
INTRODUCTION
Rapid advancements in next generation sequencing (NGS) technologies have provided a means to comprehensively determine the constitutional genome of an individual, and all genomic aberrations within a tumor that occur as a cause or consequence of malignant growth. This information, when integrated with an understanding of disease mechanism, disease behavior, and response to therapeutics underlies the concept of precision oncology: a refinement of disease taxonomy based on molecular features [1]. A practical consequence of this approach is the development of a more specific categorization of cancers with congruent behaviors and with predictable responses to therapies.
The Cancer Genome Atlas (TCGA) and other large-scale molecular profiling studies of human malignancies have identified common and rare genomic alterations, a subset of which are recurrent across different tumor types and several of which have clear therapeutic implications [2, 3]. It is now evident that carcinomas typically contain thousands of mutations and a spectrum of structural chromosomal rearrangements and epigenomic alterations, a subset of which alter gene function and influence malignant growth [4]. While only a few molecular alterations have clearly-defined implications for selecting specific therapies, these are none-the-less notable, and foreshadow the future where new treatments are developed and deployed based on targeting key causal aberrations [5–7].
The field of medical genetics is currently grappling with the opportunities and challenges of integrating genomic sequencing data into clinical practice [8, 9]. Most commonly, whole genome sequencing (WGS) or whole exome sequencing (WES) strategies have been employed to identify sequence variants shown in clinical research to cause or associate with a disease. Setting standards for methods, determining which disease-associated variants should be reported, and establishing how to communicate incidental or opportunistic findings have been the subject of several consensus panels [10, 11]. Less attention has been given to specifically establishing standards for assessing and reporting somatic events in patients with cancer, which provide many additional challenges [12, 13].
In addition to the issues faced in interpreting and reporting germline WGS/WES data, cancer genomics must consider tissue quality and quantity, intra-tumor heterogeneity, inter-tumor heterogeneity, gains and losses of chromosomal regions, and variation in the admixture of neoplastic versus benign cells in the tissue sample. The addition of complementary NGS assays such as RNA sequencing (RNAseq) for the analysis of gene expression adds additional variables. If comprehensive genome-scale assessments will be used as the basis for cancer classification and consequent treatment decisions, then accurate molecular assessments are essential.
Although providers offering cancer genomics assays have proliferated, the level of reproducibility in terms of the technical sensitivity and the resulting conclusions stemming from the data analysis has not been evaluated. In this study, we sought to determine the reproducibility of NGS-based assessments of tumor genome mutations and gene expression using WES and RNAseq, respectively. Samples of the same metastatic tumors were independently processed and subjected to WES and RNAseq at two sequencing centers. We compared the determinations of somatic DNA point mutations, indels, and copy number variants identified by WES, germline variants assessed by WES, and transcript abundance and gene rearrangements identified by RNAseq.
RESULTS
Clinical samples, pathology, and sequence analysis pipelines
Tumor and benign tissue samples were obtained from three men with widely metastatic prostate cancer [14, 15]. Tumor sections were assessed to confirm a composition of > 70% tumor cells, and benign tissues were evaluated to establish the absence of neoplastic cells. A metastasis from each patient, designated SC_9008 (liver), SC_9009 (lymph node) and SC_9010 (lymph node) was partitioned and one representative tumor sample from each metastasis with a corresponding benign tissue sample was shipped to the University of Michigan (UM) and the Broad Institute (Broad) for sequence analysis (Figure 1).
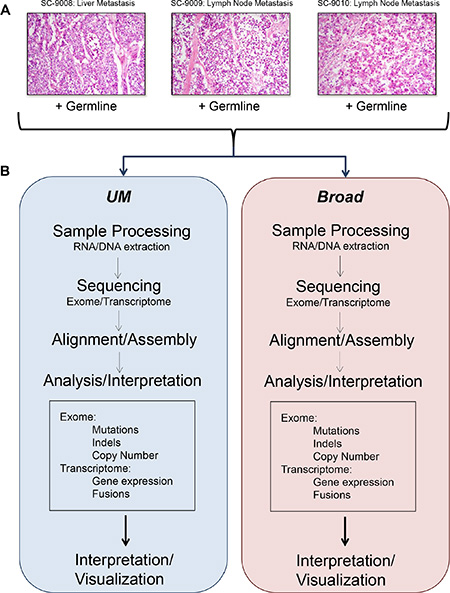
Figure 1: Flow of experiments and analyses. Representative histology images from the tumor samples included in this study (A). Overview of sample processing, sequencing, and analysis pipelines used at the two sequencing centers (B).
