
Introduction
On April 17th 2015 the Physicians Committee for Responsible Medicine (http://www.pcrm.org/) held a roundtable with expert researchers on Alzheimer disease (AD) and human-based research approaches from the United Stated and the United Kingdom, to discuss why and how the AD research community should adopt human-based research strategies to overcome the increasing prevalence of AD in the 21st century. The major goals of the roundtable were: (1) to discuss the relevance of human-based models and tools for investigating AD pathophysiology at multiple levels of biological complexity, taking human relevance into account; (2) to formulate strategic recommendations as potential guidelines for determining research funding priorities in the field of AD research. In the present document we describe the major discussion outcomes of that meeting. We also reflect on how these recommendations fit in with current, quickly evolving, scientific and public policy efforts.
It is important to note that roundtable participants sometimes expressed different opinions regarding the discussed topics. While some felt that the first step should be to reduce animal models, others felt that current techniques already offer vast, powerful and unexplored pathways to study AD, and that sufficient alternatives already exist to fully proceed with human-based research. However, all participants agreed that there is now a range of new techniques and research directions that have been under-explored and need to be supported through changes in public funding and research priorities.
The AD research paradigm is failing: main facts supporting this premise
Alzheimer disease (AD) represents the most common cause of dementia, accounting for 50-75% of all dementia cases [1, 2]. The number of persons affected by AD in the United States is expected to almost triple by 2050, reaching 13.8 million [3]. Despite intense research efforts, the mechanisms of action of both protective and causative factors for AD are still not clearly understood. In the last ten years no new drugs have been released and existing drugs only stabilize symptoms temporarily in some patients, but do not slow progression of the disease [4, 5]. This defeat is reflected by the dramatically high clinical failure rate (99.6%), which is the highest among biomedical research fields [6-8]. One bright prospect is that the occurrence of dementia has recently been reported to be stabilizing in Western Europe, but this has been primarily attributed to preventative approaches and improvements in living conditions [9]. Analogously, the Framingham Heart Study has reported a decline of both vascular risk factors and the risk of dementia associated with heart failure, stroke, or atrial fibrillation over the course of thirty years [10].
Much of the traditional AD research has been based on the use of animal models, often transgenic (Tg) and inbred mice, in an effort to recapitulate genetic and pathological traits of human disease [11]. However, Tg animals, despite presenting several of the typical AD traits, such as amyloid β (Aβ) formation, neuritic plaques, neurofibrillary tangles (NFT), gliosis, synaptic alterations and signs of neurodegeneration, do not develop the clinicopathological complexities of human AD [12-15]. Moreover, treatments that seem to work in such models have not translated to humans [11, 16-18]. This indicates the existence of a clear disconnection between the (animal) model and the human condition [17] that is not taken into sufficient account by investigators. Another issue with animal models is that they might also be generating false negative data, leading to the exclusion of compounds from clinical studies that could be effective in humans.
An examination of current methodological approaches, suggests a bias in the peer-review process in favor of using these animal models versus alternative approaches. Specifically, the number of projects - and funds - based on animal models supported by the U.S. National Institutes of Health (NIH) over the last eight years is much higher than the number of research projects focused on the use of human-based models and methods (e.g., human-derived (stem) cells, neuroimaging, computational models, prevention, clinical studies, etc.) (Figure 1). Although efforts such as the National Alzheimer’s Project Act — which has dramatically boosted resources for AD and related dementias (ADRD) research — are beginning to prioritize human relevant approaches and attempting to address the multifactorial and multi-etiology of dementia there remains a strong bias towards animal research approaches . This bias can be seen throughout reports and recommendations in the strong linking of “animal research” with “basic research”. For example, in the 2016 update of draft prioritized recommendations of the ADRD, the implementation of the recommendation from Session 6, Focus Area 1: Basic Mechanisms and Experimental Models - “Develop next generation experimental models and translational methods for VCID (vascular contributions to cognitive impairment and dementia)” six out of nine recommendations explicitly call for the development of of animal models [19]. In conflating the concepts of animal research and basic research there is a failure to recognize the important development that basic laboratory research is increasingly being performed entirely without the use of animals. Indeed, without the critical acknowledgement of the failure of past animal paradigms and the promise of new approaches, new attempts at making progress in basic AD research will be severely hampered.
Beyond laboratory models, several lifestyle-related risk factors have been shown to play key roles in the onset and progression of AD, yet research support in theses domains remains disproportionately low, with only a 3.4% of average annual funding supported by the National Institute on Aging (NIA) for prevention in 2010-2012 [20]. Although advancing age is clearly considered the main risk factor for developing AD [21-23], nutritional factors [24], low levels of physical activity [25, 26], reduced cognitive stimulation [27], socioeconomic status and educational attainment [28-30] are all directly related to AD risk. Furthermore, poor sleep quality [31-33] which is known to positively correlate to early Aβ deposition [34, 35], air pollution [36], smoking [37], intake of metals [38-40], pesticides and insecticides [41, 42] as well as metabolic-related dysfunctions [43, 44] have all been described as possible risk factors.
These data indicate that - rather than an independent health condition - AD should be reinterpreted as a complex multifactorial syndrome. Understanding this complexity is clearly critical for designing intervention strategies aimed at preventing or ameliorating early symptoms of AD. Despite this knowledge and massive potential social impact, longitudinal clinical studies focused on prevention are very poorly supported in the U.S. (only ~ 7%-9% of the $30 billion NIH total discretionary budget) [20]. For all these reasons there is an urgent need to rethink current research funding strategies to directly target human relevance and disease causation.
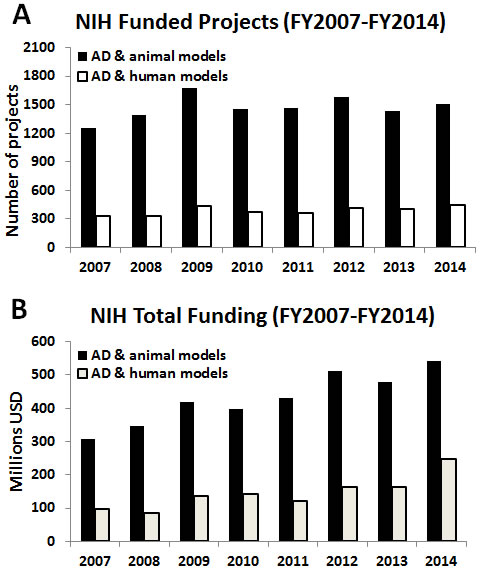
Figure 1: Bar graphs reporting the absolute numbers of AD-related projects focused on the use of animal models (black bars) vs projects accounting only for human-relevant models/methods (white bars). A. and relative funding B., provided by the NIH from fiscal year (FY) 2007 to 2014. Analysis has been done usinghttp://projectreporter.nih.gov/reporter.cfm (as of July 6th 2015), project search was limited to ‘project terms’. List of applied keywords per category: AD & animal models: Alzheimer AND (“primate” OR “primates” OR “monkey” OR “monkeys” OR “macaca” OR “macaque” OR “marmoset” OR “vervet” OR “cercopithecus” OR “cynomolgus” OR “tamarin” OR “dog” OR “dogs” OR “canine” OR “canines” OR “canis” OR “feline” OR “felines” OR “felis” OR “guinea” OR “rabbit” OR “rabbits” OR “mouse” OR “mice” OR “porcine” OR “pig” OR “pigs” OR “ovine” OR “sheep” OR “rattus” OR “rat” OR “rats” OR “mus” OR “mice” OR “mouse” OR “mammal” OR “fish” OR “zebrafish” OR “hamster” OR “rodent” OR “animal model” OR “animals” OR “animal” OR “xenopus” OR “caenorhabditis elegans” OR “c. elegans” OR “drosophila melanogaster” OR “drosophila” OR “lamprey”). AD & human models: Alzheimer AND “human” AND (“stem cells” OR “induced pluripotent stem cells” OR “iPS” OR “imaging” OR “PET” OR “MRI” OR “computational” OR “prevention” OR “preventive strategy” OR “clinical study” OR “clinical” OR “clinical trial” OR “patient”) NOT (“primate” OR “primates” OR “monkey” OR “monkeys” OR “macaca” OR “macaque” OR “marmoset” OR “vervet” OR “cercopithecus” OR “cynomolgus” OR “tamarin” OR “dog” OR “dogs” OR “canine” OR “canines” OR “canis” OR “feline” OR “felines” OR “felis” OR “guinea” OR “rabbit” OR “rabbits” OR “mouse” OR “mice” OR “porcine” OR “pig” OR “pigs” OR “ovine” OR “sheep” OR “rattus” OR “rat” OR “rats” OR “mus” OR “mice” OR “mouse” OR “mammal” OR “fish” OR “zebrafish” OR “hamster” OR “rodent” OR “animal model” OR “animals” OR “animal” OR “xenopus” OR “caenorhabditis elegans” OR “c. elegans” OR “drosophila melanogaster” OR “drosophila” OR “lamprey”)
Addressing human relevance in AD research with the use of alternatives to animal experiments
Recent developments have brought about a staggering array of research approaches that are offering bold new ways to study human brain aging and are yielding profuse and meaningful human relevant data. These techniques include: (1) several human-based models focused on the use of patient-derived cells, such as induced pluripotent stem cells (iPSCs) and neuronal and glial cultures, (2) multiple ‘omic’ technologies (e.g., genomics, proteomics, lipidomics, transcriptomics, metabolomics, etc.) resulting from overall analyses of biological samples by high-throughput analytical approaches and databases, (3) computational analytical approaches and (4) novel neuroimaging readouts [17, 18, 45].
