
INTRODUCTION
Somatic activating mutations in KRAS or NRAS are present in up to 52% of colorectal cancer (CRC) [1, 2], causing constitutive activation of the RAF/MEK/ERK signaling pathway independent of upstream receptor tyrosine kinases like the epidermal growth factor receptor (EGFR). These mutations are known predictive biomarkers of resistance in metastatic CRC to anti-EGFR therapy, such as cetuximab or panitumumab, and consequently patients whose tumors harbor KRAS or NRAS mutations have fewer therapeutic options. There are currently no known effective therapies that exploit KRAS or NRAS mutations to target malignant cells.
Though MEK inhibitors were found to have intriguing in vitro activity in KRAS mutated CRC models [3–6], efficacy was variable in different cell lines. More importantly, MEK inhibitor monotherapy proved to be largely ineffective in patient-derived xenograft (PDX) murine models [7] and in human clinical trials [8]. Myriad mechanisms of resistance have been identified [9–13], with most causing increased signaling through upstream receptor tyrosine kinases or activation of parallel signal transduction cascades to bypass or overcome MEK inhibition and reactivate ERK signaling. Indeed, unless an approximately ten-fold reduction in ERK activation is achieved, cell proliferation persists [14]. Consequently, rational drug combinations with MEK inhibitors, potentially with agents that target downstream effectors of ERK, are likely necessary to overcome compensatory responses to MEK inhibitors.
Phosphorylation and activation of ERK is well-known to cause increased proliferation and increased activity of the cell cycle by causing increased cyclin D expression [15, 16]. Cyclin D expression is the rate-limiting step in cell cycle progression from G1 into S phase [17]. Cyclin D complexes with and activates cyclin dependent kinase (CDK) 4 and 6, which phosphorylate and inactivate the tumor suppressor retinoblastoma protein (Rb) [18]. In its unphosphorylated state, Rb is bound to the transcription factor E2F, and Rb phosphorylation releases E2F, freeing it to increase transcription of genes promoting cell cycle progression into S phase. Selective CDK4/6 inhibitors have been developed with minimal off-target kinase inhibition [19–21], with the caveat that they require intact expression of Rb for antitumor efficacy [19]. However, inactivating mutations in RB1 are very uncommon in CRC and do not exceed the expected background rate of mutations [1], and in fact the majority of CRCs have higher levels of Rb than normal colon mucosa [22]. Furthermore, loss of Rb speeds cell growth in conjunction with RAS mutations [23–25], indicating that RAS mutant cell growth increases with unimpeded, dysregulated cell cycle progression. Thus, RAS mutant CRCs are a candidate for further investigation of the efficacy of CDK4/6 inhibitors. The combination of CDK4/6 and MEK inhibitors may be particularly efficacious in RAS mutated malignancies. Inhibition of CDK4 was found to be synthetically lethal in vitro and in vivo in KRAS mutant non-small cell lung cancers [26, 27]. However, CDK4/6 inhibitor monotherapy in early human clinical trials did not yield any responses in CRC [28–31], arguing that combination therapy is required. In an inducible NRAS Q61K genetically engineered mouse model of melanoma, the combination of MEK and CDK4/6 inhibitors caused tumor regression that paralleled extinction of mutant NRAS, but either monotherapy alone did not [32]. Consequently, we hypothesized that the combination of a CDK4/6 inhibitor and a MEK inhibitor would have synergistic antitumor activity in vitro and in vivo in KRAS mutant CRC.
RESULTS
Dual inhibition of MEK and CDK4/6 markedly attenuates cell growth in vitro
To examine the efficacy of dual blockade of CDK4/6 and MEK on KRAS mutant CRC cells, cell growth and colony formation were determined after treatment with the MEK inhibitor MEK162 and the CDK4/6 inhibitor palbociclib, using clinically relevant doses of palbociclib [28, 29] and doses optimized to each cell line to maximally display contrast of cell growth between monotherapies and combination therapy (See Supplementary Table S1 and Figure S1). As shown in Figure 1A–1G, the combination of MEK162 and palbociclib was markedly effective in attenuating cell growth and colony formation in a broad panel of KRAS mutant CRC cell lines. As shown in Figure 1H, the combination of MEK inhibitor and CDK4/6 inhibitor was more effective in limiting colony formation and cell growth than MEK inhibitor monotherapy in the majority of the 11 KRAS mutant CRC cell lines assayed.
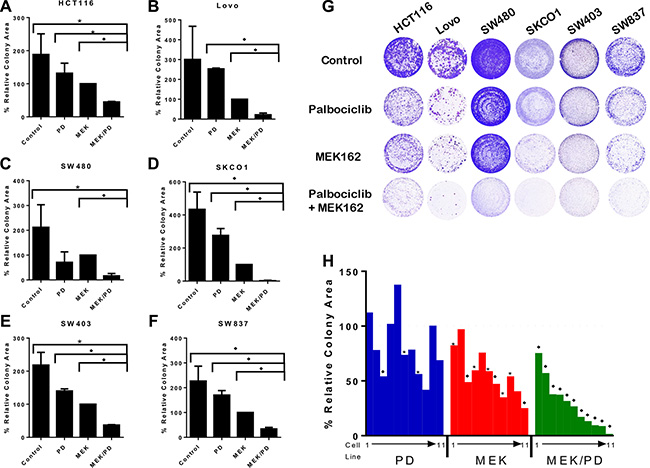
Figure 1: Combination of MEK and CDK4/6 inhibitors markedly attenuates cell growth in vitro in a panel of KRAS mutant CRC cell lines. (A–F), Colony assays for six representative cell lines treated with the CDK4/6 inhibitor palbociclib (PD), the MEK inhibitor MEK162 (MEK), the combination (MEK/PD), or with DMSO control for 10–14 days. Results are normalized to MEK162 monotherapy. The data represent mean values ± SD for 3–4 independent experiments. (G), Depiction of the stained cell wells. (H), Summary colony assay results for a panel of 11 KRAS mutant CRC cell lines treated with DMSO control, PD, MEK, or MEK/PD at concentrations approximating the IC50 for MEK monotherapy for 10–14 days. Results here are normalized to DMSO control. A waterfall plot is depicted showing values, with cell lines consistently depicted in the same order of T84, LS174T, SW1116, SW948, LS1034, HCT116, SW837, SW403, SW480, Lovo, and SKCO1. The data represent mean values for 3–4 independent experiments. *p < 0.05, *p < 0.006. All p-values were generated by Student’s t-test.