Each sequencing center processed the tissue samples using institutional protocols (see Methods). Exome libraries were sequenced using Illumina HiSeq instruments with a target of 50 million paired-end reads per sample. The actual number of reads ranged from 110 to 250 million (Table 1). RNAseq libraries were constructed from both PolyA+ selected and total RNA libraries (UM) or total RNA alone (Broad) and sequenced using Illumina HiSeq 2500 instruments with a target of 50 million paired-end reads per sample. The actual number of reads ranged from 100 to 134 million (Table 1).
Table 1: Sequencing metrics
Sequence Type |
Metric |
SC_9008 |
SC_9009 |
SC_9010 |
|||
|---|---|---|---|---|---|---|---|
|
|
Broad |
UM |
Broad |
UM |
Broad |
UM |
WES (somatic) |
|||||||
|
MTC |
177.99 |
264.10 |
147.09 |
271.13 |
122.57 |
265.04 |
|
Selected bases (%) |
0.83 |
0.75 |
0.84 |
0.76 |
0.85 |
0.72 |
|
Zero coverage targets (%) |
0.014 |
0.018 |
0.015 |
0.019 |
0.015 |
0.017 |
|
NSV |
852 |
1203 |
42 |
57 |
47 |
90 |
|
Point Mutations |
652 |
811 |
38 |
47 |
45 |
82 |
|
Insertion/Deletions |
200 |
392 |
4 |
10 |
2 |
8 |
WES (germline) |
|||||||
|
MTC |
143.40 |
226.97 |
173.84 |
236.99 |
122.57 |
188.19 |
|
Selected bases (%) |
0.85 |
0.79 |
0.86 |
0.81 |
0.85 |
0.75 |
|
Zero coverage targets (%) |
0.014 |
0.020 |
0.013 |
0.021 |
0.015 |
0.020 |
|
ACMG 56 gene coverage > 30X (%) |
100 |
100 |
100 |
100 |
100 |
100 |
RNAseq (somatic) |
|||||||
|
Aligned in pairs reads (%) |
0.976 |
0.912 |
0.973 |
0.914 |
0.974 |
0.920 |
|
PF reads aligned (%) |
0.938 |
0.821 |
0.948 |
0.819 |
0.940 |
0.812 |
MTC, mean target coverage; NSV, non-synonymous nucleotide variant.
Sequencing coverage
A mixture model was used to estimate the tumor cell content of each sample (eMethods). Histological assessments estimated that each tumor comprised > 70% neoplastic cells. The sequence-based estimates of tumor content were 86%, 76% and 58% for SC_9008, SC_9009 and SC_9010, respectively (Supplementary Figure 1). Mean target coverage and additional sequencing metrics are in Table 1 and Figure 2A–2C.
