of p-CHK1 (S345), indicative of DNA damage, were detected in LY2603618 treated cells. In addition, there was a concentration-dependent increase of γH2AX post LY2603618 treatments, further suggesting that LY2603618 induced DNA damage in the cells.
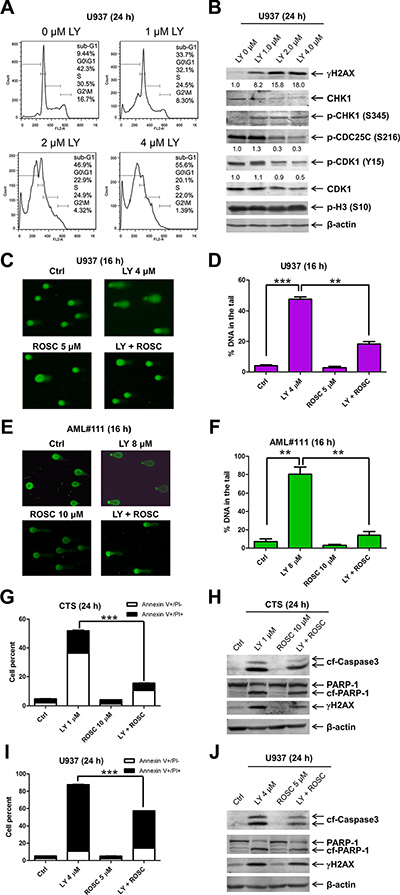
Figure 3: LY2603618 treatment results in DNA double strand breaks. (A) U937 cells were treated with LY for 24 h, then fixed with ethanol and stained with PI for cell cycle analysis. (B) U937 cells were treated with LY for 24 h. Whole cell lysates were subjected to Western blotting. The fold changes for the γH2AX, p-CDC25C, and p-CDK1 densitometry measurements, normalized to β-actin and then compared to no drug treatment control, are indicated. (C) U937 cells were treated with LY and Roscovitine (ROSC), alone or in combination, for 16 h and then subjected to alkaline comet assay analysis. Representative images are shown. (D) Comet assay results are graphed as median percent DNA in the tail from 4 replicate gels ± s.e.m. **indicates p < 0.005 and ***indicates p < 0.0005. (E) Primary AML patient sample AML#111 was treated with LY and Roscovitine (ROSC), alone or in combination, for 16 h and then subjected to alkaline comet assay analysis. Representative images are shown. (F) Comet assay results are graphed as median percent DNA in the tail from 4 replicate gels ± s.e.m. **indicates p < 0.005. (G–J) CTS and U937 cells were treated with LY and ROSC, alone or in combination for 24 h. The cells were subjected to Annexin V/PI staining and flow cytometry analysis (G and I). ***indicates p < 0.0005. Whole cell lysates were subjected to Western blotting (H&J).
To evaluate if LY2603618 treatment truly induced DNA damage and determine the relationship between the induced DNA damage and CDK activity, U937 cells were treated with LY2603618 and Roscovitine (a CDK inhibitor), alone and in combination, for 16 h and then subjected to the alkaline comet assay. As shown in Figure 3C and 3D, LY2603618 treatment resulted in significantly increased percent DNA in the tail, which was significantly attenuated by the addition of Roscovitine. Essentially the same results were also obtained with a primary AML patient sample AML#111 (AML M2, Figure 3E and 3F). In addition, Roscovitine significantly mitigated LY2603618-induced apoptosis in both CTS and U937 cells (Figure 3G and 3I), accompanied by decreased levels of γH2AX, cleaved PARP, and cleaved caspase-3 (Figure 3H and 3J). These results demonstrate that LY2603618 induces DNA damage and apoptosis in AML cells by a process which is at least partially dependent on CDK activity.
Decrease of Mcl-1 plays an important role in LY2603618-induced apoptosis in AML cell lines
It has been previously demonstrated that DNA damage can cause decreased expression of Mcl-1, which plays an important role in DNA damage-induced apoptosis [26]. It is conceivable that LY2603618 treatment induces DNA damage which causes decreased expression of Mcl-1, leading to apoptosis in AML cells. To test this possibility, we investigated the effects of LY2603618 treatment on the Bcl-2 family proteins in U937 cells. While protein levels for Bcl-2, Bcl-xL, Bak, Bax, and Bim remained unchanged post treatment with LY2603618 for 24 h, protein levels for Mcl-1 decreased significantly in cells treated with higher concentrations of LY2603618 (Figure 4A–4C), which correlated with much higher levels of apoptosis induced by the agent (Figure 2B). Interestingly, Roscovitine partially abolished the decrease of Mcl-1 induced by treatment with LY2603618 (Figure 4D), demonstrating that CDK activity contributed to the decrease of Mcl-1. To determine if Mcl-1 inhibits LY2603618-induced apoptosis, we ectopically overexpressed Mcl-1 (U937/Mcl-1) and red fluorescent protein (U937/RFP) in U937 cells (50% increase compared to U937/RFP cells, Figure 4E). As shown in Figure 4F, apoptosis induced by LY2603618 in the U937/Mcl-1 cells was significantly lower than that in U937/RFP cells, demonstrating that decrease of Mcl-1 contributes to LY2603618-induced apoptosis in AML cells.