Dual inhibition of MEK and CDK4/6 synergistically inhibits growth of KRAS mutant colon cancer cell lines
To examine the mechanism of dual blockade of CDK4/6 and MEK on KRAS mutant CRC cells, cell growth was assessed with MTS assay after treatment with MEK162 and palbociclib for 72 hours. The cell lines HCT116, Lovo, SW480, and LS174T showed improved efficacy of the combination of MEK162 and palbociclib compared to either monotherapy, with the effect reaching significance for Lovo, SW480, and LS174T at all concentration levels tested (Figure 2A–2D). On assessment for formal synergy, these three cell lines all displayed synergistic effects (combination index, CI, at EC50 for Lovo was 0.05, for SW480 was 0.68, and for LS174T was 0.22; CI values < 1 indicate synergy). An isobologram for these findings is depicted in Figure 2E.
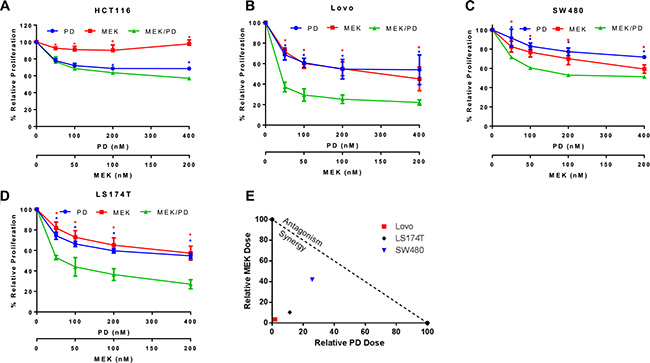
Figure 2: Combination of MEK and CDK4/6 inhibitors synergistically attenuates cell growth in a panel of KRAS mutant CRC cell lines. (A), HCT116 cells were treated with the CDK4/6 inhibitor palbociclib (PD) (400 nM, 200 nM, 100 nM, or 50 nM) and/or the MEK inhibitor MEK162 (MEK) (200 nM, 100 nM, 50 nM, or 25 nM respectively) or control for 72 hours and proliferation and cell growth was assessed using MTS assay. The data represent mean values ± SD for 3 independent experiments. (B), Lovo cells treated as described for HCT116. (C), SW480 cells treated as described for HCT116. (D), LS174T cells treated as described for HCT116. (E), Isobologram shows synergy of the combination of PD0332991 and MEK162. Plotted points farther to the bottom left of the Figure represent increasing degree of synergy. *p < 0.05, ♦p < 0.006. All p-values were generated by Student’s t-test.
Using a flow-based annexin V-FITC assay, treatment with the combination of CDK4/6 inhibitor and MEK inhibitor for 72 hours induced apoptosis in a greater proportion of cells than either monotherapy in HCT116 and Lovo cell lines, but not in SW480 and SW403 (Figure 3A–3B; SW403 not shown). The combination also induced senescence in a greater proportion of cells after 72 hours as determined by staining for senescence-associated beta-galactosidase, for Lovo and SW480 (Figure 3D–3E), while there was negligible staining in HCT116 cells regardless of treatment (not shown), and SW403 could not be assessed because of cell clumping precluding accurate counting. Overall, these results suggest that the synergy of the drug combination acts through some combination of increased apoptosis and/or senescence, at least in vitro.
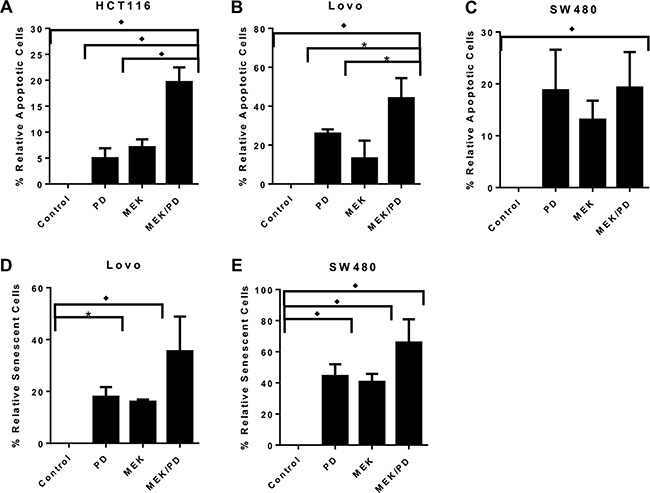
Figure 3: Combination of MEK and CDK4/6 inhibitors inhibits cell growth in KRAS mutant CRC cell lines through a variety of mechanisms. (A), HCT116 cells were treated with the CDK4/6 inhibitor palbociclib (PD, 400 nM), the MEK inhibitor MEK162 (MEK, 200 nM), the combination (MEK/PD), or with DMSO control for 72 hours, and stained with propidium iodide and annexin V-FITC prior to performing flow cytometry. The data represent mean values ± SD for 3 independent experiments. (B), Lovo cells were treated as described for HCT116. C, SW480 cells were treated as described for HCT116. *p < 0.05. ♦p < 0.01 for (A–C), with all p-values generated by Student’s t-test. (D), Lovo cells were treated with PD (400 nM), MEK (200 nM), the combination, or with DMSO control for 72 hours, and stained with X-gal to detect senescence-associated β-galactosidase. The proportion of stained cells in 10 random high-powered fields was counted. The data shows mean values from a representative experiment ± SD for the 10 fields. (E), SW480 cells were treated as described for Lovo. ♦p ≤ 0.0002 for combination compared to any other value, with all p-values generated by Student’s t-test.
Dual inhibition of MEK and CDK4/6 results in greater inhibition of phosphorylation of ribosomal protein S6
To determine mechanisms of action and inform on pharmacodynamic markers of response, we performed reverse phase protein arrays (RPPA) using protein lysates obtained from 10 KRAS mutant CRC cell lines treated for 24 hours with MEK162, palbociclib, the combination, or DMSO control (Figure 4A–4B). Protein changes after MEK inhibitor treatment were in line with previous publications using RPPA [32, 33], including downregulation of Myc and Fra1, upregulation of Bim, and a feedback increase in MEK phosphorylation.