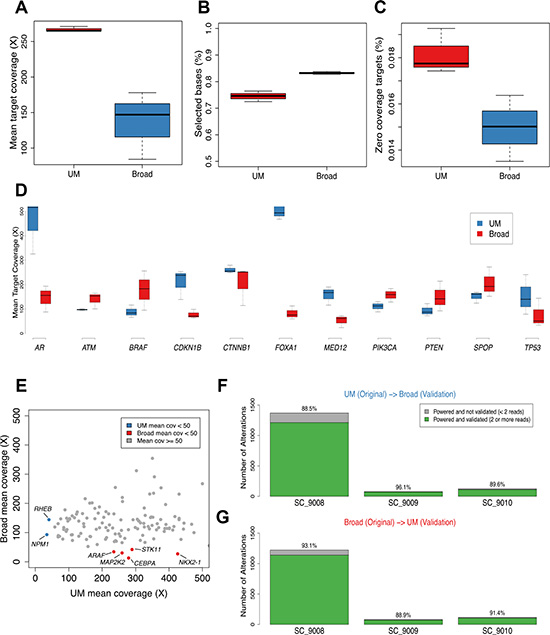
Figure 2: Sequence coverage of comparisons of mutation calls in prostate cancer across sequencing centers. The range of mean target coverage (A), selected bases (%) (B), and zero coverage targets (%) (C) for tumors sequenced at the two sequencing centers are shown. Mean target coverage for biologically relevant prostate cancer genes are from tumors sequenced in the two sites are shown (D). Using a larger panel of 130 clinically relevant genes, mean target coverage for UM and Broad tumors is plotted in (E), with designations for genes that had < 50 X mean target coverage for UM (blue) or Broad (red) platforms. The cross validation rates for UM to Broad and Broad to UM are shown in (F) and (G), respectively when accounting for whether there was adequate power to detect an alteration at both sites which corrects for the difference in sequencing depth achieved between the two centers.
To compare the biological utility of exploring prostate cancer metastasis by WES, we assessed the mean sequencing depth of coverage for 11 genes shown in previous studies of prostate carcinoma to be recurrently mutated (Figure 2D) [16–18]. Each gene had a minimum of 50 reads spanning each nucleotide across the targeted exons. For the androgen receptor (AR) the coverage exceeded 150X; the UM bait design included additional sequencing specifically for AR and FOXA1, which resulted in enhanced coverage of these genes. We also assessed a panel of 134 cancer genes that, when altered, may be clinically actionable: defined as predictive for response or resistance to therapy, and/or with prognostic or diagnostic relevance [19]. Though read depth varied substantially, with some genes exhibiting deeper coverage in UM sequencing and others exhibiting deeper coverage in Broad sequencing, for all but two genes in the UM set (NPM1, RHEB) and five genes in Broad set (NKX2-1, STK11, MAP2K2, CEBPA, ARAF), read-depths exceeded 50X (Figure 2E).
Tumor exome analysis: Mutations
The tumors evaluated in this study varied substantially in the number of non-synonymous somatic mutations ranging from 852B/1203UM in SC_9008, which harbors an MSH2 mutation that likely underlies this hypermutation phenotype [20], to 42B/57UM in SC_9009 (Supplementary Tables 1, 2). Since mean target coverage was different at the two sites, which impacts power to detect mutations at lower allelic fractions and thus can confound comparison analyses, mutation comparisons were performed with a focus on adequately powered genetic loci [19, 21]. Of the mutations originally called in the SC_9008 tumor in the UM analysis, 88.5% of adequately powered events were concordantly called in the Broad-sequenced tumor, whereas 11.5% were powered to detect a mutation but the mutation was not identified (Figure 2F). In this tumor, 93% of the mutations originally called in the Broad analysis were adequately powered and validated in the UM analysis with 7% of the mutations adequately powered but not identified (Figure 2G). Analyses of the mutations in the other two tumors yielded similar rates of reproducibility. Collectively, these results may reflect differences in capture reagents, depth of coverage, analytical methods used for variant identification, or true biology in terms of intra-tumor heterogeneity.
Examination of variant calls from each center was performed for variants with at least 30X depth and an allelic fraction of 0.1 or higher, acknowledging that a majority of alterations present in one set but not the other is a result of insufficient power to call the variant. In both cases, the majority of nonsynonymous alterations identified in one set but not the other (374/613 [61%] for UM and 206/294 [70%] for Broad) were classified as short insertion/deletion events, consistent with prior reports that note challenges in reproducibly identifying this type of variant [22].
Several mutations were detected in genes with functional roles in prostate or other cancers. The exome of SC_9008 included an AR mutation, T878A that broadens ligand specificity, an inactivating TP53 mutation, and a recurrent mutation in SPOP. A frameshifting indel disrupting MSH2 was identified which likely contributed to the hyper-mutated genome of this tumor. The exome of SC_9010 had a point mutation in ZFHX3/ATBF1, a tumor suppressor gene previously reported to be recurrently inactivated in prostate cancers [23–25, 16]. Each of these pathogenic mutations was identified in both the UM and Broad analyses. A PTEN frameshift mutation (p.L296fs; allelic fraction 0.8) that accompanied a PTEN copy loss in SC_9008 was only identified in the initial UM insertion/deletion analysis, but was confirmed following manual review in both sequence data sets.