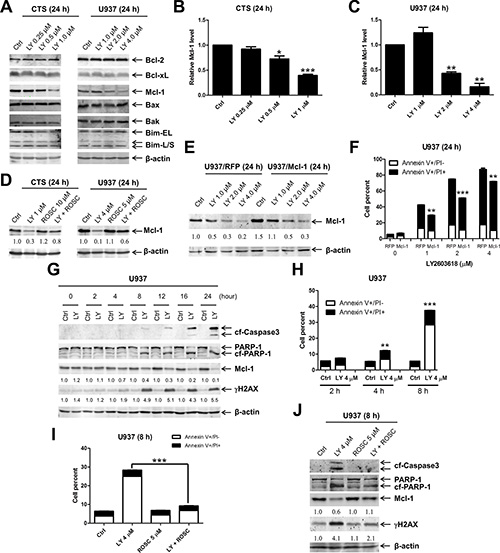
Figure 4: LY2603618 treatment results in decreased expression of Mcl-1. (A) CTS and U937 cells were treated with LY for 24 h. Whole cell lysates were subjected to Western blotting. (B and C) Relative densitometry of Mcl-1 expression were measured using Odyssey Software V3.0 and graphed as fold change compared to the no drug control ± s.e.m. *indicates p < 0.05, **indicates p < 0.005, and ***indicates p < 0.0005. (D) CTS and U937 cells were treated with LY and ROSC alone or in combination for 24 h. Whole cell lysates were subjected to Western blotting. The fold changes for the Mcl-1 densitometry measurements, normalized to β-actin and then compared to no drug treatment control, are indicated. (E) U937 cells were infected with Precision LentiORF Mcl-1 (U937/Mcl-1) and RFP control (U937/RFP) lentivirus particles overnight, then washed and incubated for 48 h prior to adding puromycin to the culture medium. The puromycin-resistant cells were treated with LY for 24 h. Whole cell lysates were subjected to Western blotting. The fold changes for the Mcl-1 densitometry measurements, normalized to β-actin and then compared to no drug treatment control, are indicated. (F) U937/RFP and U937/Mcl-1 cells were treated with LY for 24 h and then subjected to Annexin V/PI staining and flow cytometry analysis. **indicates p < 0.005 and ***indicates p < 0.0005. (G) U937 cells were treated with 4 μM LY for the indicated times. Whole cell lysates were subjected to Western blotting. Mcl-1 densitometry measurements, normalized to β-actin and then compared to no drug treatment control at the corresponding time point, are indicated. (H) U937 cells were treated with LY for 2 h, 4 h, and 8 h and then subjected to Annexin V/PI staining and flow cytometry analysis. **indicates p < 0.005 and ***indicates p < 0.0005. (I) U937 cells were treated with LY and ROSC, alone or in combination, for 8 h. Cells were then subjected to Annexin V/PI staining and flow cytometry analysis. ***indicates p < 0.0005. (J) Whole cell lysates were subjected to Western blotting. Densitometry measurements, normalized to β-actin and then compared to no drug treatment control, are indicated.
To further elucidate the relationship among DNA damage, Mcl-1 levels, and apoptosis, we treated U937 cells with LY2603618 for up to 24 h and then subjected whole cell lysates to western blotting. These experiments revealed a time-dependent increase of γH2AX, cleaved PARP and cleaved caspase3 as well as a time-dependent decrease of Mcl-1 (Figure 4G). It is important to note that obvious changes of γH2AX and Mcl-1 began at 4 h and were accompanied by a minor but statistically significant induction of apoptosis by LY2603618 (Figure 4H). To determine if CDK activity contributed to the induction of DNA damage, decrease of Mcl-1, and apoptosis by LY2603618, we treated U937 cells with LY2603618 and Roscovitine, alone or in combination, for 8 h. Roscovitine treatment abrogated LY2603618-induced apoptosis and rescued Mcl-1 protein (Figure 4I and 4J). However, Roscovitine treatment could only partially inhibit LY2603618-induced γH2AX. Similar results were obtained in CTS cells (Figure S3). These results demonstrate that LY2603618 treatment causes decreased expression of Mcl-1 and induces apoptosis in a partially CDK-dependent fashion in AML cells.
