
INTRODUCTION
Breast cancer (BC) is the most common invasive cancer in females both in the developed and developing countries, with an estimated 234,190 new cases and 40,730 deaths expected in the United States in 2015 [1]. Risk factors for developing BC include obesity, lack of physical exercise, drinking alcohol, hormone replacement therapy during menopause, older age, first menstruation at early age, and so on. A familial history of BC also increases the risk of developing BC. Several mutations in BRCA1, BRCA2 and TP53 involve in a very high risk of BC. However, these mutations account for only a small portion of the total BC burden.
Most BCs are derived from the epithelial lining of the ducts or lobules. BC has been traditionally classified based on clinical and histopathologic characteristics such as histologic grade, stage of disease, and receptor status [2]. The classifications can affect the prognosis and the response to treatment. For example, poorly differentiated cancers often have the worst prognosis [3]. Since BC leads to high mortality, the early diagnosis especially the molecular diagnosis is particularly important for the therapy. Traditionally, treatment decisions have been based on tumor histology and three receptor biomarkers including ER (estrogen receptor 1), PR (progesterone receptor), and HER2 (erb-b2 receptor tyrosine kinase 2) [2]. Cancers that do not have any of these three receptor types are called triple-negative BC. They usually express receptors for other hormones [4]. Despite significant improvements in the treatment of BC, new therapies and treatment strategies are still needed.
So far, numerous genes have been found involved in breast tumorigenesis which can be acted as biomarkers for the early diagnosis and further clinical application. Although dozens of related genes have been found, they are insufficient to elucidate the tumorigenesis of BC unless more relevant genes being identified. Therefore, it is an extremely crucial task to discover novel candidate genes. It is time-consuming and cost-spending to discover disease related genes by experiment alone, because the search space is very large. Computational approach is an alternative method which can help investigators to cope with various biological problems, such as analyzing complex biological network [5–9] and identifying novel genes [10–11]. For example, Zhu et al. developed a robust geometric approach for modeling protein-protein interaction networks [8]. Huang et al. presented a model for predicting protein-protein interactions based on protein-protein correlation using least squares regression [9]. Deng et al. proposed a method to predict novel genes associated with cervical cancer through gene co-expression networks [10]. In addition, several studies have reported prognostic gene expression signatures for BC [12–14]. However, these studies have generally been limited by specific BC subtypes. Hence, development of a more robust molecular predictor that overcomes BC subtype variability is necessary as well as identification of novel genes.
In this study, a computational method was built to discover BC candidate genes based on known BC related genes retrieved from BCGD (the Breast Cancer Gene Database, http://www.tumor-gene.org/tgdf.html). After applying the subnetwork extraction algorithms with gene co-expression and protein-protein interaction (PPI) data, we obtained three networks containing the known BC related genes (denoted as seed genes) and the candidate genes (denoted as linker genes). Through comparing these three subnetworks, we found seven common candidate genes. Further analysis suggests that all of these seven genes are consistent with previous reports that they have relationship with BC or cancer. Using this 7-genes signature, we observed significant differences in relapse-free survival between low-risk and high-risk BC patients in Kaplan–Meier analyses. This analysis provides important insight into the subnetwork biomarkers associated with BC and the identified seven genes may be readily utilized for prognostication and risk-stratification of BC patients.
RESULTS AND DISCUSSION
Strategy for prediction of breast cancer candidate genes
Our goal is to use seed genes and a common biological network between gene co-expression and PPI networks with the subnetwork extraction algorithms to identify candidate genes that are related to the pathogenesis of BC. The method was based on three steps (Figure 1).