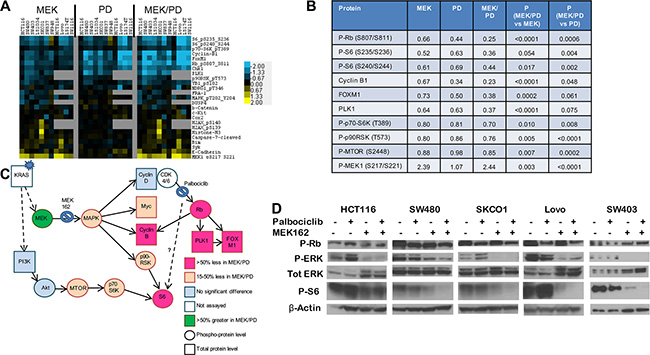
Figure 4: Combination of CDK4/6 and MEK inhibitors induces greater inhibition of phosphorylation of S6 and other growth factor signaling and cell cycle proteins. (A), Protein lysates were obtained from a panel of 9 KRAS mutant CRC cell lines after treatment with DMSO control, palbociclib (PD) 400 nM, MEK162 (MEK) 200 nM, or MEK162 200 nM + palbociclib 400 nM (MEK/PD) for 24 hours, and RPPA was performed. Protein levels were normalized to DMSO controls of each cell line, and a log2 heatmap of the most differentially expressed proteins was generated. (B), Median expression levels of proteins in KRAS mutant CRC cell lines treated with MEK162 (MEK) alone, palbociclib (PD) alone, or the combination (MEK/PD), relative to DMSO-treated control. p-values were determined using paired Student’s t-test. (C), Protein expression data from RPPA was mapped according to relevant pathways. Circles represent phosphorylated epitopes on the proteins and squares represent total proteins. Activating KRAS mutations are thought to lead to constitutive activation of downstream signaling pathways, but adding MEK inhibitor and CDK4/6 inhibitor yielded the depicted changes in protein expression. (D), Western blotting of the indicated antibodies for KRAS mutant cell lines HCT116, SW480, SKCO1, Lovo, and SW403 treated with palbociclib 400 nM and/or MEK162 200 nM for 24 hours.
Consistent with the selectivity of palbociclib for CDK4/6, only five proteins were consistently affected by palbociclib across all lines, including significant downregulation of the proliferation markers Rb, Foxm1, Plk1, and Ccnb1 (Cyclin B1). Palbociclib was consistently better than or at least as efficient as MEK162 in suppressing these markers. Importantly, the drug combination more strongly downregulated these markers than either monotherapy alone in every cell line tested, Figure 4B suggesting a cooperative antiproliferative effect.
Unexpectedly, palbociclib alone also resulted in the inhibition of phosphorylation of ribosomal protein S6 in 8 of the 10 lines, which has not been reported previously. MEK162 alone also downregulated pS6, consistent with known signaling, and the effect was more marked with the drug combination in all cell lines except HCT116. These findings were recapitulated by immunoblotting for 5 of the lines (Figure 4D) including HCT116. In SW480, SKCO1, Lovo, and SW403, pS6 was nearly or completely undetectable after combination treatment. Notably, pS6 has been identified as a marker of in vivo responsiveness to MEK inhibitors in BRAF-mutant melanoma [34]. Thus, the muted combination effect in HCT116 on pS6 is consistent with the observed lack of formal drug synergy (Figure 2A). Furthermore, the combination demonstrated slight but significant cooperative downregulation in components upstream of pS6, including phospho-mTOR, phospho-p70-S6 kinase, and p90 ribosomal S6 kinase (p90rsk) (see Figure 4B, 4C). These findings together suggest a degree of cross-talk or feedback between CDK4/6 signaling and mTOR.
As S6 also lies downstream of the PI3K pathway, we next asked whether activation of PI3K by PIK3CA mutations affected the pS6 response to combination therapy. We noted that of three PIK3CA mutant cell lines, HCT116 and SW948 had relatively less pS6 downregulation than PIK3CA wild-type cell lines, but that the third, LS174T, had strong pS6 downregulation (see Figure S2). Consequently, though PIK3CA mutation may be associated with shallower changes to phospho-S6 on treatment, it may not be completely sufficient or fully penetrant in blocking pS6 downregulation.
Dual inhibition of MEK and CDK4/6 inhibits tumor growth in vivo in KRAS mutant CRC cell line xenograft and PDX
We next tested the combination in vivo using CRC xenograft models. The combination of the MEK and CDK4/6 inhibitors trametinib (3 mg/kg QOD) and palbociclib (100 mg/kg QD) using oral gavage yielded significantly greater tumor growth inhibition in murine xenografts of the KRAS mutant Lovo cell line compared to vehicle-treated controls or treatment with monotherapy of each drug alone (see Figure 5A). The combination treatment did cause weight loss in mice, with a maximum weight loss tending to occur at days 10 through 14. Importantly though, mice with less weight lost showed weight stabilization and/or complete recovery with continued treatment (see Figure S3A), suggesting that alternative dosing schedules may improve toxicity. We also developed a trametinib/palbociclib chow calculated to deliver a similar dose to oral gavage continuously. Lovo xenografts again showed regression upon treatment with this combination chow, demonstrating similar efficacy to oral gavage (see Figure S3B). Independent cell line xenografts of HCT116 and SW480 also showed tumor regression in mice treated with the combination chow (see Figure 5B–5C). No tumors showed resistance to the combination at 30+ days of treatment. Furthermore, these mice did not demonstrate weight loss exceeding 20% or morbidity with the combination chow despite demonstrating tumor shrinkage.
