
INTRODUCTION
Doxorubicin (DOX) is an anthracycline antineoplastic drug to treat a variety of solid tumors and hematological malignancies [1]. The antitumor activity of DOX is attributable to its ability to intercalate DNA helix and inhibit topoisomerase II, causing DNA double strand breaks, G2 arrest and apoptosis in replicating cells [2]. Despite its effectiveness against cancer, the clinical use of DOX is critically limited by its cumulative cardiotoxicity [3]. A greater incidence of DOX-related congestive heart failure was found in patients after receiving a cumulative dose of 400 mg/m2 [4]. The onset of cardiac complications can occur during the treatment or up to 10 years after the cessation of DOX therapy [5]. Cardiovascular-related disease derived from the adverse effects of cancer treatments has become the leading noncancer-related cause of morbidity and mortality in long-term cancer survivors [5].
Cardiotoxic effects of DOX, distinct from its antitumor activities, develop from increasing mitochondrial iron content and cellular reactive oxygen species (ROS) levels [6]. Currently, dexrazoxane is the only agent approved by the United States FDA and the European Medicines Agency to prevent long-term cardiotoxicity caused by DOX. Dexrazoxane, structurally similar to EDTA, is a strong chelator of iron and prevents the iron-dependent ROS production induced by DOX [7]. Because ROS also contribute to the antineoplastic activity of DOX [1], dexrazoxane may interfere with the anticancer activity of DOX and leads to the higher latent risk for acute myeloid leukemia and other secondary malignancies in pediatric patients receiving dexrazoxane [8]. A more specific strategy needs to be developed to prevent DOX cardiotoxicity.
Fas ligand (FasL) is a critical mediator in DOX-associated cardiotoxicity [9]. Despite the pivotal role of FasL and its receptor Fas in cardiac injury, the activation of Fas signaling alone is not sufficient to induce apoptosis in mouse hearts or in cultured cardiomyocytes [10, 11]. CCN1, a matricellular protein, sensitizes cardiomyocytes to FasL-induced apoptosis both in mice and in cultured cells [12, 13]. CCN1 promotes various cell activities and regulates cell survival and death through interaction with distinct integrin receptors in a cell type- and context-dependent manner [14]. CCN1 is dynamically expressed in the cardiovascular system during embryogenesis and Ccn1-null mice die prenatally from cardiovascular defects [15, 16]. CCN1 expression during adulthood is largely silenced, but is redeployed upon tissue injury and in a variety of pathological conditions [14]. In the heart, CCN1 is induced in end-stage ischemic cardiomyopathy in humans and after myocardial infarction in animal models [17]. CCN1 is also induced in mice with work-overload-induced cardiac injury, and sensitizes FasL-mediated cardiomyocyte apoptosis by engaging its cell-surface receptor integrin α6β1 [12]. CCN1/α6β1 signaling triggers the accumulation of cellular ROS, activation of p38 mitogen-activated protein kinase (MAPK), and translocation of mitochondrial Smac and HtrA2 to cytosol, thereby counteracting the anti-apoptotic activity of XIAP and facilitating FasL-induced apoptosis in cardiomyocytes [13].
Because CCN1 is induced upon various cardiac insults to promote cell death, we hypothesized that CCN1 may mediate DOX-associated cardiotoxicity through engaging integrin α6β1. To test this hypothesis, we first examined the cardiac expression of CCN1 after DOX treatment in mice. Subsequently, a previously generated knockin mice (Ccn1dm/dm), expressing an α6β1-binding defective CCN1 mutant with two of its binding sites mutated (double mutant) [18], were used to assess the role of CCN1 in mediating DOX cardiotoxicity. Furthermore, cardiomyoblast H9c2 cells were used for in vitro mechanistic studies.
RESULTS
CCN1 critically mediates DOX-associated cardiomyopathy
To examine Ccn1 expression in DOX-associated cardiomyopathy, DOX was delivered to Ccn1+/lacZ mice in which an allele of Ccn1 was replaced by a lacZ reporter gene [16]. Heart tissue was collected 1 day after DOX injection (15 mg/kg; i.p.). Ccn1 expression was assessed through measuring the enzymatic activities of LacZ by X-gal staining. Ccn1 expression was not detected in the PBS-controlled hearts (Figure 1A). Ccn1 expression became evident (blue) in the cardiomyocytes 1 day after DOX treatment before any tissue lesions occurred (Figure 1A). To assess the role of CCN1 in mediating DOX cardiotoxicity, DOX was delivered to Ccn1dm/dm (DM) mice. Heart tissue was collected 14 days after DOX injection. Cardiac fibrotic lesions (blue) identified by trichrome staining were increased in the perivascular areas in the myocardium of wild-type (WT) mice receiving DOX (Figure 1B). DOX-induced fibrotic lesions were not observed in Ccn1dm/dm mice (Figure 1B). TUNEL+ apoptotic cardiomyocytes (green troponin-I+ cells with pink nuclei, arrows in Figure 1E) were increased by DOX in the myocardium of WT mice, and were not affected by DOX in Ccn1dm/dm mice (Figure 1E). The cardiac lesion created by DOX leads to deterioration of cardiac function. Prolonged electrocardiographic (ECG) PR and QT intervals were detected in the WT mice receiving DOX treatment. The ECG measurements were not altered by DOX in Ccn1dm/dm mice (Figure 1C). Consistently, left ventricular systolic function indices, ejection fraction (EF) and fractional shortening (FS) determined by echocardiography, were better maintained in Ccn1dm/dm mice after DOX treatment (Figure 1D). Together, these results demonstrated that CCN1 critically mediates DOX-associated cardiotoxicity. Disrupting the binding between CCN1 and integrin α6β1 effectively prevents the cardiotoxicity of DOX in mice.