Given the need to integrate the huge amount of incoming data, comprehensive multi-scale and systems biology approaches are becoming fundamentally important. These approaches must take into account all the different levels of biological complexity (including population, individual, organ/tissue, cellular, protein, and gene level), thereby allowing for the elucidation of disease-related adverse outcome pathways (AOPs), as already envisioned in toxicology [46] and proposed for AD research [17]. Within this new framework it is becoming increasingly possible to not only determine the effects of an exposure to a given compound (for instance, pollutants possibly implicated in the onset of AD) but also to investigate how these effects are induced [47, 48]. Defining which signaling pathways are perturbed at early stages of AD (i.e., the AD-related pathways) might help predict long-term effects and sequelae. For this reason, multiscale AOPs should become the core of the new paradigm in AD research. Investigating AD-related multiscale AOPs could allow researchers to link environmental and genetic causes with outcomes at individual/body level [17].
A number of cellular in vitro models of AD and human-based methods can already take into account different levels of biological complexity (Table 1). For example, iPSCs have been widely applied in AD research [49-53] and can be used to: (i) assess clinical candidate drugs on human brain cell types; (ii) conduct phenotypic screening of compounds that modulate or normalize disease phenotype; (iii) conduct target-based screening if candidate genes are identified; (iv) compare genetically-diverse panels; or (v) select or stratify participants of clinical trials based on their genetic backgrounds and/or phenotypic traits.
Moreover, human-based intervention trials focused on nutrition, physical activity, and cognitive training are particularly relevant to preventing AD and cognitive decline. These trials have proven to be the most effective strategies to reduce AD symptoms [20, 25, 26, 54-59].
In order to stimulate the creation of multifactorial approaches to AD, global efforts have been made to improve access and discussion online for researchers. In recent years, common platforms, such as CLIR (Collaborative Laboratory Integrated Reports), developed at the Mayo Clinic (https://clir.mayo.edu/), have been shown suitable to create groups of interest and propose multidisciplinary team approaches, allowing comparisons among different sub-populations, different ages and different treatments. At the clinical level, databases, such as the Laboratory of Neuroimaging - Image Data Archive (LONI-IDA), provide user access to de-identified data from positron emission tomography (PET), magnetic resonance imaging (MRI), cognitive data sets and biomarkers. These interfaces represent a large step forward in maximizing the impact of these data.
Table 1: Human-based studies, models and readouts suitable for AD research
Human-based models/tools |
Characteristics and applicability |
Biological complexity level |
Epidemiological studies, randomized clinical trials |
To assess the complex interrelations of risk factors and ameliorating influences including: environmental triggers, genetic susceptibility, sex, gender, diet, physical activity, co-occuring conditions (e.g., diabetes), cognitive engagement, social interactions and other cultural factors. |
Population, individual |
Human ex vivo tissue |
Healthy and diseased brain tissues with short post-mortem intervals, standardized preparation, accessible samples and data. To account for patient heterogeneity and study cellular and structural pathologies. To aid in the validation of biomarkers and to refine analysis of factors involved in disease progression |
Individual, whole brain |
Neuroimaging techniques (e.g., MRI, PET, MRI tractography) |
To study human brain anatomy through 2D and 3D images in in vivo and ex vivo studies. To refine AD diagnosis and uncover early markers of disease, understand longitudinal structural development of AD, assessment of treatment effects, construction of brain atlas/connectome |
Individual, whole brain |
Connectomics, PBPK, and PD studies, IVIVE, |
To define kinetics and dynamics of environmental factors (e.g., compounds, nutrients) exposure and to predict their long term effects in relation to AD. To assess the efficacy of compounds for AD treatment |
Individual, whole brain |
Microfluidics/organ-on-chip |
To investigate tissue complexity, assess effects of possible therapeutic compounds. |
Tissue, whole brain |
Patient-derived samples: CSF, blood/plasma, fibroblasts, lymphocytes |
To define early biomarkers of AD, to generate xeno-free iPSCs. |
Tissue |
3D models, organoid systems (e.g., iPSCs, NSCs) |
To mimic physiology of the brain tissues. Suitable depending on the research goals. |
Cell, Networks, Organoids |
Early, familial and late-onset AD patient-iPSCs and their differentiated functional derivatives (2D and 3D) |
Glutamatergic & cholinergic neurons and astrocytes. iPSC-neurons show AD phenotypic traits consistent with the Aβ tau hypotheses after limited time in culture (e.g., elevated Aβ production, increased levels of p-tau) and responsiveness to β and γ secretase inhibitors. Genome-editing technologies (e.g., ZFN, TALENs, and CRISPR/Cas9) can be used to add, disrupt or modify the sequence of specific genes related to AD, measure their impact on human iPSC-derived neurons and, ideally, design patient tailored treatments. To identify disease pathways & drug targets, & assess therapeutic compounds. |
Cell, Assemblies, Networks |
Synchrotron x-ray fluorescence imaging |
To define bio-metals distribution and concentrations in the human brain affected in AD. To characterize the metallo-relationship of plaques and tangles, volumetric reductions in brain regions in AD. |
Multi-scale: Sub-cellular to Individual, whole brain |
Omics: transcriptomics, proteomics, lipidomics, metabolomics, exposomics, nutrigenomics, nutrigenetics, genomics, epigenomics |
To assess signaling pathways, epigenetic, genetic mutations, gene expression & lifetime exposures |
Protein, gene, individual |
Computational modeling |
Can be applied at any of the above levels to investigate the causal relations, illuminate underlying mechanisms and to help predict outcomes of interventions in relation to AD at single and multiple scales |
Ranges from gene to neural population dynamics |
Abbreviations: CSF, cerebrospinal fluid; iPSCs, induced pluripotent stem cells; NSCs, neural stem cells; MRI, magnetic resonance imaging; PET, positron emission tomography; PBPK, physiologically based pharmacokinetics; IVIVE, in vitro-in vivo extrapolation; PD, pharmacodynamics; ZFN, zinc-finger-nucleases; TALENs, transcription activator-like effector nucleases; CRISPR/Cas9, clustered regularly-interspaced short palindromic repeats/CRISPR-associated protein-9 nucleases.
As alternatives to the use of traditional mammalian species, some non-mammalian/non-vertebrate models, such as Dictyostelium discoideum, have also be applied to undertake new directions in basic research [60, 61] and define the role of previously unexplored proteins/molecules. Taking into account their biological limitations (e.g., they cannot effectively be used to mimic the large-scale anatomical and behavioral aspects of an aging human brain), these non-mammalian models are relatively easy to handle, cost effective, and can be manipulated to express AD-related human genes/proteins [61]. providing innovative approaches to research in this area.
The availability of a range of approaches and collaborative tools is an important development in AD research. Obviously, the use of a given model should be driven by the specific research objectives, whether they be basic research or translational. In light of the fact that a unique model suitable to tackle all aspects of AD does not (and may never) exist, different models might be suitable to cover the many different aspects of the disease depending on the level and mechanism being investigated. For this reason, in an effort to gather a global picture of the environmental/lifestyle risk factors, the etiopathological mechanisms of the disease and define possible preventive, intervention and pharmaceutical strategies, the creation of a multidisciplinary team approach in AD research, combining different expertise, should be mandatory. In line with this, some research initiatives have been undertaken in an effort to define correlations among Aβ formation, neuroanatomy, cognitive and lifestyle factors [62, 63].
Limitations of alternative approaches and strategies to overcome these limitations
Despite the great potential of new human-based approaches and non-mammalian models, their broad applicability and reliability is currently hampered by some limitations. It is essential to clearly recognize these constraints and define strategies to overcome them (Table 2).