LY2603618 enhances ABT-199-induced apoptosis in AML cell lines
We and others have demonstrated that Mcl-1 plays an important role in the antileukemic activity of the Bcl-2-selective inhibitor ABT-199 in AML and other blood cancer cells [12, 16–18]. In our most recent study, we demonstrated that induction of Mcl-1 represents an intrinsic mechanism of resistance to ABT-199 in AML cells [19]. Thus, combining LY2603618, which can cause DNA damage and decreased expression of Mcl-1, with ABT-199 may enhance its antileukemic activity in AML cells. To investigate this, we treated the ABT-199 resistant cell line U937 (a relatively resistant AML cell line with an ABT-199 IC50 of 13.5 μM, as determined previously [12]) with LY2603618 and ABT-199 for 8 h. LY2603618 or ABT-199 treatment alone did not cause significant DNA damage after 8 h treatment, as assessed by alkaline comet assay (Figure 5A and 5B). However, the combined treatment resulted in a significant increase in DNA damage. In addition, LY2603618 treatment abrogated the increase of Mcl-1 seen with ABT-199 treatment (Figure 5C). Furthermore, the combined drug treatment resulted in synergistic induction of apoptosis in U937 cells (Figure 5D), which was accompanied by increased cleavage of PARP and caspase-3 (Figure 5C). Similar results were also obtained in THP-1 cells (another relatively resistant AML cell line with an ABT-199 IC50 of 2.4 μM, as determined previously [12], Figure 5E and 5F).
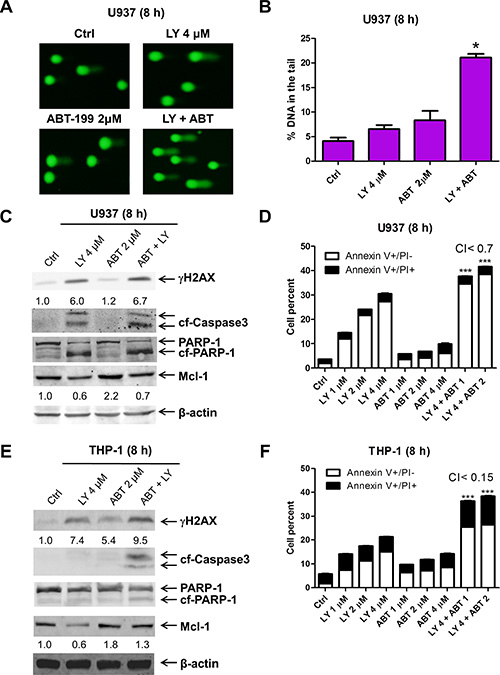
Figure 5: ABT-199 synergizes with LY2603618 in AML cells. (A) U937 cells were treated with LY and ABT-199 (ABT), alone or in combination, for 8 h and then subjected to alkaline comet assay analysis. Representative images are shown. (B) Comet assay results are graphed as median percent DNA in the tail from 3 replicate gels ± s.e.m. * indicates p < 0.05. (C) Whole cell lysates were subjected to Western blotting. The fold changes for the Mcl-1 densitometry measurements, normalized to β-actin and then compared to no drug treatment control, are indicated. (D) U937 cells were treated with LY and ABT-199, alone or in combination, for 8 h and then subjected to Annexin V/PI staining and flow cytometry analysis. ***indicates p < 0.0005. (E) THP-1 cells were treated with LY and ABT, alone or in combination, for 8 h. Whole cell lysates were subjected to Western blot analysis. (F) THP-1 cells were treated with the indicated drugs for 8 h and then subjected to Annexin V/PI staining and flow cytometry analyses. ***indicates p < 0.0005.
LY2603618 enhances ABT-199-induced apoptosis in primary AML patient samples
Then we tested the antileukemic effects of LY2603618 and ABT-199 in two primary patient samples which were relatively resistance to ABT-199 (AML#31 ABT-199 IC50 was 1 μM and AML#38 ABT-199 IC50 was 5 μM, as determined by MTT assays) by Annexin V/PI staining and flow cytometry analyses (patient characteristics are presented in Table 1). In agreement with the AML cell lines, LY2603618 combined with ABT-199 resulted in synergistic induction of cell death (Figure 6A and 6B). Finally, we tested the combined drug treatments in 10 primary AML patient samples which were relatively resistance to ABT-199 (IC50s ranging from 1 μM to 20 μM by MTT assays) by MTT assays and standard isobologram analyses. Again, the combined drug treatments resulted in synergistic antileukemic activities against the primary patient samples (Figure 6C).