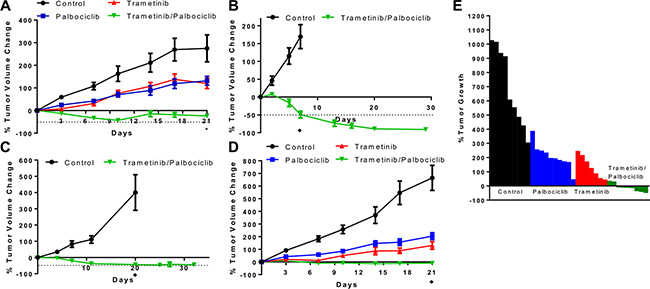
Figure 5: Combination of CDK4/6 and MEK inhibitors induces tumor regression in vivo in KRAS mutant CRC xenografts. (A), Lovo cell line xenografts were treated with vehicle control daily, palbociclib 100 mg/kg daily, trametinib 3 mg/kg every 2 days, or palbociclib 100 mg/kg daily + trametinib 3 mg/kg every 2 days, and tumor volume was measured twice per week. Data are mean ± SEM, with 10 mice/arm. *p < 0.01 on day 21 for combination vs. each other arm by Student’s t-test. (B), HCT116 cell line xenografts were treated with control chow or chow containing trametinib + palbociclib, and tumor volume was measured longitudinally. ♦p < 0.001 on day 7 by Student’s t-test. (C), SW480 cell line xenografts were treated with control chow or chow containing trametinib + palbociclib, and tumor volume was measured longitudinally. ♦p < 0.001 on day 20 by Student’s t-test. (D), Xenografts implanted with patient-derived CRC cells harboring KRAS A146T mutation were treated with vehicle control daily, palbociclib 100 mg/kg daily, trametinib 3 mg/kg every 2 days, or palbociclib 100 mg/kg daily + trametinib 3 mg/kg every 2 days, and tumor volume was measured twice per week. Data are mean ± SEM, with 9–10 mice/arm. ♦p < 0.001 on day 21 for combination vs. any other arm by Student’s t-test. Dotted lines represent 50% decrease in tumor volume. (E), Change in tumor volume for each of the patient-derived xenograft mice in D at day 21 of treatment.
We also leveraged a KRAS mutant CRC PDX model that we have recently established. Similar to the results in the Lovo cell line xenograft model, the combination of MEK and CDK4/6 inhibitors yielded significantly greater tumor growth inhibition in the PDX model compared to vehicle-treated controls or treatment with monotherapy of each drug alone (see Figure 5D). Notably, this PDX model responded to the combination despite harboring an atypical KRAS A146T mutation and an activating AKT1 E17K mutation. The combination treatment yielded regression of tumors after 21 days, unlike treatment with either monotherapy. The combination treatment again caused weight loss in mice, with a nadir weight between days 7 and 14 before stabilization and partial recovery of the lost weight (see Figure S3C).
To molecularly compare the in vivo inhibition with our earlier in vitro studies, RPPA was performed using protein lysates obtained from PDX tumors after treatment for 21 days (Figure 6A–6B). Similar to the in vitro data, treatment with the combination of MEK and CDK4/6 inhibitors downregulated Rb, Plk1, Ccnb1, and pS6 to a greater extent than either monotherapy alone. Interestingly, unlike in the in vitro model, treatment with trametinib did not show a feedback increase in phosphorylation of MEK (see Figure 6B) after 21 days of treatment. Additionally, a number of proteins showed cooperative changes upon combination therapy that were not observed in vitro including CDK1 and FASN, suggesting the influence of the tumor microenvironment on inhibitor-induced protein changes. Immunohistochemical staining from PDX samples demonstrated a marked decrease in cell staining for Ki67 after 21 days of treatment (see Figure 6C–6D). Staining for phospho-S6 was decreased in the trametinib and the combination trametinib + palbociclib treated groups compared to control in this chronically-treated cohort (see Figure S4). Collectively, these data support the hypothesis that the combination of MEK and CDK4/6 inhibitors induces tumor regression via the cooperative induction of cell cycle arrest, and that Ki67 and pS6 may serve as surrogates for combined target engagement.
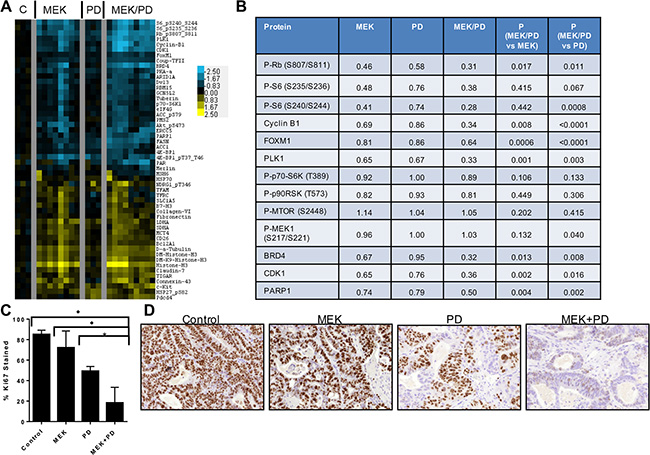
Figure 6: Combination of CDK4/6 and MEK inhibitors induces greater inhibition of phosphorylation of S6 and other growth factor signaling and cell cycle proteins in vivo in PDX models of KRAS mutant CRC. (A), Protein lysates were obtained from tumors after 21 days of treatment with vehicle control (C), trametinib (MEK), palbociclib (PD), or trametinib + palbociclib (MEK/PD), and RPPA was performed. Protein levels were normalized to the geometric mean of vehicle-treated controls, and a heatmap of the most differentially expressed proteins was generated. Values are log2-transformed fold-change compared to the geometric mean of the controls. (B), Median expression levels of selected proteins in KRAS mutant CRC PDXs treated with trametinib (MEK) alone, palbociclib (PD) alone, or the combination (MEK/PD) for 21 days, relative to vehicle-treated control. p-values were determined using paired Student’s t-test. (C), Percentage of nuclear area stained by Ki67 antibody. Data represent mean ± SD of samples from 3–4 mice. *p ≤ 0.02, ♦p < 0.003 by Student’s t-test. (D), Representative immunohistochemistry images for Ki67 staining.
DISCUSSION
Though mutations in KRAS and other RAS isoforms are widespread in many malignancies, as yet efforts to therapeutically target KRAS mutations have been unsuccessful. Previous studies investigating the use of MEK inhibitor monotherapy revealed that though resistance may arise through a variety of mechanisms, generally they result in compensatory activation of parallel signaling pathways to reactivate phosphorylation of ERK [9–13]. ERK plays a key role in promoting cell cycle progression and proliferation [35], including through increased transcription of cyclin D1 [15, 16]. Several synthetic lethality screens have been performed on KRAS mutant cancer cell lines to determine potential targets, and results have frequently included genes important in cell cycle progression or mitosis, such as CDK1 [36], cyclin A2 [37], PLK1 [37], and CDC6 [38]. Furthermore, abrogation of CDK4 was synthetically lethal in KRAS mutant non-small cell lung cancer models [26, 27]. Consequently, we hypothesized that the combination of inhibition of the MAPK pathway with a MEK inhibitor and inhibition of cell cycle progression with a CDK4/6 inhibitor would be more effective. Indeed, our studies demonstrate that combined inhibition of MEK and CKD4/6 is synergistic in vitro in a variety of KRAS mutant CRC cell lines and yields tumor regression in vivo in cell line xenografts and PDXs of KRAS mutant CRC.