Table 2: Limitations of alternative models and methods and strategies to overcome these limitations |
||
Human-based models/tools |
Limitations |
Strategies to address limitations |
Epidemiological studies, randomized clinical trials |
Inability to determine causality due to potential multiple interacting and confounding factors Difficult to compare studies designed according to different inclusion/exclusion criteria |
Comprehensive assessment of multiple behaviors and risk factors and complex multivariate analyses to address conjoint confounding and effect modification. Application of machine learning and other techniques capable of non-linear and high-dimensional pattern recognition in large data sets. |
Possibility to create multi-center collaborations, taking advantage of common platforms |
||
Multiple intervention studies to test treatment effects in different types of populations |
||
Patient-derived samples: CSF, blood/plasma, fibroblasts, and postmortem AD and control brain tissues |
Storage and analytic methods are often not standardized, preventing inter-lab comparisons Poorly preserved brain tissues and long postmortem delays Samples are often not readily available for test for reproducibility and validation |
Creation of multi-center collaborations to standardize methods & optimize distribution: e.g., 2-3 nationwide brain banks centers of excellence, with 24/7 autopsy services, short postmortem delays (2-3 hours maximum) and with standardized neuropathological protocols. Digitize neuropathology finds using standardized methods and creating an open-access database for additional analysis. |
Neuroimaging techniques (e.g., MRI, PET, MRI tractography) |
High costs; sometimes weak correlations between measures and clinical manifestations; sometimes difficult to quantify |
Consider large-scale studies to improve correlations between imaging measurements and clinical manifestations |
Synchrotron x ray fluorescence imaging |
Requires ex vivo or post mortem brain tissue |
Integrate this technology with other neuroimaging tools |
Microfluidics/organ-on-chip |
Some limitations with regard to transport and diffusion of nutrients and oxygen; individual organs, kept in isolation |
Increase investment in research and development. Complement these technologies with neuroimaging data and/or other omics data sets |
3D models (e.g., iPSCs, NPCs) |
Not applicable for all purposes |
Integrate 3D models with 2D models depending on applications and research goals |
AD patient-iPSCs and their differentiated functional derivatives |
Generating high-quality iPSCs is expensive and time consuming; a limited number of AD iPSC lines have been generated and thoroughly characterized so far |
Cost is dropping over time; several entities (e.g., CIRM, NYSCF, etc.) are funding the development of hundreds of iPSC lines from AD patients |
They might be not fully representative of the complex physiology of the brain and/or of AD pathophysiology |
Possibility to create co-culture systems with human microglial cells. Genome-editing technologies can be applied to create mutations related to the AD genetics, measure their impact on patient iPSC-derived neurons and design patient tailored treatments. |
|
Different reprogramming and QCs have been used, so comparisons between labs are difficult to make at this time |
Several entities (e.g., CIRM, NYSCF, etc.) could standardize reprogramming methods allowing inter-lab comparisons |
|
Challenges with regard to penetrance, cell purity, degree and type of differentiated cells generated from iPSCs |
Need to harmonize QC standards, which would be more feasible with the participations of dedicated entities |
|
Traditional reprogramming methods (e.g., integrating lentiviruses) and xeno-contamination might have affected the phenotype of the lines |
Develop and adopt xeno-free techniques with non-integrating reprogramming vectors |
|
Epigenetic signatures of the somatic cell of origin might be retained in the reprogrammed iPSCs (NB: evidence that epigenetic traits get lost upon long term culture) |
Possibility to directly reprogram fibroblasts into neurons |
|
Possibility to reprogram post-mitotic neurons and frozen brain tissue samples into iPSCs (to retain the neuronal epigenetic and pathologic background) |
||
iPSCs metabolic profile has not been investigated enough (which has special relevance in AD research) |
Define QC metrics to establish metabolic features of iPSCs |
|
Still not clear how long iPSC-derived neurons should be kept in culture in order to mimic late-onset AD neurons and tissue pathophysiology; possible issues with the loss of aging-related transcriptional signatures and features. |
Use AD brain tissues as benchmark models to define QC metrics suitable to assess neuronal and pathological features of differentiated iPSCs. Overexpression of aging-related genes (e.g., progerin) might help model AD in a dish. Direct conversion of aging donors' fibroblasts into neurons (iNs) can help retain aging-related transcriptional signatures. |
|
2D and 3D iPSC cultures might be characterized by different biological/cellular/molecular features and generate different responses |
Define QC metrics to establish features of 2D vs. 3D iPSC cultures |
|
Not clear if AD-derived fibroblasts might be proven as suitable as their reprogrammed counterparts (i.e. iPSCs) to define molecular/cellular features of AD (e.g., metabolic profiles) |
Define QC metrics to establish features of AD-derived fibroblasts vs AD-derived reprogrammed iPSCs |
|
| Non-mammalian/invertebrate models of AD | More phylogenetically distant from humans than mammalian species; might lead to intermediate validation steps in mammalian (non-human) species | Consider their suitability for basic research effort; less time consuming and less expensive than traditional animal models |
| Investigate directly in human ex vivo tissues/cultures (rather than animals) to assess preclinical data (applying microdosing analysis) | ||
| PBPK and PD studies, IVIVE | PBPK, PD and IVIVE are currently applied mainly in toxicology. |
Possibility to establish dedicated consortia with a multi-disciplinary approach (e.g., combining medical research and toxicology expertise). |
| Connectomics, computational analysis and modeling | Connectomics still in early development. Resolution too low. Very large data sets. Computational models are often restricted to simply mimicking observed phenomena and have no predictive value. |
Develop techniques to study both individual and large cohorts necessary to recognize significant patterns. Increases in resolution, computational power and large-scale analysis algorithms are all rapidly improving. Encourage move to foundational computational simulations that explore the basic effects of cellular, network and system factors in aging and dementia. Use to elucidate and predict previously unrecognized changes in anatomy, physiology and cognition. |
| Various other omics: transcriptomics, proteomics, lipidomics metabolomics, exposomics, nutrigenomics, nutrigenetics, genomics, epigenomics | High costs | Costs of analysis are reducing. Possibility to establish dedicated consortia with a multi-disciplinary approach (e.g., combining molecular biology and biostatics expertise) |
Abbreviations: CSF, cerebrospinal fluid; iPSCs, induced pluripotent stem cells; QC, quality control; NSCs, neural stem cells; MRI, magnetic resonance imaging; PET, positron emission tomography; PBPK, physiologically based pharmacokinetic; IVIVE, in vitro-in vivo extrapolation; PD, pharmacodynamics |
||
AD-derived iPSC models
With specific regard to iPSC-derived models of AD, it is generally recognized that generating high-quality iPSC lines is still expensive and time consuming. In addition, only a limited number of AD iPSC-derived lines have been generated and thoroughly characterized so far. These AD-related iPSC studies have used different programming and quality control methods, as well a variety of somatic cell types. These differences in protocol make inter-laboratory comparisons difficult at this time. Moreover, the reprogramming mechanism used to generate older iPSC lines, are often based on the use of integrating lentiviruses and retroviruses, which may have caused insertional mutagenesis [64, 65]. Many approaches remain xeno-contaminated. For this reason, to minimize these issues, current and future reprogramming methods should aim to be xeno-free and based on the use of non-integrating reprogramming vectors or entirely vector-free approaches.
Nevertheless, the iPSC approach holds enormous potentials and the rapidly expanding research field is already tackling these limitations. For example, the production cost is progressively dropping, and several entities, such as the California Institute for Regenerative Medicine (CIRM,https://www.cirm.ca.gov/) and the New York Stem Cell Foundation (NYSCF,http://nyscf.org/) are currently funding the development of hundreds of iPSC lines from both early- and late-onset AD patients that will be available globally at low cost to investigators. It will now be important to generate dedicated and accessible bio-banks for the collection and distribution of AD patient-derived fibroblasts or peripheral blood cells for reprogramming purposes, accounting for both late- and early-onset AD, mild cognitive impairment, and healthy elderly donors. These samples should be made available to the scientific community whenever required to facilitate inter-laboratory reproducibility, data validation and outline correlations between patients’ clinical history and patient-related cellular and molecular data sets, which might help develop novel therapies. In particular, the collection of late-onset, sporadic AD patient-derived fibroblasts for the generation of late-onset AD iPSCs will be critical in providing insight into late-onset AD pathology, which represents the majority of AD cases (~95%), as compared to early-onset AD (representing ~5% of all AD cases) [23, 66]. These collection efforts should pay particular attention to the language of the informed consent forms used in donor recruitment in order to ensure patient protection as well as the broadest possible use of the samples by both in academic and commercial research and clinical usersapplications.
Several aspects of the generation of iPSCs as well as their use in studying AD include the “epigenetic memory”, the types of reprogrammed cells used for studying the etiology of AD, and the ability of iPSCs to mimic AD pathophysiology. With regards to this “epigenetic memory” phenomenon, there is evidence that the epigenetic signatures of the somatic cells of origin might be retained in the reprogrammed iPSCs. As a consequence, iPSCs might preferentially generate derivatives of the donor somatic cell type [67, 68], inadvertently skewing results. Possible strategies to overcome this limitation might be to directly reprogram fibroblasts into nervous system cell types [69, 70] or, in order to retain the epigenetic background of neuronal cells, to reprogram post-mitotic neurons into iPSCs [71]. iPSCs have been successfully obtained by reprogramming frozen non-cryoprotected dural tissue samples (stored at −80°C for up to 11 years), which allowed for generating iPSCs with confirmed pathology even from AD patients with rare genetic variants [72]. Nevertheless, there is evidence that iPSCs lose epigenetic traits during long term culture [73], which might be considered either as a positive aspect (as the epigenetic memory of somatic cells of origin might be mitigated) or a negative aspect (in light of the fact that AD patient epigenetic signatures might also be lost over time).
Additionally, it is also unclear whether AD-derived fibroblasts might be proven as suitable as their reprogrammed iPSC counterparts to define some of the molecular/cellular features of AD. For instance, using AD patient-derived fibroblasts or other cell types, such as peripheral blood mononuclear cells, might be sufficient to detail some AD-related genetic, epigenetic and/or metabolic features, avoiding all the reprogramming steps and the overall time consuming neuronal differentiation process. For all these reasons, establishing appropriate quality control metrics, accounting for gene expression analyses and quantifications of protein/biomarker levels will help define the expandability of an iPSC model for a given purpose and harmonize data interpretations, allowing inter-laboratory comparisons, as already envisioned and practiced in toxicology studies [74].