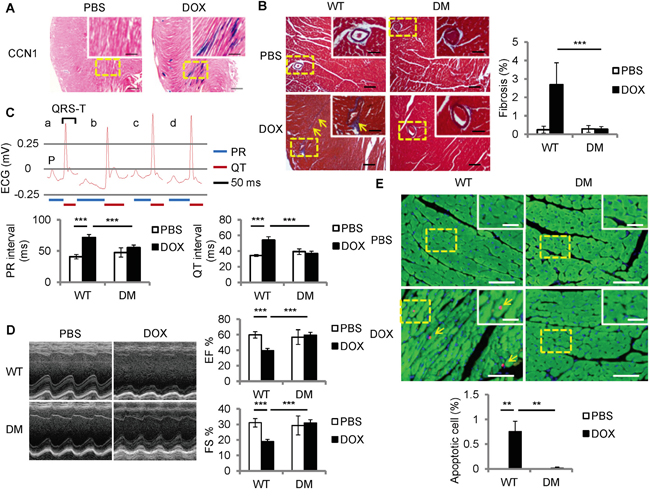
Figure 1: Ccn1dm/dm mice were resistant to Doxorubicin (DOX)-induced cardiomyopathy. A. For Ccn1 expression, the hearts from Ccn1+/lacZ mice 1 day after DOX treatment (15 mg/kg; i.p.) were stained with X-gal (blue). High power views of the dashed areas were shown in the insets. B, E. Cardiac tissue was collected from WT or Ccn1dm/dm (DM) mice 14 days after PBS or DOX administration (15 mg/kg; i.p.) (n = 6 for each group). (B) Cardiac fibrotic lesions were identified by Masson’s trichrome staining (blue, arrows). The percentage of the fibrotic area was quantified using the NIH ImageJ program. Data are means ± SEM from 8 comparable tissue sections per mouse. C, D. Cardiac physiology was assessed by electrocardiography (ECG) and echocardiography on WT or Ccn1dm/dm mice (n=4)14 days after PBS or DOX administration. (C) The lengths of PR interval and QT interval were indicated below the representative ECGs from a: WT mice/PBS; b: WT mice/DOX; c: Ccn1dm/dm mice/PBS; d: Ccn1dm/dm mice/DOX. Data are means ± SEM from 3 measurements per mouse. (D) Representative echocardiograms demonstrate the structural dynamics during left ventricular systole. Ejection fraction (EF) was computed from the measurements of the end-diastolic volume and end-systolic volume. Fractional shortening (FS) is the fraction of the diastolic dimension that is reduced in systole. Data are means ± SEM from 3 measurements per mouse. (E) The percentage of apoptotic (TUNEL staining, pink nuclei) cardiomyocytes (troponin I+ cells, green) indicated by arrows was quantified from 10 random high-power views per tissue section and 8 sections per mouse. Tissue sections were counterstained with DAPI for nuclei (blue). Bars in (A): 500 μm; in the insets of (A): 200 μm. Bars in (B): 100 μm; in the insets of (B): 50 μm. Bars in (E): 50 μm; in the insets of (E): 20 μm.
FasL is induced by DOX in cardiomyocytes
Fas signaling is required for DOX-induced cardiomyopathy in rats [19]. CCN1 promotes cardiomyocyte apoptosis through potentiating the death effect of FasL [12, 13]. We examined the induction of FasL after DOX treatment by immunohistochemical staining with anti-FasL antibody. FasL was increased in the myocardium of WT mice 14 days after DOX injection (reddish-brown staining in Figure 2A). In Ccn1dm/dm mice, FasL staining was significantly higher in DOX-treated hearts than in PBS controls, though the levels of FasL induction were lower than in WT mice (Figure 2A). To investigate whether DOX directly induces FasL expression in cardiomyocytes, we treated rat cardiomyoblast H9c2 cells with DOX in vitro. FasL mRNA levels, determined by quantitative real-time PCR (qRT-PCR), were elevated within 1 h and sustained at higher levels from 3-5 h after DOX treatment (10 μM) in H9c2 cells (Figure 2B). Consistently, FasL protein in H9c2 cells, assessed by Western blotting, was increased significantly 3 h after DOX treatment (Figure 2C). Because oxidative stress generated by DOX mediates its cardiotoxicity [20], we tested the requirement of ROS for FasL induction by combining DOX treatment with ROS-scavenger N-acetyl-L-cysteine (NAC). Cotreatment of NAC (10 mM) completely abolished the induction of FasL expression by DOX (10 μM; 5 h) (Figure 2D), indicating the requirement of ROS in the upregulation of FasL expression.
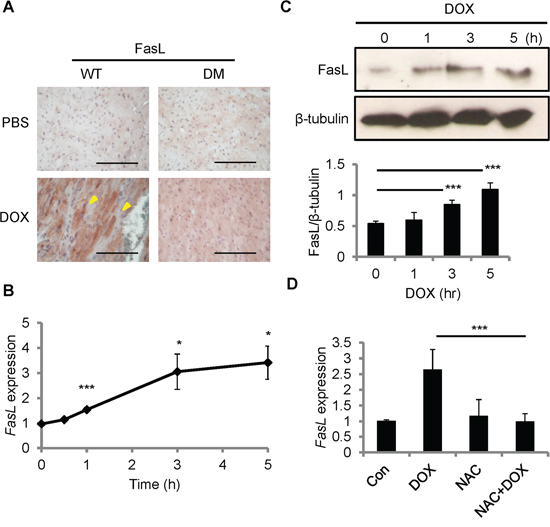
Figure 2: FasL was induced by DOX in the mouse heart and in cardiomyoblast H9c2 cells. A. Heart tissue sections from WT and Ccn1dm/dm mice 14 day after PBS or DOX treatments (15 mg/kg; i.p.) were immunohistochemically stained with anti-FasL antibody (reddish-brown). B, C. H9c2 cells were treated with DOX (10 μM) for the indicated times before total RNA were isolated for FasL qRT-PCR (B), or total cell lysates were isolated and immunoblotted with anti-FasL antibody or anti-β-tubulin antibody as a loading control (C). Protein band intensity was quantified using the ImageJ program. The statistical significance was compared between experimental and control (0 h) groups. D. Where indicated, cells were pretreated with ROS scavenger NAC (5 mM) for 30 min prior to adding DOX (10 μM) and culturing for additional 5 h. Subsequently, total RNA was isolated to measure FasL mRNA levels by qRT-PCR. Data are expressed as mean ± SEM of triplicate experiments. Bars in (A): 100 μm.