It was previously not well recognized that CDK4/6 inhibition would inhibit phosphorylation of S6, which is more typically associated with growth factor signaling through the PI3K/MTOR and MEK/ERK pathways. Our results have most notably found that both inhibition of MEK and CDK4/6 individually cause decreases in phospho-S6 levels, and the combination yields even greater decrease. Ribosomal protein S6 is a component of the 40S ribosomal subunit that can be phosphorylated at five serine residues by S6 kinase in the mTOR pathway or by p90rsk in an ERK-dependent manner [39–41]. Phosphorylated S6 is associated with increased cell size and increased translation of polypeptides encoded by mRNAs with a 5′-polypyrimidine tract [41]. Entry into the cell cycle is associated with increased phosphorylation of S6 at Ser240/244 [42], and so decrease in phospho-S6 may reflect generally decreased flux through the cell cycle. Nevertheless, decreased phospho-S6 level upon combination treatment appears to be a pharmacodynamic biomarker of therapy that warrants further evaluation.
Mechanisms of action of CDK4/6 inhibitors also remain under investigation. Decreases in the levels of cyclin B1, FOXM1, and PLK1 were observed with the combination of CDK4/6 inhibitors and MEK inhibitors. Similar changes in levels of downstream target genes have been found in vitro in estrogen receptor-positive breast cancer cell lines treated with palbociclib combined with the aromatase inhibitor letrozole [43]. Decreases of these effectors are important in mediating cellular fate. CDK4 and CDK6 phosphorylate and stabilize the transcription factor FOXM1 to maintain G1/S phase gene expression and suppress senescence [44], so decreased levels of FOXM1 likely contribute toward shifting the cell into senescence. PLK1 is a kinase important in promoting mitotic entry and proliferation [45], and its transcription is upregulated by E2F [46], and thus decreased levels of PLK1 are expected with decreased phospho-RB. Cyclin B1 complexes with CDK1 to promote mitotic entry, and accumulation of cyclin B1 depends on E2F target genes [47], so decrease in phospho-RB levels would also likely result in decreased cyclin B1 levels.
A previous study of palbociclib in KRAS mutant pancreatic cancer cell lines showed concern that though palbociclib monotherapy inhibited cell proliferation, it appeared to also increase epithelial mesenchymal transition (EMT) in cell lines with wild-type SMAD4 [48]. In that study, CDK4/6 inhibition or knockdown resulted in induction of the EMT markers beta-catenin, Slug, N-cadherin, and vimentin. However, in our experiments with both KRAS mutant CRC cell lines and KRAS mutant PDX, there were no significant changes in protein levels of E-cadherin, N-cadherin, or beta-catenin in vitro or in vivo. Notably, in vivo results were obtained after 21 days of treatment. Several of the cell lines used, including HCT116 and Lovo, were wild-type in SMAD4 [49], as was the PDX tested. In vitro, there were also no differences in vimentin or fibronectin. There was a trend toward increased fibronectin level in vivo with combination MEK and CDK4/6 inhibitors that was not observed with CDK4/6 inhibitor monotherapy. This result may be different than the in vitro result due to the longer duration of treatment in the in vivo experiment or due to stromal cells increasing fibronectin expression. While increased fibronectin is commonly associated with EMT, fibronectin levels may also be altered due to other phenomena, and increased fibronectin expression has even been associated with increased cell senescence [50], so elevated fibronectin in isolation is not sufficient to state there is an increase in EMT. Indeed, inhibition of CDK4 in a triple-negative breast cancer cell line caused decreases in the EMT-associated transcription factors Snail and Twist and decreases in phosphorylated Smad3 [51, 52]. Thus, differences in EMT or other feedback pathways in response to CDK4/6 inhibition are likely to be cell-type-specific, but warrant further investigation.
Though the combination of CDK4/6 inhibitors and MEK inhibitors has previously been found to be effective in NRAS-mutant melanoma models [32], we felt that empirically assessing the efficacy of the combination in KRAS-mutant CRC models was necessary to justify consideration of clinical trials in patients with CRC, especially given the many distinctions between RAS-mutated melanomas and CRCs. First, RAS-mutated melanoma is predominantly NRAS-mutated at codon 61, while RAS-mutated CRC is predominantly KRAS-mutated at codons 12 and 13. NRAS and KRAS mutations have distinct phenotypic effects on resulting tumors in CRC genetically engineered mouse models [53], and mutations at different codons even within NRAS cause different phenotypes [54]. Second, RAS-mutant melanoma and CRC have markedly different patterns of concomitant genomic alterations, such as CDKN2A loss [55, 56], which are likely to be associated with sensitivity to CDK4/6 inhibitors [57]. Third, MEK inhibitor monotherapy is more effective in clinical trials of NRAS-mutant melanoma than in KRAS-mutant CRC, as early phase III trial data of MEK162 (binimetinib) in NRAS-mutant melanoma showed the study met its primary endpoint of improvement in progression-free survival (unpublished data), while MEK inhibitors had no evidence of efficacy in phase I studies in RAS-mutant CRCs [58]. Finally, the tissue-specific context of oncogene mutations is clearly critical in determining sensitivity to targeted therapies; notably, single-agent BRAF inhibitors or combination of BRAF and MEK inhibitors are effective inBRAF-mutant melanomas [59, 60], while they are not effective in BRAF-mutant CRCs [61, 62]. Given these concerns, our data demonstrated preclinical efficacy of combination MEK and CDK4/6 inhibitors in RAS-mutant CRCs. This data also complements the recent report of the combination of palbociclib and trametinib in KRAS-mutant CRC PDX models [63], but demonstrates that inhibition of phospho-S6 is a potential pharmacodynamic biomarker, and uniquely demonstrates that this inhibition is synergistic with the combination of CDK4/6 and MEK inhibitors in a broader number and mutational diversity of cell lines, including “atypical” KRAS A146T mutant CRC PDX models. Our data has justified development of a planned phase II clinical trial of the combination in patients with refractory metastatic RAS-mutant CRC.