Furthermore, there are some challenges with regard to penetrance, cell purity, degree and type of differentiated cells that can be generated from iPSCs that need to be taken into account when developing new models. In terms of the future directions of iPSCs in AD, greater consideration to the metabolic profile would be advantageous. The majority of studies published so far have not extensively investigated the iPSC metabolic profile; however, several lines of evidence indicate that AD should be studied as a complex systemic/metabolic dysfunction, correlated to metabolic syndrome [43], hypometabolism, oxidative stress, and modifications of the glucose-fatty acid cycle [75].
Beyond issues of epigenetic memory and metabolism, it remains unclear how long iPSC-derived neurons should be kept in culture in order to mimic the development of late-onset AD neuronal cells and tissue pathophysiology [76]. In general, modeling aging and neurodegenerative disease, like AD, in differentiated human neurons, such as those derived from AD patient iPSCs, can be a challenging task. Often neuronal cells cultured in vitro do not retain the aging-associated transcriptional profile and phenotype, which represents a major issue when modeling late-onset disorders, such as late-onset AD. There are some possible ways to “age” human neurons in a dish. In particular, the overexpression of the premature aging-related gene s, such as progerin, has been shown suitable to model Parkinson’s disease in iPSCs and might be possibly applicable also for AD-iPSCs [77]. Alternatively, the direct conversion of aging donors’ fibroblasts into neurons (namedcalled induced neurons, or iNs), avoiding cells reprogramming toward the an embryonic phenotype, has been shown promiseing; iNs were found to retain an aging-related transcriptional signature (i.e., decline of the nuclear transport receptor RanBP17) when compared to iPSCs and their neuronal derivatives [78].
Additionally, genome-editing technologies, such as the zinc-finger-nucleases (ZFN), the transcription activator-like effector nucleases (TALENs), and the clustered regularly-interspaced short palindromic repeats/CRISPR-associated protein-9 nucleases (CRISPR/Cas9) can now be used to overcome variability in human genomes. Although still in development, these genome-editing technologies can already be used to add, disrupt or modify the sequence of specific genes related to AD and measure their impact on human iPSC-derived neurons [79]. In particular, these nucleases can induce guided DNA breaks, which can be repaired by homologous recombination with a donor vector carrying a desired point mutation or gene, in order to better model the disease in vitro and, ideally, design patient tailored treatments [80-82].
Moreover, even as researchers are using cell lines to address the complexities of developing a robust two-dimensional in vitro model for AD research, others are taking it a step further by working towards three-dimensional iPSC cultures. This is important because two-dimensional cultures of iPSCs seem to be characterized by considerably different biological, cellular, and molecular features as compared with their three-dimensional counterparts [51]. The implications of the differences in responses upon exposure to potential therapeutic compounds in two-dimensional versus three-dimensional models still need to be elucidated and their biological relevance assessed. Despite these limitations and open questions the added dimension provides an entirely new platform to investigate pathology and therapeutics (Table 2).
Microfluidics/organ-on-chip systems
Beyond iPSCs models, microfluidics/organ-on-chip systems have been created in an effort to simulate in vitro human organ and tissue biology and function. These models combine different cell types in specific 3D culture systems [83, 84] and might be useful to test novel therapeutic compounds in human physiological-like systems. However, despite their potential applicability, these technologies are still in their infancy and require further validation. Additionally, these models, regardless of their level of optimization, remain fundamentally disembodied and thus cannot capture the full complexity and physiological function of a living organism. For this reason, direct clinical studies, including neuroimaging data and various omics data sets, that incorporate the full richness of human cognitive, environmental and social interactions will be required to complement and interpret information derived from these in vitro models (Table 2).
Non-mammalian models of AD
Non-mammalian/invertebrate models of AD (e.g., Dictyostelium discoideum, Drosophila melanogaster, etc.) were previously judged poorly relevant from a biological standpoint and for this reason less worthy of funding compared to mammalian species phylogenetically closer to humans (Table 2). Nevertheless, it’s worthwhile considering the limited “return on investment” that has been gained after extensively funding translational research projects focused on the use of traditional mammalian models, in particular mice. The over-reliance on the use of animals, together with the lack of implementation and optimization of human-based models, have contributed to the current clinical attrition rate in AD translational research [6, 8, 16].
Although non-mammalian species can be used to define basic disease mechanisms, human-based cell models, such as AD-iPSC neuronal cell cultures and human-based organ-on-chip systems, could also be applied for pre-clinical drug discovery.
One approach that has been suggested is to validate data obtained in non-mammalian models in a small number of animals before moving to human tissues/cultures or clinical trials, thereby contributing to a reduction of the use of animals, according to the 3Rs principle envisioned in toxicology and biomedical research [85]. However, it should also be considered that intermediate validation steps in non-human mammals might generate false negative results, possibly invalidating results that might actually be proven valuable in human settings. Moreover, the assumption that non-mammalian species require intermediate validation steps in mammalian non-human models before translating obtained data into humans largely remains unquestioned. However, data obtained in non-mammalian and non-animal models can be validated directly and more effectively in human ex vivo tissues/cultures and/or postmortem tissue. This is already being done for some rare diseases, using microdosing analyses [86]. Most importantly, the rapidly expanding availability of direct human assays, imaging and clinical data collection techniques is increasingly rendering animal models of all scales unnecessary.
Post-mortem AD brain tissues
Post-mortem AD brain tissues are important biological resources for AD research, from which the major AD therapies were discovered [87]. It is also critical as a validation resource with which to assess discoveries from cell culture and animal models. They represent an invaluable resource to conduct neuropathology studies, spanning from morphology, connectivity, cellular, molecular and genome perspectives. Moreover, as clinical studies of AD attempt to discover earlier and more sensitive biomarkers, neuropathology studies remain the key reference for validation [88].
However, brain tissue samples are often of suboptimal quality, due to long postmortem delays and inappropriate postmortem handling and storage. High quality brain tissue from normal control subjects is particularly scarce. Some aspects of protein function, phosphorylation [89], RNA integrity and the aforementioned epigenetic modifications are strongly altered by postmortem delay, freeze-thaw cycles and even by freezing itself [90]. For these reasons, much of the currently available brain tissue, while being useful to conduct morphological studies and assess robust disease biomarkers, may be unsuitable for many molecular studies [91, 92]. Nevertheless, microRNA analysis of AD-affected temporal lobe neocortical tissues collected in short post-mortem interval of about 1 hour can provide important starting points for examining specific AD alterations [93].
It has to be considered that limited availability of post-mortem tissue and differing collection and preservation protocols make projects requiring very large subject numbers, as well as inter-laboratory replication studies difficult or impossible to perform. For these reasons, the creation of multi-center collaborations and bio-banks would greatly increase the ability of researchers to make the most of these resources (Table 2). However, it would be counter-productive to simply replicate existing brain bank networks, such as those of the NIA Alzheimer’s Disease Centers (https://www.nia.nih.gov/alzheimers/alzheimers-disease-research-centers) or BrainNet Europe (http://www.brainnet-europe.org/) as these have not been able to rapidly and systematically provide autopsies or sufficient numbers of normal control brains. A more targeted approach would be to provide proportionately greater funding to a small number of specialized centers, allowing them to meet these critical needs. These centers should allow a streamlined system of sharing, improve timing of distribution, increase the quality of the available materials, harmonize tissue collection standards and provide better correlations between the pathology and neuroimaging patient data. A more ambitious, yet potential ground-breaking step for neuropathological approaches would be to digitize research data and make them openly available along with detailed tissue collection and analysis protocols. A model for this could be the Alzheimer’s Disease Neuroimaging Initiative (ADNI), which provides in-depth information in their neuroimaging, biomarkers and genetics data collected from large multicenter collaborations [94]. This level of sharing and standardization would maximize the usage of these precious tissue resources and accelerate the improvement of neuropathologic approaches by increasing interactions between investigators.
Neuroimaging
Novel in vivo imaging readouts, such as PET and ultra-high-field MRI are currently available to diagnose AD [95, 96] and have been successfully applied to assess in vivo the effects of specific nutritional interventions [54-56]. Importantly, neuroimaging readouts have been critical to discover commonalities of neuroanatomical features shared by AD, type-2 diabetes and the metabolic syndrome [45, 97-99], pathologies that have been shown to be highly interconnected [100-102]. Moreover, human connectomics enabled by techniques such as MRI tractography, allows for the reconstruction of 3D neuronal networks, brain anatomy, and AD-related neuroanatomical modifications [103-105].
As is the case with many new approaches, future neuroimaging technologies will need to overcome the sometimes prohibitively expensive development and operational costs. In addition, researchers will need to improve the clarity of the identification of correlations between retrieved measures and AD-related clinical manifestations, as well as devise methods to deal with the complex data sets to improve quantification of imaging features. Despite current limitations, even existing technologies represent essential tools to support human-relevant AD research approaches, and significant extension of large-scale clinical studies using current techniques should be encouraged to help improve correlations between imaging measurements and clinical manifestations (Table 2).
‘Omics’ technologies and computational models
Despite their relatively high costs, high-throughput technologies, such as proteomics, lipidomics, metabolomics, epigenomics, and genomics, are currently applied to define the molecular mechanisms underlying AD pathogenesis [106-111].