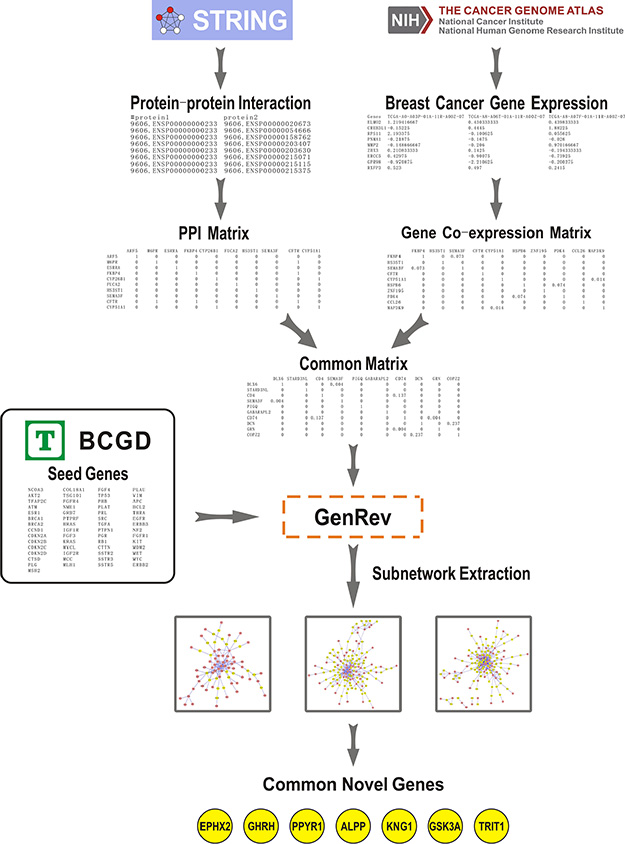
Figure 1: Summary workflow for identifying BC related candidate genes. The approach was based on three steps: (1) We obtained a PPI network derived from STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) and constructed a co-expression network using gene expression data from The Cancer Genome Atlas (TCGA). A common network was generated through comparing the two networks. (2) Seed genes and the common network were imported into GenRev which was used to extract three subnetworks with different extraction algorithms. (3) 7 common genes were found in the obtained subnetworks which were considered as key candidate genes related to BC.
(1) We obtained a PPI network derived from STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://string-db.org/), which is a database of known and predicted protein interactions [15]. Gene expression data of 531 BC tumor samples were collected from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov) [16]. The co-expression network was constructed using R software package “WGCNA” [17]. Through comparing the PPI network and the gene co-expression network, a common biological network containing both co-expression and PPI information was generated. Table 1 illustrated the number of nodes and edges in PPI network, co-expression network and their common network, respectively.
Table 1: Numbers of nodes and edges in PPI network, co-expression network and their common network were illustrated
|
PPI Network |
Co-expression Network |
Common Network |
|---|---|---|---|
Nodes |
17460 |
17325 |
9534 |
Edges |
4850628 |
2303648 |
148182 |
(2) 57 seed genes were collected from BCGD, after removing 11 genes which are not included in the common network (Table 2). Seed genes and the common network were then imported into GenRev [18], a software developed to explore the functional relevance genes. We used the subnetwork extraction algorithms in GenRev for extracting three subnetworks (see more details in the Materials and Methods section).
Table 2: Seed genes were collected from the breast cancer gene database (BCGD)
Gene Names |
|||
|---|---|---|---|
NCOA3 |
COL18A1 |
FGF4 |
PLAU |
AKT2 |
TSG101 |
TP53 |
VIM |
TFAP2C |
FGFR4 |
PHB |
APC |
ATM |
NME1 |
PLAT |
BCL2 |
ESR1 |
GRB7 |
PRL |
THRA |
BRCA1 |
PTPRF |
SRC |
EGFR |
BRCA2 |
HRAS |
TGFA |
ERBB3 |
CCND1 |
IGF1R |
PTPN1 |
NF2 |
CDKN2A |
FGF3 |
PGR |
FGFR1 |
CDKN2B |
KRAS |
RB1 |
KIT |
CDKN2C |
MYCL |
CTTN |
MDM2 |
CDKN2D |
IGF2R |
SSTR2 |
MET |
CTSD |
MCC |
SSTR3 |
MYC |
PLG |
MLH1 |
SSTR5 |
ERBB2 |
MSH2 |
|
|
|
(3) There were 7 common genes found in the three subnetworks, which were considered as key candidate genes related to BC. Then we analyzed these genes by retrieving existing literature and assessed the prognostic value of this 7-genes signature using the transcriptomic data by Kaplan Meier plotter, an online survival analysis software [19].
A summary workflow for identifying genes critical to BC is illustrated in Figure 1.
Subnetwork
As described in the Materials and Methods section, proteins in a PPI network or genes in a co-expression network may share some common or similar features. Therefore, after importing seed genes and the common network which represented both gene co-expression and PPI information into GenRev software, we searched the subnetworks connecting known BC related genes by three methods: not-weighted Klein-Ravi algorithm [20], not-weighted limited k-walk algorithm and edge-weighted limited k-walk algorithm [21].
In this work, the linker genes were considered as the candidate genes related to BC that we found in the three obtained subnetworks. The whole linker genes were listed in Table S1. Numbers of edges, seeds and linkers in each network from different methods were showed in Table 3. As an example, Figure 2 illuminated the subnetwork extracted by not-weighted Klein-Ravi algorithm which contained 136 edges and 79 nodes (57 seed and 22 linker genes). The subnetworks extracted by not-weighted and edge-weighted limited k-walk algorithm were illuminated in Figures S1 and S2, respectively. Cytoscape software was used for visualizing subnetworks [22].