CCN1 is induced by DOX in cardiomyocytes in vitro
Because CCN1 can sensitize cardiomyocytes to FasL-induced apoptosis, we examined CCN1expression in H9c2 cells after DOX treatment using qRT-PCR and Western blotting. Ccn1 gene expression was induced within 10 min after DOX treatment (10 μM), and tapered off after 1 h (Figure 3A). CCN1 protein levels were elevated 1 h after DOX treatment and the levels sustained through the 5 h treatment period (Figure 3B). Ccn1 gene expression induced by DOX (10 μM; 30 min) was completely abolished by NAC (10 mM) (Figure 3C), indicating the requirement of ROS in the CCN1 induction by DOX. It has been indicated that protein kinase C (PKC) can be activated by cellular oxidants [21] and PKC activity is essential for stress-induced myocardial CCN1 expression [17]. Here, DOX-induced CCN1 expression and caspase-3 activation (cleavage) in H9c2 cells were completely blocked by PKC inhibitor calphostin C (1 μM, 1-h pretreatment; CAL) (Figure 3D), demonstrating that PKC is required for DOX-induced CCN1 expression and cell apoptosis.
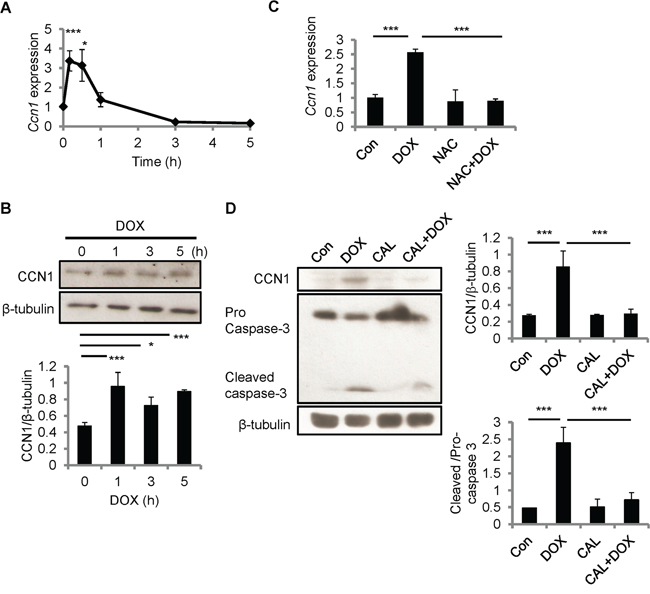
Figure 3: CCN1 was induced by DOX in H9c2 cells. A, B. H9c2 cells were treated with DOX (10 μM) for the indicated times before total RNA were isolated for Ccn1 qRT-PCR (A), or total cell lysates were isolated and immunoblotted with anti-CCN1 or anti-β-tubulin antibodies (B). C. Where indicated, cells were pretreated with NAC (5 mM) for 30 min prior to adding DOX (10 μM) and culturing for additional 5 h. Subsequently, total RNA was isolated to measure Ccn1 mRNA levels by qRT-PCR. D. Total cell lysates from cells treated with protein kinase C inhibitor calphostin C (1 μM, 1-h pretreatment; CAL) and/or DOX (10 μM, 5 h) were immunoblotted with anti-CCN1, anti-caspase 3, or anti-β-tubulin antibodies. Protein band intensity was quantified using the ImageJ program. All data are expressed as mean ± SEM of triplicate experiments.
CCN1 facilitates DOX-induced apoptosis through diminishing XIAP
To test the role of CCN1 in DOX-induced apoptosis, we knocked down Ccn1 expression by transfecting H9c2 cells with Ccn1 siRNA (siCCN1) or non-targeting (NTG) control siRNA 48 h prior to DOX treatment. The levels of CCN1 protein in siCCN1-transfected cells were largely diminished even after DOX treatment (10 μM; 5 h); whereas the control NTG-cells expressed CCN1 upon DOX treatment at the levels similar to nontransfected cells (Figure 4A, 4B). The activation of caspase-3 (cleaved caspases-3) was higher in nontransfected or NTG-cells after DOX treatment (Figure 4A, 4C). By contrast, the activation of caspase-3 was not changed by DOX in siCCN1-cells (Figure 4A, 4C), demonstrating that CCN1 is essential for apoptosis induced by DOX in cardiomyocytes. Mechanistically, CCN1 enables FasL-induced apoptosis in cardiomyocytes through dismantling caspase inhibitor XIAP [13]. Because CCN1 is induced by DOX, XIAP may be suppressed by DOX via CCN1. Indeed, the levels of XIAP were reduced after DOX treatment in nontransfected or NTG-cells (Figure 4A, 4D). By contrast, the levels of XIAP were not reduced by DOX in siCCN1-cells, thereby hindering the activation of caspase-3 (Figure 4A, 4C, 4D).
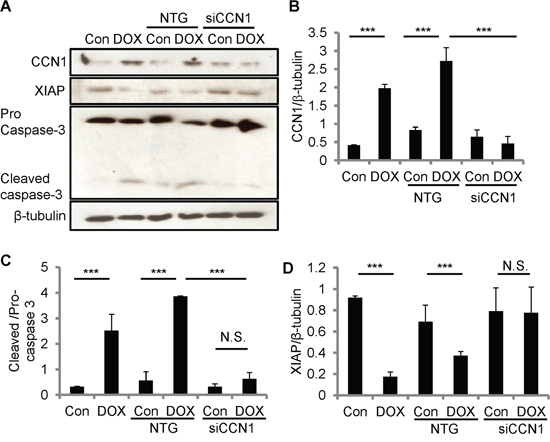
Figure 4: CCN1 critically mediated DOX cardiotoxicity through diminishing XIAP in H9c2 cells. The expression of CCN1 was knocked down by Ccn1 siRNA (siCCN1) or a non-targeting (NTG) control. Transfected cells were cultured for 48 h before further treatments. Cells with or no Ccn1-knockdown were treated with or no DOX (10 μM) for 5 h before total cell lysates were harvested and immunoblotted with anti-CCN1, anti-XIAP, anti-caspase 3, or anti-β-tubulin antibodies A. Protein band intensity was quantified using the ImageJ program. B. Reduced CCN1 band intensities in siCCN1-transfected cells demonstrated the knockdown efficiency. C. The ratio of pro-/cleaved-caspase 3 represents the level of caspase-3 activation. D. XIAP reduction by DOX was blunted by Ccn1 knockdown. All data are expressed as mean ± SEM from 3 independent experiments.