Additionally, computational models, such as in vitro-in vivo extrapolation (IVIVE), physiologically based pharmacokinetic (PBPK), and pharmacodynamics (PD) modeling, are currently applied in the field of toxicology and regulatory testing, but might be suitable to define kinetics and dynamics of compound exposure, predict their long term effects in relation to AD [47, 48, 112], and assess therapeutic potential of novel compounds for AD treatment [113-115].
While these technologies are still under development and require further optimization, establishing dedicated consortia with a multi-disciplinary approach aimed at combining medical research with toxicology expertise might prove a winning strategy to speed the drug discovery process (Table 2). More broadly, computational approaches allow for unprecedented mining of data across levels. Similarly, beyond just data mining, computational simulations can explore the correlations and underlying mechanisms at levels ranging from the molecular to cellular to network, and even social scale. These simulation techniques offer great promise but remain largely underused.
Need to prioritize human relevant research and explore alternative research avenues
Taking into account “human relevance” when addressing AD research efforts, funding agencies need to implement strategies that will encourage the use of these human-based models for AD research. The implementation of human-based methods will also contribute to minimize the use of sentient beings in biomedical research, as advocated by the NIH [116] and the public [117].
To this aim, requests for applications (RFAs) focused on the use of explicitly xeno-free human-based models and novel high-throughput technologies should be created. In particular, considering the potential of AD patient-derived iPSCs, study sections should include experts in the iPSC and reprogramming field, competent in evaluating research proposals focused on the use of iPSC models for AD.
Additionally, expansion of existing and creation of new centralized and open bio-banks providing iPSC lines and/or high quality post-mortem tissues should be encouraged and incentivized, allowing large scale distribution of biological samples to research institutes when needed. While entities distributing both healthy controls- and AD patients-derived iPSCs are already in place in Europe, the U.S. and Japan, iPSC lines are rarely fully characterized by providing entire genomic, epigenomic and patient phenotype data sets [118]. For this reason, efforts to assimilate best practice should be taken into account [118]. Interestingly, CIRM has recognized the value such genetic information adds to an iPSC line and released in early 2016 a RFA (DISC3.1) to characterize all 3000 lines in its repository. Additionally, pre-competitive and collaborative centralized distribution of iPSCs would help to increase biological sample quality and would allow for the creation of harmonized quality control standards for the use, characterization, handling and storage of biological material. Developing such guidance will accelerate inter-laboratory data comparison and validation. Again, consent forms need to allow such sharing.
Moreover, considering the important role played by lifestyle and environmental factors and the relevance of prevention to reduce the burden of AD [9], as also commented in the Leon Thal Symposium proceedings [119, 120], specific RFAs should be created to encourage investigation of early phenotypic traits of neurodegeneration and the implication of multiple networks (or “human disease pathways”) in neuronal failure at early stages of the disease. Amongst these networks, research on mitochondrial dysfunction, known to be an early event in AD progression [121-123], should be emphasized.
Learning from failure and ethics should be encouraged as a general attitude in research. Current and previous research efforts to study AD pathology in animal models and identify effective drug targets have not led to significant and effective prevention or disease modification in humans, so new avenues should be explored. In this regard, even re-evaluating the validity of traditional AD “gold standards” (i.e. the main diagnostic biomarkers of AD), such as presence of Aβ plaques and NFT, might help to hypothesize new therapeutic strategies. In particular, both Aβ and NFT are known to often appear early in time and, according to novel hypotheses, might actually not be considered as causative of AD but rather a result of AD pathology, which is often characterized by neuroinflammation as well as hypometabolism [43, 75, 124]. In this regard, negative results in science are not given enough consideration, frequently leading to their suppression during publication. However, negative results are crucial to establishing limitations of current research models [125], and defining the need for new research avenues.
The creation of pre-competitive consortia to judge the suitability of new models would be highly relevant. In this regard, pharmaceutical companies have shown a willingness to abandon obsolete models, investing resources in new human-derived paradigms involving pre-competitive consortia, such as the Cardiac Safety Research Consortium (http://cardiac-safety.org), the FDA’s Critical Path Initiative (http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/ucm076689.htm), TransCelerate BioPharma (http://www.transceleratebiopharmainc.com), the international Serious Adverse Event Consortium (http://www.saeconsortium.org/) and larger international consortia, such as the Structural Genomics Consortium (http://www.thesgc.org). Analogously, in Europe, the EU commission has allocated 1BN euro into pre-competitive consortia (e.g., the Innovative Medicines Initiative is EU’s largest public-private initiative,http://www.imi.europa.eu/). Thus, the paradigm shift to create more human-relevant standards for validation of translational research should be stimulated at all stakeholder levels.
Moreover, considering the relevance of prevention, it would be necessary to increase current research budgets allotted to preventive medical research and also to increase expertise in the field of education and nutrition in correlation to neurology.
Recommendations to guide new funding strategies
While it is important to provide appropriate funding to support research and speed the discovery process [126], currently available resources should also be better allocated, shifting the focus to prevention strategies with human relevance and to the use of human-based research methods, such as patient-derived iPSCs, computational methods, advanced brain imaging methods, epidemiological studies and human focused non-animal models. Given the important knowledge we already have regarding lifestyle-related factors (e.g., diet, exercise, environmental exposure, etc.), we recommend that public education and policy should be ramped up considerably with special attention to those who may contest these recommendation (e.g., fast food industry).
Considering the multi-dimensional nature of AD pathology, we believe that the time is ripe for a reevaluation of the current definitions of aging, cognition, and their relationship to a variety of biological, social, and environmental variables. Instead of examining a single variable or biomarker at a time, as has often been done in the past, it would be worthwhile to consider the interconnected implications of several genetic, epigenetic, morphological, environmental, behavioral and social factors in the onset and consolidation of AD.
This envisioned paradigm and the application of human-based models, in conjunction with large scale randomized clinical trials and multi-dimensional -omics readouts, will help revolutionize our knowledge of AD pathology and etiology, contributing to the creation of a more holistic perspective regarding AD in the context of aging and lifestyle.
The NIH ADRD Research Summit, held at NIH in February 2015, advocated for “a change in how the academic, biopharmaceutical and government sectors participating in Alzheimer’s research and therapy generate, share and use knowledge to propel the development of critically needed therapies” [127]. In particular, the limitations of rodent models were highlighted [127]. In line with this, we propose a list of practical recommendations possibly suitable to guide current funding priorities in AD research, addressing human relevance. These recommendations, outlined in Table 3, are meant to be applicable to the NIH as well as any AD association subsidizing AD research.
The implementation of the proposed strategies would necessarily require additional efforts to increase general public awareness regarding AD pathology and recognition of current failures in research efforts, and ways to prevention. In particular, public initiatives and national campaigns addressing the relevance of nutrition, cognitive training, and physical activity as preventive strategies to reduce the risk of AD and ameliorate AD symptoms, as the ones recently undertaken [62, 63], should be encouraged and supported.
Finally, it is important to implement education and design curricula focused on currently available human-based methods and readouts in both schools and universities, to train new generations of scientists competent in the field of alternatives to animal experimentation, and well versed in the necessity and power of multiscale human-based research approaches.
Table 3: List of recommendations to guide new funding strategies |
||
Recommendations |
Comments |
|
| R1 | Implement funding for the production and centralized distribution of AD patient-derived cells (e.g., fibroblasts, peripheral blood cells, iPSCs) | Consider establishing NIH-funded centers to provide investigators with patient-derived cells and already reprogrammed iPSCs. However, this might be proven unnecessary if other entities, such as CIRM and NYSFC, will do this on their own |
| R2 | Allocate funding for research proposals aiming at defining & validating early biomarkers of AD | Current biomarkers measure levels of Aβ (in CSF), and levels of phospho-tau and total tau (in CSF). In this regard, neuroimaging technologies by means of MRI and PET (FDG-PET and amyloid imaging) are particularly suitable to allow early detection of AD and assess therapeutic efficacy in vivo. Develop and validate additional portable and non-invasive techniques that can identify predictive biomarkers. |
| R3 | Allocate more funding to research projects focusing on the most prevalent late-onset/sporadic AD | Despite the fact that the majority of AD cases are late-onset, the current number of NIH funded active projects focused on the late-onset/sporadic AD is lower than the number of projects on early-onset and familial AD (81 vs 182, as of July 6th 2015. Data retrieved from http://projectreporter.nih.gov/reporter.cfm ) |
| R4 | Allocate funding to centers conducting omics research in human-based settings | This would be relevant considering the need for expensive high throughput technological tools and creation of multidisciplinary teams of experts |
| R5 | Create specific RFAs focused on non-animal/human-based research | One example in this direction to significantly reduce animal experimentation is provided by Europe and UK: for instance, NC3Rs rates projects considering their scientific value as 50% and their contribution to the reduction of animal tests as the remaining 50% of the final score (http://www.nc3rs.org.uk/funding). More directly, a dedicated call should be made for complete and direct alternatives that offer new perspectives and fundamentally ethical approach that do not involve animal experimentation |
| R6 | Increase funding support for basic research studies to speed the discovery process | Recognize the many types and growing applicability of non-animal models in basic research. Dedicated funding should be allocated to high-risk high innovation studies, including the development of non-animal models for research in this area. Not all projects need to be immediately translational in nature |
| R7 | Increase funding to study risk factors and evidence-based prevention approaches to slow the progression of AD | There is an urgent need to increase funding for epidemiological and clinical studies, focused on the impact of specific nutrition, level of physical activity, and level of educational attainment in the onset and progression of AD. Also, increase resources for examining factors across multiple risk and ameliorating variables including: environmental exposure, access to health care, sex and gender, ongoing social and cognitive engagement. Design intervention strategies in large scale cohorts. Dedicate resources to disseminate knowledge of known lifestyle factors to the public at large as well as new incoming information. Randomized clinical trials of individual dietary practices as well as nutritional supplements. Begin with individuals who have low or insufficient nutrient levels and for whom the highest beneficial effects have been observed (Morris, Tangney et al. 2015) |
| R8 | Consider ethno-cultural factors | Epidemiological studies addressing ethnic, cultural variations and implication of lifestyle risk factors would be highly relevant both to smaller communities and lessons that can be extended to the population at large. Collaboration with epidemiological studies in other clinical domains, such as vascular research (Satizabal, Beiser et al. 2016) will be critical for unmasking these complex relationships. |
| Abbreviations: CIRM, California Institute for Regenerative Medicine; NYSCF, New York Stem Cell Foundation; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; PET, positron emission tomography; FDG-PET, fluorodeoxyglucose-PET; RFAs, Requests for Applications. | ||
Conflicts of Interests
Gillian Langley is a consultant to Humane Society International. Francesca Pistollato, Ann Lam, Neal Barnard, Mei-Chun Lai, Ryan Merkley and P. Charukeshi Chandrasekera work at the Physicians Committee for Responsible Medicine, a non-profit medical advocacy organization. Thomas J. Novak is an employee of Cellular Dynamics International (CDI) and the PI on CDI’s grant with the California Institute for Regenerative Medicine (CIRM) to generate 3000 patient-derived iPSC lines, including those from patients with late-onset AD.