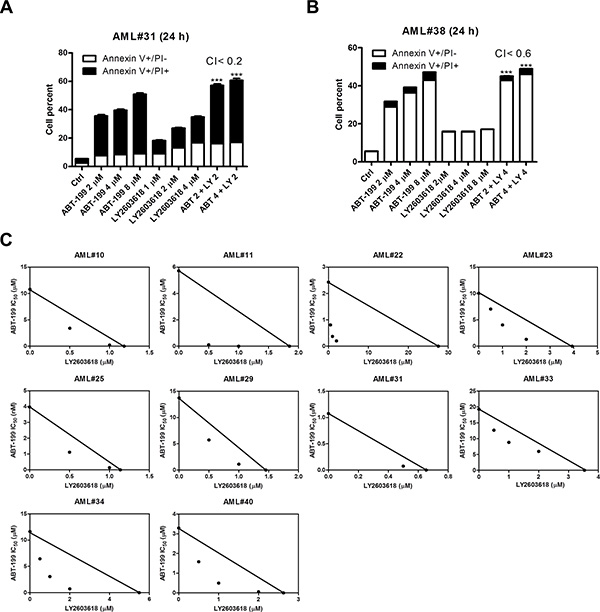
Figure 6: LY2603618 synergizes with ABT-199 in primary AML patient samples. (A and B) Patient samples were treated as indicated for 24 h and then subjected to Annexin V/PI staining and flow cytometry analyses. CI values were calculated using CompuSyn software. ***indicates p < 0.0005. (C) Primary AML patient samples were treated with ABT-199 and LY for 72 h and then viable cells were determined using MTT reagent. The IC50 values are means of duplicates from one experiment due to limited sample. Standard isobologram analysis of antileukemic interactions was performed to determine the extent and direction of the antileukemic interaction. The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergism whereas those above the line indicate antagonism.
DISCUSSION
In this study, we investigated using a CHK1 inhibitor as a DNA damaging agent to decrease Mcl-1 protein levels and enhance ABT-199 sensitivity in AML cells, based on a previous report that DNA damage can cause decreased expression of Mcl-1 in cancer cells [26]. We found that primary patient samples obtained at initial diagnosis and relapse had similar sensitivities to the CHK1 inhibitor LY2603618, suggesting that this agent could be used for treating relapsed AML. We also found that LY2603618 sensitivity did not correlate with CHK1 transcript levels, which is in agreement with Bryant et al. who found that CHK1 protein levels did not correlate with CHK1 inhibitor V158411 sensitivity in leukemia, lymphoma, and lung cancer cell lines [30]. Therefore, CHK1 expression levels probably are not biomarkers for predicting anti-tumor activities of CHK1 inhibitors. Although ectopic overexpression of CHK1 in THP-1 AML cell line had no impact on LY2603618 sensitivity, shRNA knockdown of CHK1 in the cells resulted in significantly increased sensitivity to the agent. These results suggest that there is a minimum amount of CHK1 that is required for cellular functions and anything above that threshold seems unnecessary.
As expected, LY2603618 treatment of AML cells caused DNA damage, along with decreased Mcl-1 protein levels and apoptosis. Interestingly, our time course experiment demonstrated that induction of DNA damage and decrease of Mcl-1 protein occurred at the same time, which was associated with the initiation of apoptosis. While CHK1 inhibitor-induced DNA damage and cell death is expected, decrease of Mcl-1 is an interesting finding which may be useful for designing combinations with CHK1 inhibitors. As one could expect, this property of LY2603618 rendered its ability to overcome intrinsic resistance to the Bcl-2-selective inhibitor ABT-199, mediated by Mcl-1 in AML cells, leading to synergistic antileukemic interactions between the two agents in both AML cell lines and primary AML patient samples. Although we found that overexpression of Mcl-1 only partially inhibited LY2603618-induced apoptosis, Nijhawan et al. revealed that overexpression of Mcl-1 inhibited UV-induced apoptosis [26]. This difference may be due to the difference in the extent of Mcl-1 overexpression or could suggest that additional factors are also involved in LY2603618-induced apoptosis.
A surprising finding of this study was that LY2603618-induced DNA damage was enhanced by ABT-199 in AML cells, which was potentially responsible for the abolishment of ABT-199-induced increase of Mcl-1. Though the exact mechanism by which ABT-199 treatment enhances DNA damage induced by LY2603618 remains unknown, Bcl-2 has been demonstrated to be associated with numerous DDR proteins, such as APE1, PARP1, Ku70 and BRCA1 (reviewed in [31]), therefore it is plausible that ABT-199 treatment inhibits or enhances Bcl-2’s role in the DDR which can then increase CHK1 inhibitor-induced DNA damage. Studies are underway investigating the molecular mechanism responsible for the enhancement of ABT-199 on LY2603618-induced DNA damage. However, this is not in the scope of this paper.