Disrupting CCN1-α6β1 engagement by T1 peptide ameliorates DOX cardiotoxicity
The resistance to DOX cardiotoxicity in Ccn1dm/dm mice suggests the involvement of integrin α6β1 in CCN1 signaling. To investigate whether α6β1 is necessary for facilitating apoptosis induced by DOX, we used T1 peptide to block the binding between CCN1 and α6β1. T1, a 17-residue peptide sequence derived from an α6β1-binding site of CCN1, specifically antagonizes integrin α6β1 [22]. Though DOX can be absorbed by cells and works intracellularly, the incubation with T1 peptide (20 μM) in the media 1 h prior to the addition of DOX (10 μM; 5 h) completely blunted caspase-3 activation in H9c2 cells (Figure 5A), conceivably through blocking the effect of CCN1. Consistently, DOX-induced apoptosis was repressed by T1 peptide (Figure 5B). Apoptosis was assessed using flow cytometry analysis after annexin V/propidium iodide (PI) double staining. Notably, the increase of FasL and activation of caspase-8 (cleaved caspase-8) by DOX were not affected by T1 peptide (Figure 5A), suggesting that CCN1 is not involved in the induction of FasL by DOX.
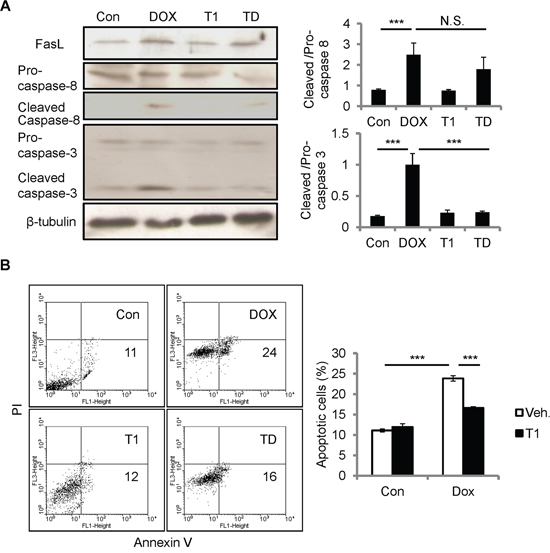
Figure 5: Disrupting CCN1/α6β1 engagement inhibited DOX-induced apoptosis but not FasL induction or caspase-8 activation in H9c2 cells. A, B. Where indicated, cells were pre-incubated with T1 peptide (20 μM) for 1 h prior to DOX treatment (10 μM, 5 h). (A) Total cell lysates were immunoblotted with anti-caspase-3, anti-caspase-8, or anti-β-tubulin antibodies. Protein bands were quantified and expressed as in Figure 2. (B) Apoptosis was assessed by annexin V/PI staining using flow cytometry. The percentage of apoptotic cells (annexin V+/PI-) was scored from the lower right quadrant of the histogram. Data are expressed as mean ± SEM of triplicate experiments.
T1 peptide partially reduces ROS accumulation by DOX
To further understand how T1 peptide affects the cellular responses to DOX, we first examined the levels of ROS accumulation after DOX treatment. H9c2 cells were treated with DOX (10 μM) and/or T1 peptide (20 μM) for the time indicated, and then fluorescent ROS indicator CM-H2DCFDA was administered 15 min prior to the end of treatment. ROS fluorescence was measured using flow cytometry. Cellular ROS accumulation induced by DOX was not affected by the combination of T1 peptide within 1 h of treatment (Figure 6A). Interestingly, while the elevation of ROS levels by DOX continued until 5 h of treatments, the levels of ROS reached a plateau after 1 h of T1+DOX treatment (Figure 6A), suggesting that the initial ROS induction by DOX does not require CCN1. The extension of ROS increase beyond 1 h of DOX treatment can be attributed to the newly synthesized CCN1 protein. To assess the oxidative stress induced by DOX in vivo, we examined the mouse hearts 14 days after DOX treatment by immunohistochemical staining with anti-8-OHdG antibody, a marker of oxidative DNA damage [23]. No detectable 8-OHdG+ cells were found in the mice receiving PBS. 15% of cardiomyocytes in DOX-treated WT mice were 8-OHdG+ (Figure 6B, brown nuclei indicated by arrows). ~3% of the cardiomyocytes in DOX-treated Ccn1dm/dm mice were 8-OHdG+ (Figure 6B). Together, these results demonstrated that DOX can generate oxidative stress independent of CCN1.
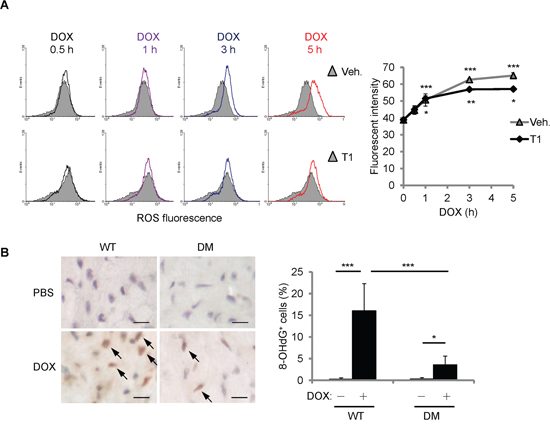
Figure 6: DOX increased cellular ROS partially independent of CCN1. A. Where indicated, H9c2 cells were pretreated with T1 peptide (20 μM) for 1 h before being treated with DOX (10 μM) for the indicated times. Fluorescent ROS indicator CM-H2DCFDA was delivered to the cells 15 min prior to the end of DOX treatment. The level of ROS was measured using flow cytometry. Cells with no DOX treatment were used as a control for basal ROS levels (grey area in the histogram overlays). Quantified ROS fluorescent intensities were expressed as means ± SEM from triplicate experiments. The statistical significance was compared between experimental and control (0 h) groups. B. The cardiac tissue from WT or Ccn1dm/dm mice 14 days after PBS control or DOX treatments was immunostained with anti-8-OHdG antibody (brown; arrows). The percentages of 8-OHdG+ cells were determined from 10 random high-power views per tissue section and 4 sections per mouse (n = 6 for each group), and presented as means ± SEM. Bars: 10 μm.