References
1. Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimer’s & dementia. 2015; 11:332-384.
2. Gotz J, Ittner LM and Schonrock N. Alzheimer’s disease and frontotemporal dementia: prospects of a tailored therapy? The Medical journal of Australia. 2006; 185:381-384.
3. Hebert LE, Weuve J, Scherr PA and Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013; 80:1778-1783.
4. Bezprozvanny I. The rise and fall of Dimebon. Drug news & perspectives. 2010; 23:518-523.
5. Corbett A and Ballard C. New and emerging treatments for Alzheimer’s disease. Expert opinion on emerging drugs. 2012; 17:147-156.
6. Perrin S. Preclinical research: Make mouse studies work. Nature. 2014; 507:423-425.
7. Mak IW, Evaniew N and Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. American journal of translational research. 2014; 6:114-118.
8. Cummings JL, Morstorf T and Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimer’s research & therapy. 2014; 6:37.
9. Wu YT, Fratiglioni L, Matthews FE, Lobo A, Breteler MM, Skoog I and Brayne C. Dementia in western Europe: epidemiological evidence and implications for policy making. The Lancet Neurology. 2016; 15:116-124.
10. Satizabal CL, Beiser AS, Chouraki V, Chene G, Dufouil C and Seshadri S. Incidence of Dementia over Three Decades in the Framingham Heart Study. The New England journal of medicine. 2016; 374:523-532.
11. Cavanaugh SE, Pippin JJ and Barnard ND. Animal models of Alzheimer disease: historical pitfalls and a path forward. Altex. 2014; 31:279-302.
12. Dodart JC, Mathis C, Bales KR and Paul SM. Does my mouse have Alzheimer’s disease? Genes, brain, and behavior. 2002; 1:142-155.
13. Duyckaerts C, Potier MC and Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta neuropathologica. 2008; 115:5-38.
14. Manich G, del Valle J, Cabezon I, Camins A, Pallas M, Pelegri C and Vilaplana J. Presence of a neo-epitope and absence of amyloid beta and tau protein in degenerative hippocampal granules of aged mice. Age. 2014; 36:151-165.
15. Porquet D, Andres-Benito P, Grinan-Ferre C, Camins A, Ferrer I, Canudas AM, Del Valle J and Pallas M. Amyloid and tau pathology of familial Alzheimer’s disease APP/PS1 mouse model in a senescence phenotype background (SAMP8). Age. 2015; 37:9747.
16. Sabbagh JJ, Kinney JW and Cummings JL. Animal systems in the development of treatments for Alzheimer’s disease: challenges, methods, and implications. Neurobiology of aging. 2013; 34:169-183.
17. Langley GR. Considering a new paradigm for Alzheimer’s disease research. Drug discovery today. 2014; 19:1114-1124.
18. Pistollato F, Cavanaugh SE and Chandrasekera PC. A Human-Based Integrated Framework for Alzheimer’s Disease Research. Journal of Alzheimer’s disease. 2015; 47:857-868.
19. NINDS. (2016). Draft Prioritized Recommendations. Alzheimer’s disease and related dementias (ADRD) Summit 2016: NINDS), pp. 1-21.
20. Calitz C, Pollack KM, Millard C and Yach D. National institutes of health funding for behavioral interventions to prevent chronic diseases. American journal of preventive medicine. 2015; 48:462-471.
21. Chai CK. The genetics of Alzheimer’s disease. American journal of Alzheimer’s disease and other dementias. 2007; 22:37-41.
22. Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A and Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Archives of general psychiatry. 2006; 63:168-174.
23. Rossor MN, Fox NC, Freeborough PA and Harvey RJ. Clinical features of sporadic and familial Alzheimer’s disease. Neurodegeneration. 1996; 5:393-397.
24. Gillette Guyonnet S, Abellan Van Kan G, Andrieu S, Barberger Gateau P, Berr C, Bonnefoy M, Dartigues JF, de Groot L, Ferry M, Galan P, Hercberg S, Jeandel C, Morris MC, Nourhashemi F, Payette H, Poulain JP, et al. IANA task force on nutrition and cognitive decline with aging. The journal of nutrition, health & aging. 2007; 11:132-152.
25. Hernandez SS, Sandreschi PF, da Silva FC, Arancibia BA, da Silva R, Gutierres PJ and Andrade A. What are the Benefits of Exercise for Alzheimer’s Disease? A Systematic Review of the Past 10 Years. Journal of aging and physical activity. 2015; 23:659-668.
26. Okonkwo OC, Schultz SA, Oh JM, Larson J, Edwards D, Cook D, Koscik R, Gallagher CL, Dowling NM, Carlsson CM, Bendlin BB, LaRue A, Rowley HA, Christian BT, Asthana S, Hermann BP, et al. Physical activity attenuates age-related biomarker alterations in preclinical AD. Neurology. 2014; 83:1753-1760.
27. Gates NJ and Sachdev P. Is cognitive training an effective treatment for preclinical and early Alzheimer’s disease? Journal of Alzheimer’s disease. 2014; 42:S551-559.
28. Russ TC, Stamatakis E, Hamer M, Starr JM, Kivimaki M and Batty GD. Socioeconomic status as a risk factor for dementia death: individual participant meta-analysis of 86 508 men and women from the UK. The British journal of psychiatry. 2013; 203:10-17.
29. Sattler C, Toro P, Schonknecht P and Schroder J. Cognitive activity, education and socioeconomic status as preventive factors for mild cognitive impairment and Alzheimer’s disease. Psychiatry research. 2012; 196:90-95.
30. Paradise M, Cooper C and Livingston G. Systematic review of the effect of education on survival in Alzheimer’s disease. International psychogeriatrics. 2009; 21:25-32.
31. Lim MM, Gerstner JR and Holtzman DM. The sleep-wake cycle and Alzheimer’s disease: what do we know? Neurodegenerative disease management. 2014; 4:351-362.
32. Videnovic A, Lazar AS, Barker RA and Overeem S. ‘The clocks that time us’-circadian rhythms in neurodegenerative disorders. Nature reviews Neurology. 2014; 10:683-693.
33. Peter-Derex L, Yammine P, Bastuji H and Croisile B. Sleep and Alzheimer’s disease. Sleep medicine reviews. 2015; 19:29-38.
34. Lucey BP and Bateman RJ. Amyloid-beta diurnal pattern: possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiology of aging. 2014; 35 Suppl 2:S29-34.
35. Ju YE, Lucey BP and Holtzman DM. Sleep and Alzheimer disease pathology—a bidirectional relationship. Nature reviews Neurology. 2014; 10:115-119.
36. Moulton PV and Yang W. Air pollution, oxidative stress, and Alzheimer’s disease. Journal of environmental and public health. 2012; 2012:472751.
37. Cataldo JK, Prochaska JJ and Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. Journal of Alzheimer’s disease. 2010; 19:465-480.
38. Shcherbatykh I and Carpenter DO. The role of metals in the etiology of Alzheimer’s disease. Journal of Alzheimer’s disease. 2007; 11:191-205.
39. Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. BioFactors. 2012; 38:107-113.
40. Tong Y, Yang H, Tian X, Wang H, Zhou T, Zhang S, Yu J, Zhang T, Fan D, Guo X, Tabira T, Kong F, Chen Z, Xiao W and Chui D. High manganese, a risk for Alzheimer’s disease: high manganese induces amyloid-beta related cognitive impairment. Journal of Alzheimer’s disease. 2014; 42:865-878.