Tumor exome analysis: Genome structural alterations
To identify regions of the genome with allelic copy loss or gain, we assessed the exome sequence data using segmentation derived from copy ratios (See Supplementary Methods). Overall, there were substantial regions of copy gain and loss in each of the tumors. Overlays of the chromosome plots showed nearly identical patterns across the tumor genomes (Figure 3A; Supplementary Figure 3). Notable alterations in SC_9008 included a single copy loss of APC, PTEN loss, RB1 loss, and focal amplifications of 8q that included the MYC locus. In SC_9010 notable alterations included JAK2 loss on Chr9 and a copy gain of the AR. Each of these alterations was called in the UM and Broad analyses.
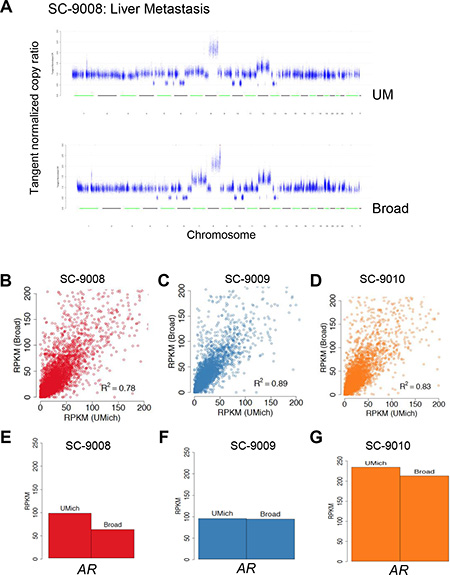
Figure 3: Comparison of DNA copy number assessments and RNAseq between sequencing centers. A representative copy number profile obtained from UM and Broad from one case is show in (A). Reads per kilobase per million (RPKM) values from transcriptome data derived at each sequencing center for the three tumors are shown in (B–D) RPKM values for AR from each of the tumors is shown in (E–G).
Tumor RNAseq
RNAseq was performed to assess gene expression and identify gene rearrangements that encode fusion transcripts. Globally, gene expression concordance was significant between the transcriptomes from each tumor sample (r2 ≥ 0.78; Pearson) (Figure 3B–3D). Each tumor was found to exhibit high levels of transcripts encoding the AR (Figure 3E–3G) and AR-regulated genes such as KLK3/PSA and TMPRSS2, indicating an active AR transcriptional program, an important clinical finding for prioritizing therapeutic options.
Paired-end sequencing reads were used to identify fusion transcripts. Tumor SC_9009 expressed a fusion transcript involving the 5′ exons of TMPRSS2 and 3′ exons of ERG, consistent with the commonly observed TMPRSS2-ETS family rearrangements that occur in 40– 60% of prostate cancers [26]. Both sequencing centers identified this rearrangement. To further investigate the concordance of fusion detection within and between samples, secondary multi-caller analysis was performed on transcriptome data from each tumor sample (see Methods). There was minimal overlap of fusion transcripts between two fusion callers applied to the same sample (See Supplementary Figure 4). Among putative fusions identified by both algorithms within a given sample from Broad (n = 12), the cross validation rate for the corresponding UM sample was 75% (9/12); among fusions identified by both callers in the UM samples (n = 18), the cross-validation rate for the corresponding Broad sample was 50% (9/18). However, in each tumor, numerous fusions were detected, the vast majority of which were unique to each tumor, have not been previously reported, and are of unclear significance.
Exome assessments of germ line variants
To facilitate the accurate determination of somatic mutations and copy number alterations in a tumor sample, sequencing of germline DNA is often performed in a parallel assay. Incidental but clinically-useful findings unrelated to the intended assessment of cancer-associated alterations may be identified. To address how these incidental or secondary findings are disseminated, the American College of Medical Genetics and Genomics (ACMG) has established a list of 56 genes associated with 24 inherited conditions that should be reported [10]. Of these genes, 21 have clear causal roles in the inherited predisposition to neoplastic disease including BRCA2 and the Lynch Syndrome DNA mismatch repair genes MLH1, MSH2, and MSH6 [27–29].