DOX diminishes anti-apoptotic XIAP through CCN1 signaling
ROS generated by CCN1 leads to the activation of p38 MAPK and following mitochondrial pathways in H9c2 cells [13]. We investigated whether p38 MAPK activation is regulated by DOX by Western blotting with antibodies specifically against phosphorylated (active) p38 (pp38) or total p38 (tp38). Pp38 was significantly increased by DOX (10 μM; 5 h) (Figure 7A). The activation of p38 by DOX was completely blocked by the combination of T1 peptide (20 μM; 1-h pretreatment), suggesting that CCN1 mediates the activation of p38 by DOX. To confirm the activation of p38 in vivo, heart tissue from DOX-treated WT or Ccn1dm/dm mice was immunohistochemically stained with anti-pp38 antibody. Pp38+ cardiomyocytes were markedly increased after DOX treatment in WT mice (reddish-brown staining in Figure 7B; arrows), but were not found in Ccn1dm/dm mice. We next examined the release of mitochondrial Smac and HtrA2, a downstream event after p38 activation [13], by immunoblotting the fractional cell lysates after DOX treatment. The levels of HtrA2 and Smac were elevated in the cytosolic fractions of H9c2 cells by DOX (Figure 7C). The translocation of HtrA2 and Smac induced by DOX was blunted by T1 peptide (Figure 7C). COX-IV served as a mitochondrial contamination control. Smac promotes apoptosis through binding to XIAP and neutralizing its anti-apoptotic activity [24]. HtrA2, a serine protease, diminishes XIAP through catalytic cleavage [25]. Indeed, the levels of XIAP were lowered after DOX treatment, and were not reduced in DOX+T1-treated cells (Figure 7D). Consistently, DOX failed to lower XIAP levels in CCN1-knockdown (siCCN1) cells (Figure 4A).
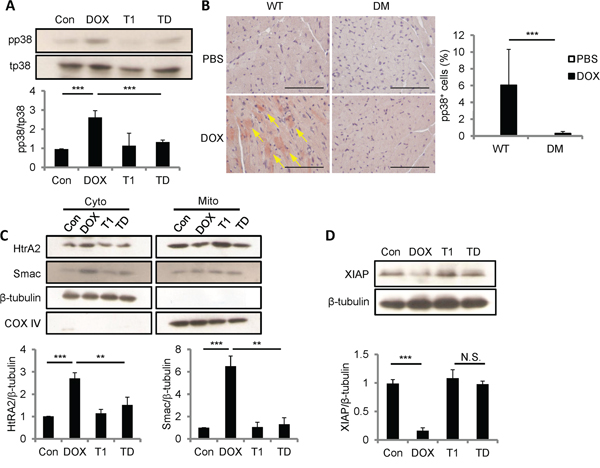
Figure 7: DOX promoted p38 activation, Smac and HtrA2 translocation to cytosol, and XIAP reduction through CCN1/α6β1 signaling. A, C, D. H9c2 Cells were pretreated with or no T1 peptide (20 μM) for 1 h before being treated with or no DOX (10 μM, 5 h). Total cell lysates were immunoblotted with anti-total (t)p38 or anti-phospho (p)p38 antibodies (A); or with anti-XIAP and anti-β-tubulin antibodies (D). Cytosolic and mitochondrial fractional lysates were immunoblotted with anti-HtrA2, anti-Smac, anti-β-tubulin, or anti-COX IV antibodies (C). Protein band intensity was quantified and expressed as in Figure 2 from 3 independent experiments. B. Cardiac tissue from WT or Ccn1dm/dm mice 14 days after PBS or DOX treatment was immunostained with anti-pp38 antibody (reddish-brown; arrows). The percentages of pp38+ cells were determined from 10 random high-power views per tissue section and 4 sections per mouse (n = 6 for each group), and presented as means ± SEM. Bars: 100 μm.
DISCUSSION
The clinical use of DOX is limited by its cardiotoxicity. Here we showed that matricellular protein CCN1 mediates the cardiotoxicity of DOX. CCN1 enables FasL-induced apoptosis in cardiomyocytes. We found that CCN1 and FasL were upregulated by DOX in the mouse heart or in cultured cardiomyocytes. Knockin mice expressing an α6β1-binding defective CCN1 mutant (CCN1-DM) became resistant to DOX-induced cardiomyopathy. In cultured cardiomyoblasts, CCN1 mediated the reduction of XIAP by DOX to facilitate apoptosis through activating p38 MAPK and the release of Smac and HtrA2 from mitochondria. Disrupting the binding between CCN1 and α6β1 by T1 peptide effectively abolished apoptosis induced by DOX (Figure 8).
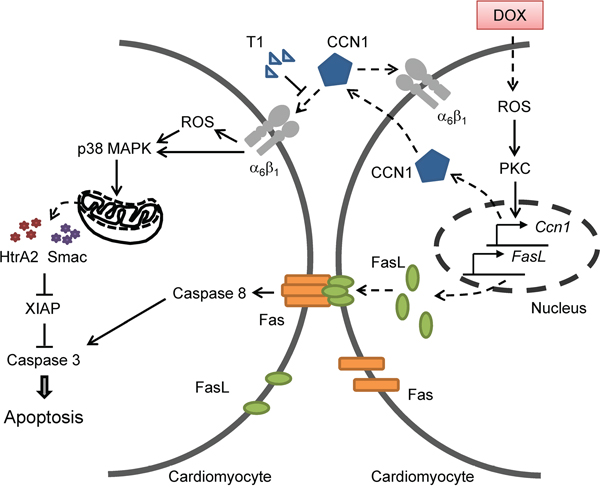
Figure 8: A diagram illustrates the mechanism by which CCN1 mediates DOX cardiotoxicity through autocrine and paracrine signaling by engaging integrin α6β1 to elicit signaling to dismantle the inhibition of XIAP and facilitate apoptosis.