In summary, we evaluated a novel combination of MEK and CDK4/6 inhibitors in KRAS mutant CRCs and found synergistic inhibition of cell growth in vitro, with contributions of decreased proliferation, increased apoptosis, and increased senescence; and we subsequently found the combination of MEK and CDK4/6 inhibitors caused tumor regression in vivo in cell line and patient-derived xenografts. We also described inhibition of phosphorylation of ribosomal protein S6 to a greater extent with the combination of MEK and CDK4/6 inhibitors compared to either alone. Though our studies were focused on KRAS mutant CRC, this does not rule out activity in RAS wild-type or BRAF mutant CRC, and further study is warranted. Given the more limited number of options in treatment of KRAS mutant CRC compared to KRAS wild-type CRC, the development of effective therapies for this patient population is an area of unmet need. Combinatorial approaches targeting both the MAPK pathway and cell cycle regulatory pathways merit further preclinical and clinical investigation.
MATERIALS AND METHODS
Cell culture and drugs
A panel of KRAS mutant CRC cell lines was cultured in 1:1 DMEM/Ham’s F12 media with 10% fetal bovine serum, 1% penicillin/streptomycin, and 2 mM L-glutamine. Palbociclib/PD0332991 (Selleck Chemicals) and MEK162 (Selleck Chemicals) were dissolved in dimethyl sulfoxide (DMSO) as 10 mM and 30 mM stocks, respectively. Lovo cells were obtained from ATCC. HCT116, SW480, SKCO1, SW403, SW837, LS174T, SW948, SW1116, LS1034, and T84 cell identity was verified by short tandem repeat (STR) analysis.
Cell proliferation and colony formation assays
Cells were seeded in a 96-well plate at 2000–6000 cells/well, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 72 hours. Cell proliferation was determined using MTS solution (Promega), and formal assessment for synergy performed per the Chou-Talalay method [64, 65] using CompuSyn (ComboSyn, Inc, Paramus, NJ). For colony formation assay, cells were seeded in a 6-well plate at 8000–80,000 cells/well, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 2–3 weeks. Cell colonies were fixed with ice-cold methanol and stained with 1% crystal violet. The density of colonies over the plate area was quantified by ImageJ (NIH) [66]. For detection of exposed phosphatidylserine residues reflective of apoptosis, Annexin V-FITC apoptosis assay kit (BD Biosciences) was used. Cells were seeded at 500,000 cells/well in 6-well plates, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 72 hours. Cells were washed and stained with annexin V-FITC and propidium iodide, and flow cytometry was performed using the Gallios flow cytometer (Beckman Coulter) and analyzed with Kaluza flow analysis software (Beckman Coulter). To measure cell senescence, treated cells were stained for senescence-associated beta-galactosidase (Chemicon). Cells were seeded at 500,000 cells/well in 6-well plates, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 72 hours. Cells were washed with PBS, fixed with glutaraldehyde and methanol, washed twice more, and then incubated overnight in the dark at 37°C under ambient atmospheric conditions with X-Gal. Subsequently, cells were washed, and 10 high-powered light microscopy images of each well were captured, and cells with blue staining were manually counted.
Protein analysis
Reverse-phase protein array analysis (RPPA) was performed as previously described [67]. Briefly, cells were seeded at 500,000 cells/well in 6-well plates, incubated overnight at 37°C to allow adhesion, and then treated with inhibitors for 24 hours. Cellular proteins were collected and denatured by 1% sodium dodecyl sulfate (SDS) with β-mercaptoethanol. The ratio of values for each treatment within each cell line compared to control was determined. To construct a heatmap, these ratios were log2 transformed, and proteins with at least two cell lines with at least 1.74-fold increase or decrease of protein expression compared to control were filtered. Protein values were clustered using Gene Cluster 3.0 [68], and heatmap was created using Java Treeview version 1.1.6r4 (http://jtreeview.sourceforge.net) [69].
For immunoblotting, phospho-p42/44 ERK T202/Y204, p42/44 ERK1/2, phospho-RB Ser780, RB, and phospho-S6 Ser235/236 were purchased from Cell Signaling Technology (Danvers, MA). Anti-beta-actin was obtained from Sigma-Aldrich. Anti-mouse and anti-rabbit horseradish peroxidase-linked secondary antibodies were obtained from Cell Signaling Technology.
In vivo murine xenografts
CD-1 nude 6-week old female mice (Charles River) were injected with 0.2 ml Lovo cell suspension subcutaneously in the right flank and monitored for tumor growth. After tumors were established with median tumor volume exceeding 100 mm3, treatment of 10 mice/arm was commenced via oral gavage with either vehicle control per os daily, trametinib 3 mg/kg per os every 2 days, palbociclib 100 mg/kg per os daily, or combination of trametinib 3 mg/kg per os every 2 days and palbociclib 100 m/kg per os daily for 21 days. Tumor size and mouse weight were measured every 3–4 days. After 21 days, treatment was discontinued and mice were sacrificed.
Additionally, immunodeficient female nude mice were injected with one million cells of HCT116 or SW480 subcutaneously in the flank and monitored for tumor growth. After tumors were established with median tumor volume exceeding 100 mm3, treatment of 5–8 mice/arm was commenced with chow containing combination trametinib and palbociclib or control chow. The chow was designed to deliver a dose equivalent to 0.75 mg/kg trametinib and 75 mg/kg palbociclib. Tumor size and mouse weight were measured every 3–5 days. Control mice were sacrificed after 11 days due to tumor enlargement causing morbidity. After 29–32 days, treatment was discontinued and mice were sacrificed.
Patient-derived xenografts
Primary human-tumor xenograft models were established as previously described [70]. Tumor specimens were obtained from patients with metastatic colorectal cancer at the University of Texas MD Anderson Cancer Center, and all patients provided informed written consent for specimens to be used for research purposes including implantation in xenografts. Samples were obtained with approval of the institutional review board. Xenografts were propagated in NU/J 6-week old female mice (Jackson Laboratory). After tumors were established with median tumor volume exceeding 300 mm3, treatment of 10 mice/arm was commenced via oral gavage with either vehicle control daily, trametinib 3 mg/kg every 2 days, palbociclib 100 mg/kg daily, or combination of trametinib 3 mg/kg every 2 days and palbociclib 100 m/kg daily for 21 days. Tumor size and mouse weight were measured every 3–4 days. After 21 days, treatment was discontinued and mice were sacrificed. Tumors from 3–4 mice per arm were excised, segmented, and immediately flash frozen in liquid nitrogen. Frozen tumor tissue was homogenized in lysis buffer (1% Triton X-100, 50 mM HEPES at pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 100 mM NaF, 10 mM sodium pyrophosphate, 1 mM Na3VO4, 10% glycerol, protease and phosphatase inhibitors) and centrifuged. Protein concentration was determined, and lysates were denatured with 4 × SDS sample buffer with 10% beta-mercaptoethanol, and a final volume of 50 μl of 1 μg/μl denature protein lysate was sent for RPPA as described above.