We assessed each of the 56 ACMG genes in the whole exome data obtained from the corresponding benign tissue, and compared the read depth and sequence calls between the two sequencing centers (Table 1 and Supplementary Figure 2). Of the 56 genes, both the UM and Broad germline exomes provided > 50X coverage for all 56 ACMG genes, with only one exception (SDHC in UM exomes) (Supplementary Figure 2D). Since germline variant detection power does not require as substantial depth as tumor sequencing, this would not impact germline-focused clinical variant detection. One heterozygous variant, p.E1317Q, in the APC gene associated with a very modest 1.4-fold increased risk of colorectal cancer [30] was identified in the germline of SC_9010 by both sequencing centers (Supplementary Table 3).
DISCUSSION
Comprehensive genomic assessments are increasingly used in clinical oncology in order to provide an appraisal of molecular alterations that have the potential to influence therapeutic decisions involving the selection of treatment [15]. Though the concept of genomic sequencing is understood at a fundamental level by providers and well-informed patients, there are numerous variations in the actual methodology that can influence reportable results. These include depth of sequencing coverage, the type of capture reagents used for exome and RNA analyses, whether target-based approaches are employed, disparities in tumor purity, and the integrity of DNA and RNA.
Our objective was to assess the consistency of ascertaining genomic information from tumors and corresponding germline DNA across different sequencing centers. We did not pre-specify the type of sequencing technology, the depth of sequencing, or any other parameter. Each of the two centers followed their standard operating procedures without an attempt to follow a common protocol.
Overall, the concordance in identifying key putative oncogenic aberrations was extremely high with only a PTEN frameshift mutation and a MSH2 inactivating gene rearrangement identified by one center that accompanied a PTEN copy loss and MSH2 inactivating indel, respectively, identified by both centers (Table 2 and Supplementary Table 4, 5). The consistency of reporting non-synonymous mutations ranged from 88.5% in a hypermutated tumor to 96.1% in a tumor with 54 non-synonymous mutations. Of the discordant mutations, the vast majority had sufficient depth of coverage to identify a mutation if present, and thus likely represents intratumoral heterogeneity. As two different portions of each particular metastasis were evaluated, rather than precisely the same tumor fragment, some degree of heterogeneity was expected. However, a focused analysis of 13 genes recurrently mutated in prostate cancer and 150 others with known oncogenic roles across human cancers determined 100% concordant mutation calls, indicating that the practical implications of intratumoral heterogeneity in terms of driver mutations and actionable variants, may be limited, at least in the context of metastatic disease [18].
Table 2: Comparative summary of cancer-associated findings from tumor SC_9008
EVENT |
UM |
BROAD |
|---|---|---|
Gene Copy Number |
||
APC |
Copy Loss |
Copy Loss |
AR |
Amplification |
Amplification |
8q |
Copy Gain |
Copy Gain |
PTEN |
Copy Loss |
Copy Loss |
RB1 |
Copy Loss |
Copy Loss |
Mutation |
||
Mutations |
1203 NSVs |
852 NSVs |
AR |
p.T878A |
p.T878A |
TP53 |
p.R273C |
p.R273C |
SPOP |
p.F102C |
p.F102C |
NCOR2 |
p.E1431K;indel |
p.E1431K |
ASXL2 |
p.R591C |
p.R591C |
PBRM1 |
p.Y1009H |
p.Y962H |
ARID1B |
p.R1885H |
p.R1885H |
ARID2 |
p.A1773V |
p.A1773V |
MSH2 |
indel |
indel |
APC |
indel |
indel |
NCOR1 |
indel |
N.D. |
Expression |
||
AR |
High |
High |
KLK2 |
High |
High |
MSH2 |
MSH2-FSHR-fusion |
N.D. |
Germline |
||
56 AGMC Genes |
No Pathological Variants |
No Pathological Variants |
N.D.; not detected; NSV; non-synonymous nucleotide variant.