As a matricellular protein, CCN1 does not induce mitogenesis or apoptosis on its own but acts through enhancing other mitogenic or apoptotic factors [14]. Thus, the physiological roles of CCN1 depend not only on the expression profile of target-cell-surface integrin receptors but on the environmental cues as well. Here we found that both CCN1 and FasL were induced in the myocardium of mice treated with DOX. DOX directly upregulated CCN1 and FasL expression to promote apoptosis in cultured H9c2 cells. Ccn1dm/dm mice were resistant to DOX-induced cardiomyopathy despite the persistent induction of FasL, demonstrating the essential role of CCN1/α6β1 signaling in DOX cardiotoxicity. Consistently, T1 peptide prevented DOX-induced apoptosis in H9c2 cells by maintaining XIAP protein levels to inhibit caspase-3 activation, while the induction of FasL by DOX remained. Notably, higher FasL induction was observed in the myocardium of WT than of Ccn1dm/dm mice after DOX treatment. Because the tissue was examined 14 days after DOX injection, higher FasL expression possibly was induced by the cardiac injury and inflammation developed in WT mice.
Though apoptosis induced by DOX was completely abrogated by T1 peptide in H9c2 cells, DOX-induced ROS was only partially reduced (Figure 6A), suggesting that DOX can increase ROS independent of CCN1. Because ROS are required for the induction of both CCN1 and FasL, the initial ROS elevation directly by DOX is responsible for the induction of CCN1 and FasL and will not be blocked by T1 peptide (Figure 8). The CCN1-independent ROS elevation by DOX can activate PKC to induce the expression of CCN1, which further increases ROS through 5-lipoxygenase and NADPH oxidases after engaging integrin α6β1 [13]. Consistently, 8-OHdG+ cells were increased even in Ccn1dm/dm mice. The overly increased numbers of 8-OHdG+ cells in WT mice may be caused by a combination of direct effects from DOX and secondary effects from tissue injury and inflammation. Though ROS can be induced by both DOX (independent of CCN1) and CCN1, CCN1/α6β1 evidently triggers a distinct set of events that cannot be replaced by ROS alone. A potential candidate is integrin-linked kinase (ILK), which binds to the cytoplasmic tail of β1 and β3 integrins [26]. ILK interacts with β1 integrins to promote p38 MAPK activation and cell cycle arrest of epithelial cells in renal tubulogenesis [27]. It is tempting to speculate that ILK is involved in DOX cardiotoxicity through transducing CCN1/α6β1 signaling. This CCN1-α6β1-ILK-p38 signaling axis underlying DOX cardiotoxicity is distinct from the mechanism of the anticancer activity of DOX, and serves as a more specific target for chemoprotection.
To establish CCN1 as a chemoprotective target, the role of CCN1 in tumorigenesis needs to be considered. CCN1 overexpression is associated with the progression of various tumors, including prostate cancer, breast cancer, glioma, colorectal cancer, and osteosarcoma, through its ability to promote angiogenesis, cell migration or proliferation [28, 29]. In contrast to in cardiomyocytes, overexpression of CCN1in human breast cancer MCF-7 cells upregulates XIAP expression to promote chemoresistance to paclitaxel, Adriamycin, and β-lapachone through integrin αvβ3/αvβ5 and NFκB signaling pathway [30]. Thus, blocking CCN1/α6β1 signaling is not expected to impose any negative impact to patients receiving DOX treatment. However, CCN1 can also serve as a tumor suppressor for endometrial and lung cancers [31, 32]. Moreover, despite conflicting reports regarding the role of CCN1 in hepatocarcinogenesis [33, 34], CCN1 has been suggested to suppress hepatocellular carcinoma by inhibiting carcinogen-induced compensatory hepatocyte proliferation through integrin α6β1 [35]. The risks of latent tumorigenesis resulting from disrupting CCN1/α6β1 signaling long term need to be further evaluated. Nevertheless, because replicating cells are more sensitive to DOX [2], cotreatment of DOX likely will compensate the loss of the antineoplastic activities from disrupting CCN1/α6β1 signaling in hepatocytes.
In summary, we identified matricellular protein CCN1 as a novel downstream effector for DOX cardiotoxicity. CCN1 is induced by DOX in cultured cardiomyoblast H9c2 cells and in mouse hearts to promote cell death and cardiac dysfunction. CCN1 acts through engaging integrin α6β1 to trigger the activation of p38 MAPK, translocation of mitochondrial Smac and HtrA2, thereby diminishing anti-apoptotic XIAP and facilitating apoptosis in cardiomyocytes. Disrupting CCN1/α6β1 engagement prevents DOX-induced cardiomyopathy in mice.
MATERIALS AND METHODS
Mouse model of DOX-induced cardiomyopathy
All animal experiments conformed to the guidelines of the Institutional Animal Care and Use Committee of the National Chen Kung University. Two knockin transgenic mouse lines—Ccn1+/lacZ and Ccn1dm/dm—on the C57BL/6 background were obtained from Lester Lau, PhD (University of Illinois at Chicago, IL). Cardiomyopathy was induced in mice of 3- to 6-month-old males by a single injection of DOX (15 mg/kg; i.p.) (Sigma), as previously described [36]. Mice were euthanized by cervical dislocation before the hearts were collected from Ccn1+/lacZ 1 day after DOX treatment, or from WT and Ccn1dm/dm mice 14 days after DOX for histological analysis (n=6 for each group).
Histology
For CCN1 expression, 14 μm cryo sections of heart tissue from Ccn1+/lacZ mice were stained with X-gal as previously described [16]. For histological analysis, 8 μm paraffin sections of heart tissue from WT or Ccn1dm/dm mice were subjected to Masson’s trichrome (Sigma) staining or TUNEL staining (Millipore) following standard procedures. For immunohistochemical staining, paraffin tissue sections were antigen-retrieved before being incubated with primary antibodies, then secondary antibodies, then visualized with AEC detection IHC kit (Abcam) or M.O.M. Immunodetection kit (Vector Laboratories). Primary antibodies used include anti-FasL (Abcam 15285, 1:100), anti-8-OHdG (JaICA, MOG-020P, 1:100), and anti-phospho-p38 (pp38) (Cell Signaling 4631, 1:50).