41. Tanner CM, Goldman SM, Ross GW and Grate SJ. The disease intersection of susceptibility and exposure: chemical exposures and neurodegenerative disease risk. Alzheimer’s & dementia. 2014; 10:S213-225.
42. Yegambaram M, Manivannan B, Beach TG and Halden RU. Role of environmental contaminants in the etiology of Alzheimer’s disease: a review. Current Alzheimer research. 2015; 12:116-146.
43. Rios JA, Cisternas P, Arrese M, Barja S and Inestrosa NC. Is Alzheimer’s disease related to metabolic syndrome? A Wnt signaling conundrum. Progress in neurobiology. 2014; 121:125-146.
44. Rosendorff C, Beeri MS and Silverman JM. Cardiovascular risk factors for Alzheimer’s disease. The American journal of geriatric cardiology. 2007; 16:143-149.
45. Pistollato F, Sumalla Cano S, Elio I, Masias Vergara M, Giampieri F and Battino M. The Use of Neuroimaging to Assess Associations Among Diet, Nutrients, Metabolic Syndrome, and Alzheimer’s Disease. Journal of Alzheimer’s disease. 2015; 48:303-318.
46. Gibson JE. An integrated summary of commentary on the National Academy of Sciences report on ’’Toxicity testing in the 21st century: a vision and a strategy’’. Human & experimental toxicology. 2010; 29:33-35.
47. Poet TS, Timchalk C, Hotchkiss JA and Bartels MJ. Chlorpyrifos PBPK/PD model for multiple routes of exposure. Xenobiotica. 2014; 44:868-881.
48. Santamaria AB. Manganese exposure, essentiality & toxicity. The Indian journal of medical research. 2008; 128:484-500.
49. Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LN, Walsh DM, Selkoe DJ and Young-Pearse TL. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Human molecular genetics. 2014; 23:3523-3536.
50. Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, Santa-Maria I, Zimmer M, Aubry S, Steele JW, Kahler DJ, Dranovsky A, Arancio O, Crary JF, Gandy S and Noggle SA. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PloS one. 2014; 9:e84547.
51. Zhang D, Pekkanen-Mattila M, Shahsavani M, Falk A, Teixeira AI and Herland A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials. 2014; 35:1420-1428.
52. Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, Takahashi K, Asaka I, Aoi T, Watanabe A, Watanabe K, Kadoya C, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell stem cell. 2013; 12:487-496.
53. Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012; 482:216-220.
54. Blasko I, Hinterberger M, Kemmler G, Jungwirth S, Krampla W, Leitha T, Heinz Tragl K and Fischer P. Conversion from mild cognitive impairment to dementia: influence of folic acid and vitamin B12 use in the VITA cohort. The journal of nutrition, health & aging. 2012; 16:687-694.
55. Mosconi L, Murray J, Tsui WH, Li Y, Davies M, Williams S, Pirraglia E, Spector N, Osorio RS, Glodzik L, McHugh P and de Leon MJ. Mediterranean Diet and Magnetic Resonance Imaging-Assessed Brain Atrophy in Cognitively Normal Individuals at Risk for Alzheimer’s Disease. The journal of prevention of Alzheimer’s disease. 2014; 1:23-32.
56. Titova OE, Ax E, Brooks SJ, Sjogren P, Cederholm T, Kilander L, Kullberg J, Larsson EM, Johansson L, Ahlstrom H, Lind L, Schioth HB and Benedict C. Mediterranean diet habits in older individuals: associations with cognitive functioning and brain volumes. Experimental gerontology. 2013; 48:1443-1448.
57. Frederiksen KS, Sobol N, Beyer N, Hasselbalch S and Waldemar G. Moderate-to-high intensity aerobic exercise in patients with mild to moderate Alzheimer’s disease: a pilot study. International journal of geriatric psychiatry. 2014; 29:1242-1248.
58. Paillard T, Rolland Y and de Souto Barreto P. Protective Effects of Physical Exercise in Alzheimer’s Disease and Parkinson’s Disease: A Narrative Review. Journal of clinical neurology. 2015; 11:212-219.
59. Morris MC, Tangney CC, Wang Y, Sacks FM, Barnes LL, Bennett DA and Aggarwal NT. MIND diet slows cognitive decline with aging. Alzheimer’s & dementia. 2015; 11:1015-1022.
60. Williams RS, Boeckeler K, Graf R, Muller-Taubenberger A, Li Z, Isberg RR, Wessels D, Soll DR, Alexander H and Alexander S. Towards a molecular understanding of human diseases using Dictyostelium discoideum. Trends in molecular medicine. 2006; 12:415-424.
61. Ludtmann MH, Otto GP, Schilde C, Chen ZH, Allan CY, Brace S, Beesley PW, Kimmel AR, Fisher P, Killick R and Williams RS. An ancestral non-proteolytic role for presenilin proteins in multicellular development of the social amoeba Dictyostelium discoideum. Journal of cell science. 2014; 127:1576-1584.
62. Ellis KA, Rowe CC, Villemagne VL, Martins RN, Masters CL, Salvado O, Szoeke C, Ames D and group Ar. Addressing population aging and Alzheimer’s disease through the Australian imaging biomarkers and lifestyle study: collaboration with the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s & dementia. 2010; 6:291-296.
63. Carrillo MC, Sanders CA and Katz RG. Maximizing the Alzheimer’s Disease Neuroimaging Initiative II. Alzheimer’s & dementia. 2009; 5:271-275.
64. Kim JB, Zaehres H, Wu G, Gentile L, Ko K, Sebastiano V, Arauzo-Bravo MJ, Ruau D, Han DW, Zenke M and Scholer HR. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008; 454:646-650.
65. Miyazaki S, Yamamoto H, Miyoshi N, Takahashi H, Suzuki Y, Haraguchi N, Ishii H, Doki Y and Mori M. Emerging methods for preparing iPS cells. Japanese journal of clinical oncology. 2012; 42:773-779.
66. Reitz C, Brayne C and Mayeux R. Epidemiology of Alzheimer disease. Nature reviews Neurology. 2011; 7:137-152.
67. Vaskova EA, Stekleneva AE, Medvedev SP and Zakian SM. “Epigenetic memory” phenomenon in induced pluripotent stem cells. Acta naturae. 2013; 5:15-21.
68. Nashun B, Hill PW and Hajkova P. Reprogramming of cell fate: epigenetic memory and the erasure of memories past. The EMBO journal. 2015; 34:1296-1308.
69. Qiang L, Inoue K and Abeliovich A. Instant neurons: directed somatic cell reprogramming models of central nervous system disorders. Biological psychiatry. 2014; 75:945-951.
70. Qiang L, Fujita R and Abeliovich A. Remodeling neurodegeneration: somatic cell reprogramming-based models of adult neurological disorders. Neuron. 2013; 78:957-969.
71. Kim J, Lengner CJ, Kirak O, Hanna J, Cassady JP, Lodato MA, Wu S, Faddah DA, Steine EJ, Gao Q, Fu D, Dawlaty M and Jaenisch R. Reprogramming of postnatal neurons into induced pluripotent stem cells by defined factors. Stem cells. 2011; 29:992-1000.
72. Sproul AA, Vensand LB, Dusenberry CR, Jacob S, Vonsattel JP, Paull DJ, Shelanski ML, Crary JF and Noggle SA. Generation of iPSC lines from archived non-cryoprotected biobanked dura mater. Acta neuropathologica communications. 2014; 2:4.
73. Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H and Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS genetics. 2011; 7:e1002085.
74. Pistollato F, Bremer-Hoffmann S, Healy L, Young L and Stacey G. Standardization of pluripotent stem cell cultures for toxicity testing. Expert opinion on drug metabolism & toxicology. 2012; 8:239-257.
75. Heininger K. A unifying hypothesis of Alzheimer’s disease. IV. Causation and sequence of events. Reviews in the neurosciences. 2000; 11 Spec No:213-328.
76. Young JE and Goldstein LS. Alzheimer’s disease in a dish: promises and challenges of human stem cell models. Human molecular genetics. 2012; 21:R82-89.
77. Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, Taldone T, Fusaki N, Tomishima MJ, Krainc D, Milner TA, Rossi DJ, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell stem cell. 2013; 13:691-705.
78. Mertens J, Paquola AC, Ku M, Hatch E, Bohnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, Goncalves JT, Toda T, Kim Y, Winkler J, Yao J, Hetzer MW, et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell stem cell. 2015; 17:705-718.
79. Mungenast AE, Siegert S and Tsai LH. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Molecular and cellular neurosciences. 2015:S1044-7431(1015)30037-30033.
80. Byrne SM, Mali P and Church GM. Genome editing in human stem cells. Methods in enzymology. 2014; 546:119-138.
81. Hendriks WT, Warren CR and Cowan CA. Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions. Cell stem cell. 2016; 18:53-65.
82. Sproul AA. Being human: The role of pluripotent stem cells in regenerative medicine and humanizing Alzheimer’s disease models. Molecular aspects of medicine. 2015; 43-44:54-65.
83. Pamies D, Hartung T and Hogberg HT. Biological and medical applications of a brain-on-a-chip. Experimental biology and medicine. 2014; 239:1096-1107.