The CDK inhibitor Roscovitine partially inhibited LY2603618-induced cell death, decrease of Mcl-1, and DNA damage, suggesting that CDK-independent mechanisms of DNA damage induced by LY2603618 treatment also contributed to cell death. As a central player in the DDR, besides regulating the cell cycle checkpoints, CHK1 also plays important roles in DNA repair and stabilization of DNA replication forks [32, 33]. Thus, targeting CHK1 with LY2603618 could inhibit CHK1’s involvement in these biological processes, leading to DNA damage independent of CDK activity.
Taken together, our results as well as those previously reported by others, provide insight into the mechanism of action for the synergistic antileukemic activity of LY2603618 and ABT-199. Our proposed mechanism is presented in Figure 7. LY2603618 treatment inhibits CHK1 resulting in accumulation of DNA damage. DNA damage decreases Mcl-1 protein levels, which decreases its inhibitory effect on pro-apoptotic proteins, ultimately resulting in the initiation of apoptosis. In ABT-199 resistant AML cells, ABT-199 treatment results in increased Mcl-1 protein levels. However, when combined with LY2603618, enhanced DNA damage occurs and results in abolishment of the increase of Mcl-1 protein, leading to synergistic induction of apoptosis. In summary, LY2603618 enhances ABT-199 treatment in AML cells. The toxicities in patients associated with either drug have been reported [27, 34–37], and do not appear to overlap. Further, the toxicities that are associated with the individual treatments are manageable; suggesting that combination therapy associated toxicities would be manageable. Although our study used a limited number of patient samples, and therefore may not necessarily represent the full spectrum of AML, our data supports the further development of CHK1 inhibition in combination with ABT-199.
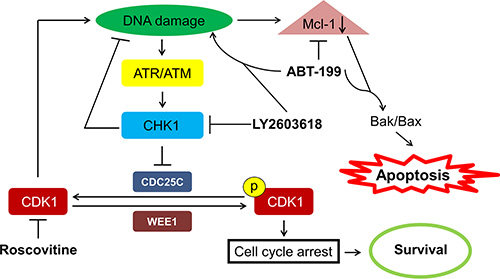
Figure 7: Proposed mechanism of action for LY2603618 alone or in combination with ABT-199 in AML cells. LY2603618 treatment inhibits CHK1, which results in CDK-dependent and CDK-independent DNA damage. DNA damage leads to a decrease of Mcl-1 protein levels and apoptosis. ABT-199 treatment in resistant cells leads to increased levels of Mcl-1 protein which can be abrogated by combination with LY2603618 due to enhanced DNA damage induced by the combination of LY2603618 and ABT-199, resulting in synergistic induction of apoptosis.
MATERIALS AND METHODS
Drugs
LY2603618, Roscovitine, and ABT-199 were purchased from Selleck Chemicals (Houston, TX).
Cell culture
U937, THP-1, MV4-11, and HL60 cell lines were purchased from the American Type Culture Collection (Manassas, VA). The CMY, CMS, and CTS cell lines were gifts from Dr. A Fuse from the National Institute of Infectious Diseases, Tokyo, Japan. The CMK, NB4, and OCI-AML3 cell lines were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany). MOLM-13 cells were purchased from AddexBio (San Diego, CA). The cell lines were cultured in RPMI 1640 (except OCI-AML3, which was cultured in alpha-MEM) with 10–15% fetal bovine serum (Life Technologies, Grand Island, NY), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. All cells were cultured in a 37°C humidified atmosphere containing 5% CO2/95% air.
Diagnostic AML blast samples derived from patients either at initial diagnosis or at relapse were purified by standard Ficoll-Hypaque density centrifugation, then cultured in RPMI 1640 with 20% fetal bovine serum supplemented with ITS solution (Sigma-Aldrich, St. Louis, MO, USA) and 20% supernatant of the 5637 bladder cancer cell line (as a source of granulocyte-macrophage colony-stimulating factor [12, 38, 39]).
Clinical samples
Diagnostic AML blast samples were obtained from the First Hospital of Jilin University. Written informed consent was provided according to the Declaration of Helsinki. This study was approved and carried out in accordance with the guidelines set forth by the Human Ethics Committee of the First Hospital of Jilin University. Clinical samples were screened for gene mutations by PCR amplification and automated DNA sequencing and for fusion genes by real-time RT-PCR, as described previously [12, 40].
In vitro cytotoxicity assays
In vitro cytotoxicities of the AML cells were measured by using MTT (3-[4, 5-dimethyl-thiazol-2-yl]-2, 5-diphenyltetrazoliumbromide, Sigma-Aldrich), as previously described [41, 42]. Briefly, the cells were treated with variable concentrations of LY2603618, ABT-199, or in combination for 72 hours. MTT was added to a final concentration of 1 mM and cells were incubated for 4 hours at 37°C. The cells were lysed overnight using 10% SDS in 10 mM HCl and plates were read at 590 nm using a microplate reader. IC50 values were calculated as drug concentrations necessary to inhibit 50% growth compared to vehicle control treated cells. The IC50 values for the cell lines are presented as mean values ± standard errors from at least three independent experiments. The IC50 values for the patient samples are means of duplicates from one experiment, due to limited sample. Patient samples for the combined drug treatments were chosen solely based on sample availability.