Electrocardiography and echocardiography
Electrocardiograms were recorded using a PowerLab 8/30 (ADInstruments) on mice (n=4) anesthetized with chloral hydrate (300 mg/kg; i.p.) in the supine position. Echocardiography was performed using a Vevo 770 (Visual Sonics) on mice (n=4) anesthetized with 2% isoflurane. Left ventricular ejection fraction (EF) and fractional shortening (FS) were computed from echocardiographic measurements by the Vevo software.
Cell culture
Rat cardiomyoblast H9c2 cells were purchased from American Type Culture Collection and maintained at 37°C in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum (Invitrogen). H9c2 cells were serum starved for overnight before treated with 10 μM DOX and/or 20 μM T1 peptide (Angene Biotech, Taiwan) for the time indicated in the figure legends. To block PKC, cells were pretreated with 1 μM calphostin C (Sigma) for 1 h.
RNA interference
H9c2 cells were transfected with 100 nM Ccn1 siRNA or a non-targeting control (Invitrogen) using Lipofectamine 3000 (Invitrogen). Cells after transfection were incubated for 2 days before following treatments. Ccn1 siRNA sequences: 5′-GCA GCA AGA CCA AGA AAT CC-3′; 5′-TTC TGG TCT GCA GAG GTG TG-3′.
Western blotting
Cytoplasmic and mitochondrial fractional extracts were isolated using Mitochondria/Cytosol Fractionation Kit (BioVision). Total cell lysates or fractional extracts were electrophoresed, transblotted, and immunoblotted following standard procedures. Primary antibodies used include anti-FasL (Santa Cruz sc6237, 1:2000), anti-CCN1 (1:5000) (a generous gift from Shr-Jeng Leu, PhD; National Yang Ming University, Taiwan), anti-XIAP (R&D AF8221, 1:4000), anti-Caspase-3 (Cell Signaling 9662, 1:4000), anti-Caspase-8 (Cell Signaling 9429, 1:1000), anti-HtrA2 (R&D AF1458, 1:4000), anti-Smac (R&D AF7891, 1:1000), anti-pp38 (Cell Signaling 9211, 1:4000), anti-tp38 (Cell Signaling 9212, 1:4000), anti-COX IV (Cell Signaling 4844, 1:1000), and anti-β-tubulin (Abcam ab6046, 1:8000). General reagents and chemicals were from Sigma.
RNA isolation and qRT-PCR
Total RNA was extracted from H9c2 cells using TRIzol Reagent (Invitrogen). RT-PCR was carried out using the iScript cDNA synthesis kit (Bio-Rad) following the manufacturer’s protocol. qRT-PCR was performed using SYBR Green-based (Invitrogen) StepOnePlus™ Real-Time PCR System. Primer pairs were: Ccn1 forward primer: 5′-GCA GCA AGA CCA AGA AAT CC-3′, reverse primer: 5′-TTC TGG TCT GCA GAG GTG TG-3′; FasL forward primer: 5′-AAA GAC CAC AAG GTC CAA CA-3′, reverse primer: 5′-AGT CTC TAG CTT ATC CAT GA-3′; and Cyclophilin forward primer: 5′-GGC AAA TGC TGG ACC AAA CAC-3′, reverse primer: 5′-TTC CTG GAC CCA AAA CGC TC-3′.
Annexin V/PI staining
Treated H9c2 Cells were harvested and incubated with FITC-conjugated annexin V antibody (BD Biosciences) and PI at room temperature for 20 min before they were analyzed using flow cytometry. The results were further analyzed using WinMDI software.
ROS measurements
Live cells were loaded with ROS dye CM-H2DCFDA (5 μM) (Sigma) 15 min prior to the end of DOX treatment. Subsequently, the cells were harvested and resuspended in cold PBS containing 5% FBS. Fluorescence was measured using flow cytometry and analyzed with WinMDI.
Statistical analysis
All assays were repeated at least 3 times and yielded similar patterns. Data are mean ± SEM. Statistical analysis was carried out using one-way (cell-based assays) or two-way (animal studies) ANOVA and post hoc tests. Significance was set at p<0.05 and indicated as N.S.: no significant difference; *: p<0.05; **: p<0.01; or ***: p<0.001.
ACKNOWLEDGMENTS
The authors thank Dr Lester Lau for providing the transgenic mice.
CONFLICTS OF INTEREST
All the authors of this paper declare that they have no conflicts of interest.
GRANT SUPPORT
This work was supported by the Ministry of Science and Technology of Taiwan [grant 103-2320-B-006-016-MY3].
REFERENCES
1. Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, Moreira PI. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009; 16:3267-3285.
2. Hawtin RE, Stockett DE, Wong OK, Lundin C, Helleday T, Fox JA. Homologous recombination repair is essential for repair of vosaroxin-induced DNA double-strand breaks. Oncotarget. 2010; 1:606-619. doi: 10.18632/oncotarget.195.
3. Von Hoff DD, Layard MW, Basa P, Davis HL, Jr., Von Hoff AL, Rozencweig M, Muggia FM. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979; 91:710-717.
4. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003; 97:2869-2879.
5. Lipshultz SE, Franco VI, Miller TL, Colan SD, Sallan SE. Cardiovascular disease in adult survivors of childhood cancer. Annu Rev Med. 2015; 66:161-176.
6. Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, Mutharasan RK, Naik TJ, Ardehali H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014; 124:617-630.
7. Hasinoff BB, Hellmann K, Herman EH, Ferrans VJ. Chemical, biological and clinical aspects of dexrazoxane and other bisdioxopiperazines. Curr Med Chem. 1998; 5:1-28.
8. Tebbi CK, London WB, Friedman D, Villaluna D, De Alarcon PA, Constine LS, Mendenhall NP, Sposto R, Chauvenet A, Schwartz CL. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin's disease. J Clin Oncol. 2007; 25:493-500.