Immunohistochemical stains
Five-micron thick sections were cut and mounted from a formalin fixed paraffin-embedded tissue block on microscope slides and baked at 60°C for 1 hour. Slides were deparaffinized in xylene 3 times for 5 minutes each, washed with 100% ethanol 3 times for 5 minutes each, and rehydrated in a series of 95%, 70%, 50% ethanol dilutions in distilled water for 5 minutes each. Slides were heated in 10 mM sodium citrate, pH 6.0, with 0.05% Tween-20 in a Decloaker chamber (Biocare Medical, Concord, CA) at 95°C for 30 minutes, then cooled to 90°C for 10 seconds. For each stain, slides were washed with 3% H2O2 in 1 × TBS for 10 minutes, then in Rodent Block M (Biocare Medical) for 20 min, and then with primary antibody for 30 minutes at room temperature. Primary antibodies were rabbit anti-Ki67 (Thermo Scientific) at 1:500 dilution or rabbit anti-phospho S6 ribosomal protein (Ser235/236) (Cell Signaling) at 1:200 dilution. Afterward, slides were incubated with Rabbit-on-Rodent HRP-polymer (Biocare Medical) for 25 minutes, stained with DAB for 5 minutes, and washed with deionized water. Slides were counterstained with Harris hematoxylin for 60 seconds and washed under water for 1 minute, and then dehydrated in 2 changes of 95% ethanol, 3 changes of 100% ethanol, and 3 changes of xylene. Images of representative areas of the slides were captured. The percentage of nuclear area stained for Ki67 was quantitated using the ImmunoRatio plugin for ImageJ [71]. The percentage of cells with cytoplasmic staining of phospho-S6 was manually counted.
Statistical analyses
Statistical calculations were performed using GraphPad Prism version 6.07. Independent samples t-tests were used to compare results between two groups. Additional details are provided in Figure legends.
CONFLICTS OF INTEREST
There are no significant conflicts of interest to disclose.
GRANT SUPPORT
ASCO Conquer Cancer Foundation Young Investigator Award (M.S.L.); generous philanthropic contributions to the University of Texas MD Anderson Moon Shots Program, including support from the Robert J. and Helen C. Kleberg Foundation (C.T., T.P.H.); NIH R01 CA172670 (G.P., S.K.); NIH R01 CA185504 (G.P.); and NIH P50 CA093459 (L.N.K.)
REFERENCES
1. The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7.
2. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013; 369:1023–34.
3. Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, Sandler RS, Kim HJ, Keku TO, Der CJ. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther. 2009; 8:834–43.
4. Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K, Ricarte Filho JC, Persaud Y, Levine DA, Fagin JA, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010; 70:5901–11.
5. Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB, Rosen N. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010; 70:6804–14.
6. Jing J, Greshock J, Holbrook JD, Gilmartin A, Zhang X, McNeil E, Conway T, Moy C, Laquerre S, Bachman K, Wooster R, Degenhardt Y. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012; 11:720–9.
7. Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012; 18:2515–25.
8. Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, Peddareddigari VG, Le NT, Hart L, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012; 13:773–81.
9. Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, Schlicker A, Wessels LF, Smit EF, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014; 7:86–93.
10. Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, Soriano R, Forrest WF, Heldens S, Chen H, Toy K, Ha C, Zhou W, et al. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012; 11:1143–54.
11. Lamba S, Russo M, Sun C, Lazzari L, Cancelliere C, Grernrum W, Lieftink C, Bernards R, Di Nicolantonio F, Bardelli A. RAF suppression synergizes with MEK inhibition in KRAS mutant cancer cells. Cell Rep. 2014; 8:1475–83.
12. Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, Engelman JA. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012; 72:3228–37.
13. Van Schaeybroeck S, Kalimutho M, Dunne PD, Carson R, Allen W, Jithesh PV, Redmond KL, Sasazuki T, Shirasawa S, Blayney J, Michieli P, Fenning C, Lenz HJ, et al. ADAM17-dependent c-MET-STAT3 signaling mediates resistance to MEK inhibitors in KRAS mutant colorectal cancer. Cell Rep. 2014; 7:1940–55.
14. Albeck JG, Mills GB, Brugge JS. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol Cell. 2013; 49:249–61.
15. Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996; 271:20608–16.
16. Winston JT, Coats SR, Wang YZ, Pledger WJ. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996; 12:127–34.
17. Resnitzky D, Reed SI. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995; 15:3463–9.
18. Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993; 7:331–42.
19. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004; 3:1427–38.
20. Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, Torres R, Ajamie RT, Wishart GN, Flack RS, Neubauer BL, Young J, Chan EM, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. 2014; 32:825–37.
21. Rader J, Russell MR, Hart LS, Nakazawa MS, Belcastro LT, Martinez D, Li Y, Carpenter EL, Attiyeh EF, Diskin SJ, Kim S, Parasuraman S, Caponigro G, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013; 19:6173–82.
22. Yamamoto H, Soh JW, Monden T, Klein MG, Zhang LM, Shirin H, Arber N, Tomita N, Schieren I, Stein CA, Weinstein IB. Paradoxical increase in retinoblastoma protein in colorectal carcinomas may protect cells from apoptosis. Clin Cancer Res. 1999; 5:1805–15.
23. Collins MJ, Napoli I, Ribeiro S, Roberts S, Lloyd AC. Loss of Rb cooperates with Ras to drive oncogenic growth in mammalian cells. Curr Biol. 2012; 22:1765–73.
24. Carriere C, Gore AJ, Norris AM, Gunn JR, Young AL, Longnecker DS, Korc M. Deletion of Rb accelerates pancreatic carcinogenesis by oncogenic Kras and impairs senescence in premalignant lesions. Gastroenterology. 2011; 141:1091–101.
25. Dean JL, McClendon AK, Stengel KR, Knudsen ES. Modeling the effect of the RB tumor suppressor on disease progression: dependence on oncogene network and cellular context. Oncogene. 2010; 29:68–80.
26. Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaria D, Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010; 18:63–73.