Transcript levels measured by RNAseq were highly concordant across the sequencing centers. All tumors demonstrated high levels of AR-regulated transcripts including KLK3/PSA, TMPRSS2 and NKX3.1 in addition to the AR itself. RNAseq analyses in both centers identified fusion transcripts including a common rearrangement between TMPRSS2 and ERG in one tumor, though the detection of other fusion transcripts varied substantially depending on the algorithm used to identify such transcripts. Of interest, an MSH2 mutation was identified in one tumor by both sequencing centers and likely contributed to the hypermutated genome. One sequencing center also identified a rearrangement involving MSH2 predicted to inactivate the second MSH2 allele. While alterations in MSH2 occur as a heritable influence on cancer development in Lynch Syndrome [31], a germline MSH2 mutation was not identified in the exome analysis from this patient.
In contrast to assessments of somatic genomic events in tumors where variation in tumor purity and tumor heterogeneity have the potential for influencing sequencing results, the analyses of germline DNA should consistently identify genomic variants. Of the 56 genes with pathogenicity as determined by the ACMG, all exons were covered to 30X for single nucleotide variant discovery. One likely pathogenic mutation in the APC gene was identified. This result is consistent with the anticipated rate of reportable incidental findings approximating 1–3% for this cohort of genes [10, 32].
Collectively, the findings from this study demonstrate that the results of somatic and germline sequencing are highly concordant across sequencing centers that have substantial experience in the technological requirements for preparing, sequencing and annotating DNA and RNA from human biospecimens. An aspect of genomic sequencing distinct from other assays used in clinical medicine is the breadth of data produced that encompasses anticipated drivers of disease as well as important incidental findings that may have health implications beyond the intended use of the original test. Further, a distinctive feature of oncology involves the iterative sequencing of tumor DNA and RNA, either directly from biopsies or potentially from circulating tumor cells or cell-free DNA, repeatedly over time to evaluate mechanisms of treatment resistance and the emergence of new targets. Establishing the reproducibility of genome-based assays is an essential step in routine use of this technology in research and clinical care.
MATERIALS AND METHODS
Tissue acquisition and preparation
Metastatic tumor samples were obtained from patients with castration resistant prostate cancer following written consent [14]. All samples were reviewed by pathologists with expertise in interpreting prostate cancer histology (Xiaotun Zhang and Lawrence True). Frozen tumor pieces containing > 70% tumor cells were processed as follows: for each frozen tumor block, a frozen section was cut, stained with hematoxylin and eosin and the percentage of tumor cells was ascertained by microscopy. The tumor block was then trisected and a second frozen section was taken from the bottom of each specimen, stained with hematoxylin and eosin and tumor cell percentage was confirmed. One tumor sample (approximately, 1/3 of the original specimen) was then sent to the Broad Institute, one tumor block (1/3 of the original specimen) was sent to the University of Michigan, and the remainder was retained at the University of Washington.
Library preparations and sequencing
Whole exome – Broad Institute
The preparation of libraries for massively parallel sequencing was performed as previously described [15, 33]. Detailed methods are provided in eMethods online. Each pool of whole exome libraries was subjected to paired 76 bp runs on a HiSeq 2000 sequencer. A BAM file was produced with the Picard pipeline (http://picard.sourceforge.net/), which aligned sequences to the hg19 human genome build.
Whole exome – University of Michigan
Tumor genomic DNA and total RNA were purified from the same sample using the AllPrep DNA/RNA/miRNA kit (QIAGEN). Libraries were sequenced with 100 bp paired reads on an Illumina HiSeq 2500 and aligned to the hg19 human genome reference.
Transcriptome – Broad Institute
RNA was extracted from frozen tissue using the miRNeasy Mini kit (Qiagen). An automated variant of the Illumina Tru Seq™ RNA Sample Preparation protocol (Revision A, 2010) was used. Flowcell cluster amplification and sequencing were performed according to the manufacturer’s protocols using either the HiSeq 2000 v3 or HiSeq 2500. Each run was a 76 bp paired-end with an eight-base index barcode read.