Quantification of gene expression by real-time RT-PCR
Total RNA was extracted using TRIzol (Life Technologies) and cDNAs were prepared from 2 μg total RNA using random hexamer primers and a RT-PCR kit (Life Technologies), and purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA) as previously described [12, 42, 43]. CHK1 (Hs00967506_m1) transcripts were quantitated using TaqMan probes (Life Technologies) and a LightCycler® 480 real-time PCR machine (Roche Diagnostics, Indianapolis, IN), based on the manufacturer’s instructions. Real-time PCR data are presented as means of duplicates from one experiment, due to limited sample, and results were normalized to GAPDH (4333764-1007034) transcripts. Fold changes were calculated using the comparative Ct method [44].
Western blot analysis
Cells were lysed in the presence of protease and phosphatase inhibitors (Roche Diagnostics). Whole cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL) and immunoblotted with anti-p-CDK1 (Y15) (9111), -CDK1 (9112), -p-CDC25C (S216) (9528), -PARP (9542), -Bax (2774), -Bak (3814), -p-H3 (S10) (9701), -cleaved caspase-3 (9661), -Bcl-2 (2876), -Bcl-xL (2764), -Mcl-1 (4572), -Bim (2819), -CHK1 (2345), -p-CHK1 (S345) (2341), -γH2AX (2577, Cell Signaling Technology, Danvers, MA, USA) or -β-actin antibody (Sigma-Aldrich), as previously described [45, 46]. Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, USA), as described by the manufacturer. Western blots were repeated at least three times and one representative blot is shown.
Apoptosis
AML cells were treated with LY2603618 and Roscovitine, alone or in combination, or with LY and ABT-199, alone or in combination, and subjected to flow cytometry analysis to determine drug-induced apoptosis using an Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis Kit (Beckman Coulter; Brea, CA, USA), as previously described [41, 43]. Apoptotic events are presented as percentage of AnnexinV+/PI- and Annexin V+/PI+ ± s.e.m. Experiments with AML cell lines were performed 3 independent times in triplicates, while patient sample experiments were performed once in triplicate due to limited sample. Data are presented as mean ± standard errors from one representative experiment. Patient samples were chosen based on availability of adequate sample for the assay. The extent and direction of antileukemic interaction were determined by calculating the combination index (CI) values using CompuSyn software (Combosyn Inc., Paramus, NJ). CI < 1, CI = 1, and CI > 1 indicate synergistic, additive, and antagonistic effects, respectively [41, 47].
Cell cycle progression
Cells were treated with the indicated drugs for up to 48 h. The cells were harvested and fixed with ice-cold 80% (v/v) ethanol for 24 h. The cells were pelleted, washed with PBS, and resuspended in PBS containing 50 μg/mL PI, 0.1% Triton X-100 (v/v), and 1 μg/mL DNase-free RNase. DNA content was determined by flow cytometry analysis using a FACS Calibur flow cytometer (Becton Dickinson), as previously described [48]. Cell cycle analysis was performed using ModFit LT 3.0 (Becton Dickinson). Histograms were created using FlowJo v7.6.5 (Tree Star, Ashland, OR, USA).
Production of lentivirus particles and transduction of AML cells
The pMD-VSV-G and delta 8.2 plasmids were gifts from Dr. Dong at Tulane University. Bak, Bax, CHK1, and non-target control (NTC) shRNA lentiviral vectors were purchased from Sigma-Aldrich. Red fluorescent protein (RFP), CHK1, and Mcl-1 cDNA constructs were purchased from Thermo Fisher Scientific Biosciences (Lafayette, CO). Lentivirus production and transduction were carried out as previously described [28]. Briefly, TLA-HEK293T cells were transfected with pMD-VSV-G, delta 8.2, and lentiviral shRNA constructs using Lipofectamine and Plus reagents (Life Technologies) according to the manufacturer’s instructions. Virus containing culture medium was harvested 48 h post-transfection. Cells were transduced overnight using 1 mL of virus supernatant and 4 μg of polybrene and then cultured for an additional 48 h prior to selection with puromycin.