9. Niu J, Azfer A, Wang K, Wang X, Kolattukudy PE. Cardiac-targeted expression of soluble fas attenuates doxorubicin-induced cardiotoxicity in mice. J Pharmacol Exp Ther. 2009; 328:740-748.
10. Nitobe J, Yamaguchi S, Okuyama M, Nozaki N, Sata M, Miyamoto T, Takeishi Y, Kubota I, Tomoike H. Reactive oxygen species regulate FLICE inhibitory protein (FLIP) and susceptibility to Fas-mediated apoptosis in cardiac myocytes. Cardiovasc Res. 2003; 57:119-128.
11. Wollert KC, Heineke J, Westermann J, Ludde M, Fiedler B, Zierhut W, Laurent D, Bauer MK, Schulze-Osthoff K, Drexler H. The cardiac Fas (APO-1/CD95) Receptor/Fas ligand system : relation to diastolic wall stress in volume-overload hypertrophy in vivo and activation of the transcription factor AP-1 in cardiac myocytes. Circulation. 2000; 101:1172-1178.
12. Hsu PL, Su BC, Kuok QY, Mo FE. Extracellular matrix protein CCN1 regulates cardiomyocyte apoptosis in mice with stress-induced cardiac injury. Cardiovasc Res. 2013; 98:64-72.
13. Su BC, Mo FE. CCN1 enables Fas ligand-induced apoptosis in cardiomyoblast H9c2 cells by disrupting caspase inhibitor XIAP. Cell Signal. 2014; 26:1326-1334.
14. Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011; 10:945-963.
15. Mo FE, Lau LF. The matricellular protein CCN1 is essential for cardiac development. Circ Res. 2006; 99:961-969.
16. Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol. 2002; 22:8709-8720.
17. Hilfiker-Kleiner D, Kaminski K, Kaminska A, Fuchs M, Klein G, Podewski E, Grote K, Kiian I, Wollert KC, Hilfiker A, Drexler H. Regulation of proangiogenic factor CCN1 in cardiac muscle: impact of ischemia, pressure overload, and neurohumoral activation. Circulation. 2004; 109:2227-2233.
18. Chen CC, Young JL, Monzon RI, Chen N, Todorovic V, Lau LF. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J. 2007; 26:1257-1267.
19. Nakamura T, Ueda Y, Juan Y, Katsuda S, Takahashi H, Koh E. Fas-mediated apoptosis in adriamycin-induced cardiomyopathy in rats: In vivo study. Circulation. 2000; 102:572-578.
20. Singh P, Sharma R, McElhanon K, Allen CD, Megyesi JK, Benes H, Singh SP. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic Biol Med. 2015; 86:90-101.
21. Gopalakrishna R, Jaken S. Protein kinase C signaling and oxidative stress. Free Radic Biol Med. 2000; 28:1349-1361.
22. Leu SJ, Liu Y, Chen N, Chen CC, Lam SC, Lau LF. Identification of a novel integrin alpha 6beta 1 binding site in the angiogenic inducer CCN1 (CYR61). J Biol Chem. 2003; 278:33801-33808.
23. Toyokuni S, Tanaka T, Hattori Y, Nishiyama Y, Yoshida A, Uchida K, Hiai H, Ochi H, Osawa T. Quantitative immunohistochemical determination of 8-hydroxy-2'-deoxyguanosine by a monoclonal antibody N45.1: its application to ferric nitrilotriacetate-induced renal carcinogenesis model. Lab Invest. 1997; 76:365-374.
24. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000; 102:33-42.
25. Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003; 17:1487-1496.
26. Legate KR, Fassler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci. 2009; 122:187-198.
27. Smeeton J, Zhang X, Bulus N, Mernaugh G, Lange A, Karner CM, Carroll TJ, Fassler R, Pozzi A, Rosenblum ND, Zent R. Integrin-linked kinase regulates p38 MAPK-dependent cell cycle arrest in ureteric bud development. Development. 2010; 137:3233-3243.
28. Li J, Ye L, Owen S, Weeks HP, Zhang Z, Jiang WG. Emerging role of CCN family proteins in tumorigenesis and cancer metastasis. Int J Mol Med. 2015; 36:1451-1463.
29. Sarkar S, Quinn BA, Shen XN, Dash R, Das SK, Emdad L, Klibanov AL, Wang XY, Pellecchia M, Sarkar D, Fisher PB. Therapy of prostate cancer using a novel cancer terminator virus and a small molecule BH-3 mimetic. Oncotarget. 2015; 6:10712-10727. doi: 10.18632/oncotarget.3544.
30. Lin MT, Chang CC, Chen ST, Chang HL, Su JL, Chau YP, Kuo ML. Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-kappaB-dependent XIAP up-regulation. The Journal of biological chemistry. 2004; 279:24015-24023.
31. Chien W, Kumagai T, Miller CW, Desmond JC, Frank JM, Said JW, Koeffler HP. - Cyr61 suppresses growth of human endometrial cancer cells. J Biol Chem. 2004; 279:53087-53096.
32. Tong X, Xie D, O'Kelly J, Miller CW, Muller-Tidow C, Koeffler HP. Cyr61, a member of CCN family, is a tumor suppressor in non-small cell lung cancer. J Biol Chem. 2001; 276:47709-47714.
33. Jia Q, Dong Q, Qin L. CCN: core regulatory proteins in the microenvironment that affect the metastasis of hepatocellular carcinoma? Oncotarget. 2016; 7:1203-1214. doi: 10.18632/oncotarget.6209.
34. Kim KH, Lau LF. CCN1 in hepatobiliary injury repair. Oncotarget. 2015; 6:34053-34054. doi: 10.18632/oncotarget.6079.
35. Chen CC, Kim KH, Lau LF. The matricellular protein CCN1 suppresses hepatocarcinogenesis by inhibiting compensatory proliferation. Oncogene. 2016; 35:1314-1323.
36. Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, Kanamori H, Khai NC, Maruyama R, Ogino A, Minatoguchi S, Fujiwara T, Fujiwara H. Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation. 2006; 113:535-543.