Transcriptome – University of Michigan
Transcriptome libraries were prepared using Agilent SureSelect Human All Exon V4 reagents and protocols. Libraries were sequenced using 100 bp paired-end reads on an Illumina HiSeq 2500.
Nucleotide variant detection
Broad institute
MuTect [21] was used to identify somatic single-nucleotide variants. Indelocator (http://www.broadinstitute.org/cancer/cga/indelocator) was applied to identify small insertions or deletions. Annotation of identified variants was done using Oncotator (http://www.broadinstitute.org/cancer/cga/oncotator).
University of Michigan
Paired-end reads were aligned using Novoalign v 3.02.00 and sorted using Novosort (Novocraft Technologies). Variants in both normal and tumor libraries were identified using the local realignment haplotype-based caller FreeBayes [34].
RNA/transcript abundance
Broad Institute
Gene expression was quantified using RNASeqQC [35].
University of Michigan
Gene expression, as fragments per kilobase of exon per million fragments mapped was calculated using Cufflinks [36].
Gene rearrangements/fusion transcripts
Broad Institute
Fusion transcripts were originally identified using Prada [37]. Resulting putative fusion transcripts were manually reviewed.
University of Michigan
Paired-end transcriptome sequencing reads were aligned to the human reference genome (GRCh37/hg19) using a RNA-Seq spliced read mapper Tophat2 [38] (Tophat 2.0.4). Fusion candidates were manually reviewed.
Multi-caller comparisons were subsequently performed using STAR Fusion and Tophat-Fusion [39].
Copy number alterations
Broad Institute
Copy ratios were calculated by dividing the tumor coverage by the median coverage obtained in a set of reference normal samples. The resulting copy ratios were segmented using the circular binary segmentation algorithm (36). Genes in copy ratio regions with segment means of greater than log2 (4) were evaluated for focal amplifications, and genes in regions with segment means of less than log2 (0.5) were evaluated for deletions.
University of Michigan
Copy number aberrations were quantified and reported for each gene as the segmented normalized log2-transformed exon coverage ratios between each tumor sample and matched normal sample [40]. The resulting copy ratios were segmented using the circular binary segmentation algorithm [41].
Germline mutation calls
Broad institute
Germline variants were identified using Unified-Genotyper [42].
University of Michigan
Germline variants were identified using FreeBayes [34].
ACKNOWLEDGMENTS AND FUNDING
We thank the patients and their families for their altruistic participation in this study. The authors declare they have no conflicts of interest and financial disclosures that are relevant to this publication. We thank Dr. Xiaotun Zhang and Lawrence True for assistance with histological analyses of the tumors. All authors are supported by a Stand Up To Cancer - Prostate Cancer Foundation Prostate Dream Team Translational Cancer Research Grant. Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research (SU2C-AACR-DT0712). A.M.C. is supported by NIH Prostate Specialized Program of Research Excellence grant P50CA69568 and the Early Detection Research Network grant UO1 CA111275. EMV and CCP are supported by PCF Young Investigator Awards. The project was also supported by Awards from the Department of Defense Prostate Cancer Research Program PC131820, PC140799, PC140519 and NIH awards: P50CA097186, P01CA163227, and P01CA085859.
CONFLICTS OF INTEREST
The authors declare they have no conflicts of interest relating to the content of this study.
REFERENCES
1. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. (Washington (DC). 2011.
2. Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013; 45:1113–1120.
3. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, et al. Pan-cancer patterns of somatic copy number alteration. Nat Genet. 2013; 45:1134–1140.
4. Garraway Levi A, Lander Eric S. Lessons from the Cancer Genome. Cell. 2013; 153:17–37.
5. Chen Y, McGee J, Chen X, Doman TN, Gong X, Zhang Y, Hamm N, Ma X, Higgs RE, Bhagwat SV, Buchanan S, Peng SB, Staschke KA, et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS One. 2014; 9:e98293.
6. Sparano JA, Golden AA, Montagna C. Translating the TCGA breast cancer results into clinical practice: searching for therapeutic clues. Oncology (Williston Park). 2013; 27:1284, 1286.
7. Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, Barrette T, Everett J, Siddiqui J, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011; 3:111ra121.
8. Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014; 371:1170.