84. Alepee N, Bahinski A, Daneshian M, De Wever B, Fritsche E, Goldberg A, Hansmann J, Hartung T, Haycock J, Hogberg H, Hoelting L, Kelm JM, Kadereit S, McVey E, Landsiedel R, Leist M, et al. State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. Altex. 2014; 31:441-477.
85. O’Connor MD. The 3R principle: advancing clinical application of human pluripotent stem cells. Stem cell research & therapy. 2013; 4:21.
86. Graul AI. Promoting, improving and accelerating the drug development and approval processes. Drug news & perspectives. 2008; 21:36-43.
87. Beach TG. Alzheimer’s disease and the “Valley Of Death”: not enough guidance from human brain tissue? Journal of Alzheimer’s disease. 2013; 33 Suppl 1:S219-233.
88. Harper L, Fumagalli GG, Barkhof F, Scheltens P, O’Brien JT, Bouwman F, Burton EJ, Rohrer JD, Fox NC, Ridgway GR and Schott JM. MRI visual rating scales in the diagnosis of dementia: evaluation in 184 post-mortem confirmed cases. Brain. 2016; 139:1211-1225.
89. Oka T, Tagawa K, Ito H and Okazawa H. Dynamic changes of the phosphoproteome in postmortem mouse brains. PloS one. 2011; 6:e21405.
90. Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, Lue L, Roher AE, Dugger BN, Maarouf C, Birdsill AC, Intorcia A, Saxon-Labelle M, Pullen J, Scroggins A, Filon J, et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology. 2015; 35:354-389.
91. Ferrer I, Martinez A, Boluda S, Parchi P and Barrachina M. Brain banks: benefits, limitations and cautions concerning the use of post-mortem brain tissue for molecular studies. Cell and tissue banking. 2008; 9:181-194.
92. Ferrer I, Santpere G, Arzberger T, Bell J, Blanco R, Boluda S, Budka H, Carmona M, Giaccone G, Krebs B, Limido L, Parchi P, Puig B, Strammiello R, Strobel T and Kretzschmar H. Brain protein preservation largely depends on the postmortem storage temperature: implications for study of proteins in human neurologic diseases and management of brain banks: a BrainNet Europe Study. Journal of neuropathology and experimental neurology. 2007; 66:35-46.
93. Sethi P and Lukiw WJ. Micro-RNA abundance and stability in human brain: specific alterations in Alzheimer’s disease temporal lobe neocortex. Neuroscience letters. 2009; 459:100-104.
94. Hendrix JA, Finger B, Weiner MW, Frisoni GB, Iwatsubo T, Rowe CC, Kim SY, Guinjoan SM, Sevlever G and Carrillo MC. The Worldwide Alzheimer’s Disease Neuroimaging Initiative: An update. Alzheimer’s & dementia. 2015; 11:850-859.
95. Kerchner GA. Ultra-high field 7T MRI: a new tool for studying Alzheimer’s disease. Journal of Alzheimer’s disease. 2011; 26 Suppl 3:91-95.
96. Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VM, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013; 79:1094-1108.
97. Willette AA, Johnson SC, Birdsill AC, Sager MA, Christian B, Baker LD, Craft S, Oh J, Statz E, Hermann BP, Jonaitis EM, Koscik RL, La Rue A, Asthana S and Bendlin BB. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer’s & dementia. 2015; 11:504-510 e501.
98. Kenna H, Hoeft F, Kelley R, Wroolie T, DeMuth B, Reiss A and Rasgon N. Fasting plasma insulin and the default mode network in women at risk for Alzheimer’s disease. Neurobiology of aging. 2013; 34:641-649.
99. Cherbuin N, Sachdev P and Anstey KJ. Higher normal fasting plasma glucose is associated with hippocampal atrophy: The PATH Study. Neurology. 2012; 79:1019-1026.
100. Pistollato F and Battino M. Role of plant-based diets in the prevention and regression of metabolic syndrome and neurodegenerative diseases. Trends in Food Science & Technology. 2014; 40:62-81.
101. Dai Y and Kamal MA. Fighting Alzheimer’s disease and type 2 diabetes: pathological links and treatment strategies. CNS & neurological disorders drug targets. 2014; 13:271-282.
102. de la Monte SM and Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochemical pharmacology. 2014; 88:548-559.
103. Jbabdi S and Johansen-Berg H. Tractography: where do we go from here? Brain connectivity. 2011; 1:169-183.
104. Gutman B, Leonardo C, Jahanshad N, Hibar D, Eschenburg K, Nir T, Villalon J and Thompson P. Registering cortical surfaces based on whole-brain structural connectivity and continuous connectivity analysis. Medical image computing and computer-assisted intervention. 2014; 17:161-168.
105. Prescott JW, Guidon A, Doraiswamy PM, Roy Choudhury K, Liu C, Petrella JR and Alzheimer’s Disease Neuroimaging I. The Alzheimer structural connectome: changes in cortical network topology with increased amyloid plaque burden. Radiology. 2014; 273:175-184.
106. Rhinn H, Fujita R, Qiang L, Cheng R, Lee JH and Abeliovich A. Integrative genomics identifies APOE epsilon4 effectors in Alzheimer’s disease. Nature. 2013; 500:45-50.
107. Zahid S, Oellerich M, Asif AR and Ahmed N. Phosphoproteome profiling of substantia nigra and cortex regions of Alzheimer’s disease patients. Journal of neurochemistry. 2012; 121:954-963.
108. Graff-Radford J and Kantarci K. Magnetic resonance spectroscopy in Alzheimer’s disease. Neuropsychiatric disease and treatment. 2013; 9:687-696.
109. Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD and Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiology of aging. 2011; 32:1161-1180.
110. Millan MJ. The epigenetic dimension of Alzheimer’s disease: causal, consequence, or curiosity? Dialogues in clinical neuroscience. 2014; 16:373-393.
111. Trushina E, Dutta T, Persson XM, Mielke MM and Petersen RC. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PloS one. 2013; 8:e63644.
112. Chetty M, Rose RH, Abduljalil K, Patel N, Lu G, Cain T, Jamei M and Rostami-Hodjegan A. Applications of linking PBPK and PD models to predict the impact of genotypic variability, formulation differences, differences in target binding capacity and target site drug concentrations on drug responses and variability. Frontiers in pharmacology. 2014; 5:258.
113. Janson J, Eketjall S, Tunblad K, Jeppsson F, Von Berg S, Niva C, Radesater AC, Falting J and Visser SA. Population PKPD modeling of BACE1 inhibitor-induced reduction in Abeta levels in vivo and correlation to in vitro potency in primary cortical neurons from mouse and guinea pig. Pharmaceutical research. 2014; 31:670-683.
114. Rao PP, Mohamed T, Teckwani K and Tin G. Curcumin Binding to Beta Amyloid: A Computational Study. Chemical biology & drug design. 2015; 86:813-20.
115. Huang HJ, Chen HY, Lee CC and Chen CY. Computational design of apolipoprotein E4 inhibitors for Alzheimer’s disease therapy from traditional Chinese medicine. BioMed research international. 2014; 2014:452625.
116. NIH. (2013). NIH to reduce significantly the use of chimpanzees in research. News & Events: NIH).
117. Goodman J. (2012). Public Opinion on Animal Testing.
118. McKernan R and Watt FM. What is the point of large-scale collections of human induced pluripotent stem cells? Nature biotechnology. 2013; 31:875-877.
119. Khachaturian ZS, Petersen RC, Snyder PJ, Khachaturian AS, Aisen P, de Leon M, Greenberg BD, Kukull W, Maruff P, Sperling RA, Stern Y, Touchon J, Vellas B, Andrieu S, Weiner MW, Carrillo MC, et al. Developing a global strategy to prevent Alzheimer’s disease: Leon Thal Symposium 2010. Alzheimer’s & dementia. 2011; 7:127-132.
120. Khachaturian ZS, Barnes D, Einstein R, Johnson S, Lee V, Roses A, Sager MA, Shankle WR, Snyder PJ, Petersen RC, Schellenberg G, Trojanowski J, Aisen P, Albert MS, Breitner JC, Buckholtz N, et al. Developing a national strategy to prevent dementia: Leon Thal Symposium 2009. Alzheimer’s & dementia. 2010; 6:89-97.
121. Devall M, Mill J and Lunnon K. The mitochondrial epigenome: a role in Alzheimer’s disease? Epigenomics. 2014; 6:665-675.
122. Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. Journal of Alzheimer’s disease. 2006; 9:119-126.
123. Picone P, Nuzzo D, Caruana L, Scafidi V and Di Carlo M. Mitochondrial dysfunction: different routes to Alzheimer’s disease therapy. Oxidative medicine and cellular longevity. 2014; 2014:780179.
124. Heppner FL, Ransohoff RM and Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nature reviews Neuroscience. 2015; 16:358-372.
125. Holbrook JB, Barr KR and Brown KW. We need negative metrics too. Nature. 2013; 497:439.
126. Kaiser J. BIOMEDICAL RESEARCH. Spending bills put NIH on track for biggest raise in 12 years. Science. 2015; 349:12-13.
127. NIH Alzheimer’s disease Research Summit 2015: Path to Treatment and Prevention. https://www.nia.nih.gov/about/events/2014/alzheimers-disease-research-summit-2015.