Alkaline comet assay
U937 cells were treated with the indicated drugs for 8 or 16 h and then subjected to alkaline comet assay, as previously described [28]. Slides were stained with SYBR Gold (Life Technologies), and then imaged on an Olympus BX-40 microscope equipped with a DP72 microscope camera and Olympus cellSens Dimension software (Olympus America Inc., Center Valley, PA). Approximately 50 comets per gel were scored using CometScore (TriTek Corp, Sumerduck, VA). The median percent DNA in the tail was calculated and graphed ± s.e.m.
Statistical analysis
Differences in cell apoptosis between treated (individually or combined) and untreated cells were compared using the pair-wise two-sample t-test. The p value for the differences between LY IC50s for the groups of patient samples was calculated using the Mann-Whitney two-sample U test. The nonparametric Spearman rank correlation coefficient was used to analyze the relationship between LY IC50s and CHK1 transcript levels in the primary AML patient samples. Statistical analyses were performed with GraphPad Prism 5.0. Error bars represent ± s.e.m. The level of significance was set at p < 0.05.
ACKNOWLEDGMENTS
This study was supported by Jilin University, Changchun, China, The Decerchio/Guisewite Family, and the Barbara Ann Karmanos Cancer Institute.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
GRANT SUPPORT
Grants from the National Natural Science Foundation of China, NSFC 31271477 (YG) and NSFC 31471295 (YG), the Graduate Innovation Fund of Jilin University (NX), Hyundai Hope On Wheels (JWT/YG), and the Ring Screw Textron Endowed Chair for Pediatric Cancer Research (JWT). The funders had no role in study design, data collection, analysis and interpretation of data, decision to publish, or preparation of the manuscript.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5–29.
2. Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, Pounds S, Razzouk BI, Lacayo NJ, Cao X, Meshinchi S, Degar B, Airewele G, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010; 11:543–552.
3. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014; 64:83–103.
4. Lichtman MA. A historical perspective on the development of the cytarabine (7 days) and daunorubicin (3 days) treatment regimen for acute myelogenous leukemia: 2013 the 40th anniversary of 7+3. Blood Cells Mol Dis. 2013; 50:119–130.
5. Lauria F, Raspadori D, Rondelli D, Ventura MA, Fiacchini M, Visani G, Forconi F, Tura S. High bcl-2 expression in acute myeloid leukemia cells correlates with CD34 positivity and complete remission rate. Leukemia. 1997; 11:2075–2078.
6. Schimmer AD, O’Brien S, Kantarjian H, Brandwein J, Cheson BD, Minden MD, Yee K, Ravandi F, Giles F, Schuh A, Gupta V, Andreeff M, Koller C, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res. 2008; 14:8295–8301.
7. Davids MS, Letai A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J Clin Oncol. 2012; 30:3127–3135.
8. Saxena A, Viswanathan S, Moshynska O, Tandon P, Sankaran K, Sheridan DP. Mcl-1 and Bcl-2/Bax ratio are associated with treatment response but not with Rai stage in B-cell chronic lymphocytic leukemia. Am J Hematol. 2004; 75:22–33.
9. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19:202–208.
10. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, Dohner H, Gaidzik VI, Galinsky I, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Discov. 2014; 4:362–375.
11. Touzeau C, Dousset C, Le Gouill S, Sampath D, Leverson JD, Souers AJ, Maiga S, Bene MC, Moreau P, Pellat-Deceunynck C, Amiot M. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014; 28:210–212.
12. Niu X, Wang G, Wang Y, Caldwell JT, Edwards H, Xie C, Taub JW, Li C, Lin H, Ge Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014; 28:1557–1560.
13. Vogler M, Dinsdale D, Dyer MJ, Cohen GM. ABT-199 selectively inhibits BCL2 but not BCL2L1 and efficiently induces apoptosis of chronic lymphocytic leukaemic cells but not platelets. Br J Haematol. 2013; 163:139–142.
14. Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, Smyth GK, Christie M, Phillipson LJ, Burns CJ, Mann GB, Visvader JE, Lindeman GJ. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer cell. 2013; 24:120–129.
15. Vandenberg CJ, Cory S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood. 2013; 121:2285–2288.
16. Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Death Dis. 2015; 6:e1593.
17. Alford SE, Kothari A, Loeff FC, Eichhorn JM, Sakurikar N, Goselink HM, Saylors RL, Jedema I, Falkenburg JH, Chambers TC. BH3 Inhibitor Sensitivity and Bcl-2 Dependence in Primary Acute Lymphoblastic Leukemia Cells. Cancer Res. 2015; 75:1366–1375.
18. Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi CX, Mantei J, Ahmann G, Gonzales IM, Choudhary A, Valdez R, Camoriano J, Fauble V, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014; 28:1657–1665.
19. Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, Caldwell JT, Xiang S, Zhang X, Chu R, Wang Z, Lin H, Taub JW, et al. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Cancer Res. 2016.
20. Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clinical Cancer Res. 2010; 16:376–383.
21. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078.
22. Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007; 28:739–745.
23. Powell SN, Bindra RS. Targeting the DNA damage response for cancer therapy. DNA repair. 2009; 8:1153–1165.
24. Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001; 21:4129–4139.
25. Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol. 2005; 25:3553–3562.
26. Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003; 17:1475–1486.
27. Calvo E, Chen VJ, Marshall M, Ohnmacht U, Hynes SM, Kumm E, Diaz HB, Barnard D, Merzoug FF, Huber L, Kays L, Iversen P, Calles A, et al. Preclinical analyses and phase I evaluation of LY2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Invest New Drugs. 2014; 32:955–968.
28. Xie C, Drenberg C, Edwards H, Caldwell JT, Chen W, Inaba H, Xu X, Buck SA, Taub JW, Baker SD, Ge Y. Panobinostat Enhances Cytarabine and Daunorubicin Sensitivities in AML Cells through Suppressing the Expression of BRCA1, CHK1, and Rad51. PloS one. 2013; 8:e79106.
29. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015; 22:58–73.
30. Bryant C, Scriven K, Massey AJ. Inhibition of the checkpoint kinase Chk1 induces DNA damage and cell death in human Leukemia and Lymphoma cells. Mol Cancer. 2014; 13:147.
31. Laulier C, Lopez BS. The secret life of Bcl-2: apoptosis-independent inhibition of DNA repair by Bcl-2 family members. Mutat Res. 2012; 751:247–257.
32. Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005; 7:195–201.
33. Petermann E, Caldecott KW. Evidence that the ATR/Chk1 pathway maintains normal replication fork progression during unperturbed S phase. Cell cycle. 2006; 5:2203–2209.
34. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016; 374:311–322.
35. Jones J, Mato AR, Coutre S, Wierda W, Choi MY, Davids MS, Lamanna N, Barr P, Burns K, Montalvo N, Zhu M, Busman T, Potluri J, et al. Preliminary Results of a Phase 2, Open-Label Study of Venetoclax (ABT-199/GDC-0199) Monotherapy in Patients with Chronic Lymphocytic Leukemia Relapsed after or Refractory to Ibrutinib or Idelalisib Therapy. Blood. 2015; 126:715–715.
36. Eichhorst BF, Schetelig J, Coutre S, Seymour JF, Munir T, Puvvada SD, Wendtner C-M, Roberts AW, Jurczak W, Mulligan S, Boettcher S, Mobasher M, Zhu M, et al. Venetoclax (ABT-199/GDC-0199) Monotherapy Induces Deep Remissions, Including Complete Remission and Undetectable MRD, in Ultra-High Risk Relapsed/Refractory Chronic Lymphocytic Leukemia with 17p Deletion: Results of the Pivotal International Phase 2 Study. Blood. 2015; 126:LBA-6-LBA-6.
37. Weiss GJ, Donehower RC, Iyengar T, Ramanathan RK, Lewandowski K, Westin E, Hurt K, Hynes SM, Anthony SP, McKane S. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest New Drugs. 2013; 31:136–144.
38. Taub JW, Matherly LH, Stout ML, Buck SA, Gurney JG, Ravindranath Y. Enhanced metabolism of 1-beta-D-arabinofuranosylcytosine in Down syndrome cells: a contributing factor to the superior event free survival of Down syndrome children with acute myeloid leukemia. Blood. 1996; 87:3395–3403.
39. Quentmeier H, Zaborski M, Drexler HG. The human bladder carcinoma cell line 5637 constitutively secretes functional cytokines. Leuk Res. 1997; 21:343–350.
40. Qi W, Xie C, Li C, Caldwell JT, Edwards H, Taub JW, Wang Y, Lin H, Ge Y. CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J Hematol Oncol. 2014; 7:53.
41. Xie C, Edwards H, Xu X, Zhou H, Buck SA, Stout ML, Yu Q, Rubnitz JE, Matherly LH, Taub JW, Ge Y. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin Cancer Res. 2010; 16:5499–5510.
42. Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PloS one. 2011; 6:e17138.
43. Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, Taub JW, Matherly LH, Ge Y. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009; 114:2744–2752.
44. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408.
45. Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, Buck SA, Massey G, Becton DL, Weinstein HJ, Ravindranath Y, Matherly LH, Taub JW. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006; 107:1570–1581.
46. Ge Y, Stout ML, Tatman DA, Jensen TL, Buck S, Thomas RL, Ravindranath Y, Matherly LH, Taub JW. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005; 97:226–231.
47. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006; 58:621–681.
48. Wang G, He J, Zhao J, Yun W, Xie C, Taub JW, Azmi A, Mohammad RM, Dong Y, Kong W, Guo Y, Ge Y. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PloS one. 2012; 7:e52095.