9. Biesecker LG. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: lessons from the ClinSeq project. Genet Med. 2012; 14:393–398.
10. Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, et al. American College of Medical G and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013; 15:565–574.
11. Green RC, Berg JS, Berry GT, Biesecker LG, Dimmock DP, Evans JP, Grody WW, Hegde MR, Kalia S, Korf BR, Krantz I, McGuire AL, Miller DT, et al. Exploring concordance and discordance for return of incidental findings from clinical sequencing. Genet Med. 2012; 14:405–410.
12. Parsons DW, Roy A, Plon SE, Roychowdhury S, Chinnaiyan AM. Clinical tumor sequencing: an incidental casualty of the American College of Medical Genetics and Genomics recommendations for reporting of incidental findings. J Clin Oncol. 2014; 32:2203–2205.
13. Bombard Y, Robson M, Offit K. Revealing the incidentalome when targeting the tumor genome. JAMA. 2013; 310:795–796.
14. Morrissey C, Roudier MP, Dowell A, True LD, Ketchanji M, Welty C, Corey E, Lange PH, Higano CS, Vessella RL. Effects of androgen deprivation therapy and bisphosphonate treatment on bone in patients with metastatic castration-resistant prostate cancer: results from the University of Washington Rapid Autopsy Series. J Bone Miner Res. 2013; 28:333–340.
15. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015; 161:1215–1228.
16. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012; 44:685–689.
17. Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, Beltran H, Abida W, Bradley RK, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015; 161:1215–1228.
18. Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, Lucas JM, Brown LG, Dumpit RF, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016.
19. Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, McKenna A, Sivachenko A, Rosenberg M, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014; 20:682–688.
20. Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, Salipante SJ, Milbank J, Yu M, Grady WM, Tait JF, Corey E, Vessella RL, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014; 5:4988.
21. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013.
22. Spencer DH, Tyagi M, Vallania F, Bredemeyer AJ, Pfeifer JD, Mitra RD, Duncavage EJ. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J Mol Diagn. 2014; 16:75–88.
23. Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, Balk SP. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997; 3:1383–1388.
24. Sun X, Frierson HF, Chen C, Li C, Ran Q, Otto KB, Cantarel BL, Vessella RL, Gao AC, Petros J, Miura Y, Simons JW, Dong JT. Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat Genet. 2005; 37:407–412.
25. Chen E, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, Loda M, True LD, Ye H, Troncoso P, Lis RT, Kantoff P, Montgomery B, et al. Abiraterone Treatment in Castration-Resistant Prostate Cancer Selects for Progesterone Responsive Mutant Androgen Receptors. Clin Cancer Res. 2014.
26. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, Lee C, Montie JE, Shah RB, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005; 310:644–648.
27. Gallagher DJ, Gaudet MM, Pal P, Kirchhoff T, Balistreri L, Vora K, Bhatia J, Stadler Z, Fine SW, Reuter V, Zelefsky M, Morris MJ, Scher HI, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010; 16:2115–2121.
28. Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2014.
29. Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014; 23:437–449.
30. Liang J, Lin C, Hu F, Wang F, Zhu L, Yao X, Wang Y, Zhao Y. APC polymorphisms and the risk of colorectal neoplasia: a HuGE review and meta-analysis. Am J Epidemiol. 2013; 177:1169–1179.
31. Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009; 76:1–18.
32. Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, Kim JH, Rosenthal EA, Kim DS, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013; 93:631–640.
33. Fisher S, Barry A, Abreu J, Minie B, Nolan J, Delorey TM, Young G, Fennell TJ, Allen A, Ambrogio L, Berlin AM, Blumenstiel B, Cibulskis K, et al. A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 2011; 12:R1.
34. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. arVIX. 2012; 1207.3907.
35. DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012; 28:1530–1532.
36. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012; 7:562–578.
37. Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014; 30:2224–2226.
38. Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011; 12:R72.
39. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21.
40. Lonigro RJ, Grasso CS, Robinson DR, Jing X, Wu YM, Cao X, Quist MJ, Tomlins SA, Pienta KJ, Chinnaiyan AM. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011; 13:1019–1025.
41. Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004; 5:557–572.
42. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303.