27. Mao CQ, Xiong MH, Liu Y, Shen S, Du XJ, Yang XZ, Dou S, Zhang PZ, Wang J. Synthetic lethal therapy for KRAS mutant non-small-cell lung carcinoma with nanoparticle-mediated CDK4 siRNA delivery. Mol Ther. 2014; 22:964–73.
28. Flaherty KT, Lorusso PM, Demichele A, Abramson VG, Courtney R, Randolph SS, Shaik MN, Wilner KD, O’Dwyer PJ, Schwartz GK. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin Cancer Res. 2012; 18:568–76.
29. Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD, Courtney R, O’Dwyer PJ. Phase I study of PD 0332991, a cyclin-dependent kinase inhibitor, administered in 3-week cycles (Schedule 2/1). Br J Cancer. 2011; 104:1862–8.
30. Infante JR, Shapiro G, Witteveen P, Gerecitano JF, Ribrag V, Chugh R, Issa I, Chakraborty A, Matano A, Zhao X, Parasuraman S, Cassier P. A phase I study of the single-agent CDK4/6 inhibitor LEE011 in pts with advanced solid tumors and lymphomas. J Clin Oncol. 2014; 32:abstr. 2528^.
31. O’Hara MH, Edmonds C, Farwell M, Perini RF, Pryma DA, Teitelbaum UR, Giantonio BJ, Damjanov N, Lal P, Feldman MD, Zhang PJ, Mankoff DA, Gallagher M, et al. Phase II pharmacodynamic trial of palbociclib in patients with KRAS mutant colorectal cancer. J Clin Oncol. 2015; 33:abstr 626.
32. Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, Bender RP, Chu GC, Flaherty KT, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012; 18:1503–10.
33. Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, Davies MA. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010; 70:8736–47.
34. Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, Brown RD, Godfrey JT, Winokur D, Walsh J, Mino-Kenudson M, Maheswaran S, Settleman J, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. 2013; 5:196ra98.
35. Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007; 1773:1299–310.
36. Sarthy AV, Morgan-Lappe SE, Zakula D, Vernetti L, Schurdak M, Packer JC, Anderson MG, Shirasawa S, Sasazuki T, Fesik SW. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther. 2007; 6:269–76.
37. Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009; 137:835–48.
38. Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, Kelly G, Saunders B, Howell M, Downward J, Hancock DC. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012; 22:1227–45.
39. Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004; 24:3112–24.
40. Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006; 31:342–8.
41. Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007; 282:14056–64.
42. Rosner M, Hengstschlager M. Evidence for cell cycle-dependent, rapamycin-resistant phosphorylation of ribosomal protein S6 at S240/244. Amino Acids. 2010; 39:1487–92.
43. Koehler M, VanArsdale TL, Shields D, Arndt K, Yuan J, Lee N, Eisele K, Chionis J, Cao J, Painter CL. Mechanism of action for combined CDK4/6 and ER inhibition in ER positive breast cancer. Ann Oncol. 2014; 25:i21–i2.
44. Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, Sicinski P. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011; 20:620–34.
45. Cholewa BD, Liu X, Ahmad N. The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res. 2013; 73:6848–55.
46. Tategu M, Nakagawa H, Sasaki K, Yamauchi R, Sekimachi S, Suita Y, Watanabe N, Yoshid K. Transcriptional regulation of human polo-like kinases and early mitotic inhibitor. J Genet Genomics. 2008; 35:215–24.
47. Lukas C, Sorensen CS, Kramer E, Santoni-Rugiu E, Lindeneg C, Peters JM, Bartek J, Lukas J. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature. 1999; 401:815–8.
48. Liu F, Korc M. Cdk4/6 inhibition induces epithelial-mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther. 2012; 11:2138–48.
49. Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF, Tomlinson IP. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci U S A 2001; 98:9719–23.
50. Kumazaki T, Robetorye RS, Robetorye SC, Smith JR. Fibronectin expression increases during in vitro cellular senescence: correlation with increased cell area. Exp Cell Res. 1991; 195:13–9.
51. Tarasewicz E, Rivas L, Hamdan R, Dokic D, Parimi V, Bernabe BP, Thomas A, Shea LD, Jeruss JS. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle. 2014; 13:3191–201.
52. Zelivianski S, Cooley A, Kall R, Jeruss JS. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol Cancer Res. 2010; 8:1375–87.
53. Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008; 40:600–8.
54. Burd CE, Liu W, Huynh MV, Waqas MA, Gillahan JE, Clark KS, Fu K, Martin BL, Jeck WR, Souroullas GP, Darr DB, Zedek DC, Miley MJ, et al. Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov. 2014; 4:1418–29.
55. Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015; 161:1681–96.
56. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7.
57. Young RJ, Waldeck K, Martin C, Foo JH, Cameron DP, Kirby L, Do H, Mitchell C, Cullinane C, Liu W, Fox SB, Dutton-Regester K, Hayward NK, et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014; 27:590–600.
58. Messersmith WA, Falchook GS, Fecher LA, Gordon MS, Vogelzang NJ, DeMarini DJ, Peddareddigari VG, Xu Y, Bendell JC, Infante JR. Clinical activity of the oral MEK1/MEK2 inhibitor GSK1120212 in pancreatic and colorectal cancer. J Clin Oncol. 2011; 29:suppl; abstr 246.
59. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–16.
60. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA, 3rd, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367:1694–703.
61. Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, Morris V, Janku F, Dasari A, Chung W, Issa JP, Gibbs P, James B, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J Clin Oncol. 2015; 33:4032–8.
62. Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, Pierobon M, Sun P, Cunningham E, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J Clin Oncol. 2015; 33:4023–31.
63. Ziemke EK, Dosch JS, Maust JD, Shettigar A, Sen A, Welling TH, Hardiman KM, Sebolt-Leopold JS. Sensitivity of KRAS Mutant Colorectal Cancers to Combination Therapy that Co-Targets MEK and CDK4/6. Clin Cancer Res. 2015.
64. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010; 70:440–6.
65. Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006; 58:621–81.
66. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9:671–5.
67. Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006; 5:2512–21.
68. de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004; 20:1453–4.
69. Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004; 20:3246–8.
70. Fiebig HH, Maier A, Burger AM. Clonogenic assay with established human tumour xenografts: correlation of in vitro to in vivo activity as a basis for anticancer drug discovery. Eur J Cancer. 2004; 40:802–20.
71. Tuominen VJ, Ruotoistenmaki S, Viitanen A, Jumppanen M, Isola J. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 2010; 12:R56.