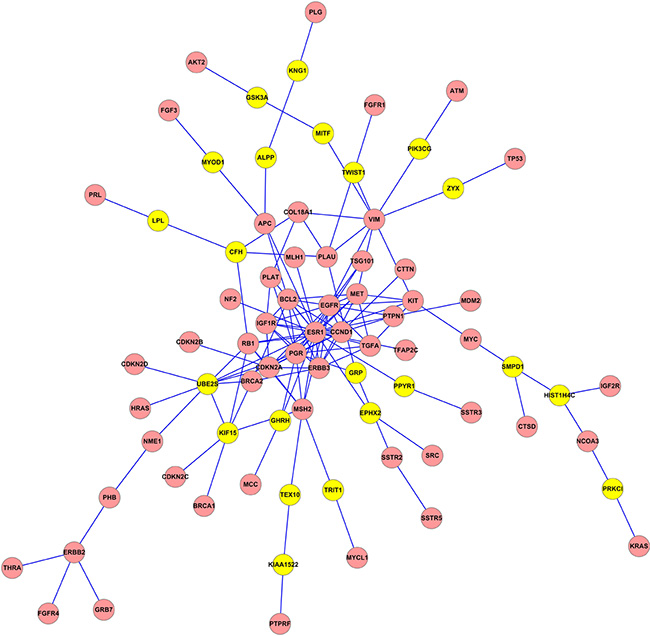
Figure 2: The subnetwork extracted by not-weighted Klein-Ravi algorithm.
Table 3: Numbers of edges, seeds and linkers in the subnetworks obtained from different methods
Algorithm |
Edges |
Seeds |
Linkers |
|---|---|---|---|
Steiner |
136 |
57 |
22 |
Kwalk (not-weighted) |
268 |
57 |
87 |
Kwalk (edge-weighted) |
348 |
57 |
83 |
Linkers refer to the novel genes. Steiner and Kwalk refer to the Klein-Ravi algorithm and the limited k-walk algorithm, respectively.
Analysis of candidate genes
After comparing the genes from the three extracted subnetworks, we obtained 7 overlapping genes which might have a strong relationship with BC. Subsequent analysis indicates that six of these seven key genes (EPHX2, GHRH, ALPP, KNG1, GSK3A and TRIT1) have been shown as BC associated genes, and one (PPYR1) as cancer associated gene based on the existing literature. In the following we will discuss them one by one.
The EPHX2 is the gene encodes a member of the epoxide hydrolase family which is found in both the cytosol and peroxisomes. It binds to specific epoxides and converts them to the corresponding dihydrodiols. Previous study has revealed that it can be targeted by tamoxifen in the treatment of BC [23] and downregulated by GW9662, a potent antagonist of PPARgamma that inhibits growth of breast tumor cells [24]. The protein encoded by GHRH is a member of the glucagon family of proteins. The encoded preproprotein is cleaved to generate somatoliberin, which acts to stimulate growth hormone release from the pituitary gland. Antagonists of this gene inhibit growth of various human cancers including BC. Splice variants of GHRH receptors could mediate the responses to GHRH and GHRH antagonists in BC through Ca2+-, cAMP- and PKC-dependent mechanisms [25]. The PPYR1 gene encodes a member of neuropeptide Y family which is one of the most relevant neuropeptides related to tumor progression [26–27]. The protein encoded by ALPP is an alkaline phosphatase, a metalloenzyme that catalyzes the hydrolysis of phosphoric acid monoesters. Overexpression of this enzyme has been detected at the surface of various solid tumors [28]. Particularly, this gene is significantly higher expressed in the trastuzumab treated than in the untreated human HER2-amplified breast cancer cell line BT474 and can be an indicator for drug sensitivity [29]. The gene KNG1 uses alternative splicing to generate two different proteins, low molecular weight kininogen and high molecular weight kininogen which is essential for blood coagulation. It has been reported that this gene is up-regulated by proteasome inhibitor in BC [30]. The protein encoded by GSK3A is a multifunctional Ser/Thr protein kinase that is implicated in the control of several regulatory proteins and transcription factors. It also plays a role in the WNT and PI3K signaling pathways which are closely associated with cancer. Induced expression of PTEN in PTEN deficient BC cells, was associated with a marked decrease in the basal phosphorylation of GSK3A and other downstream components of the PI3K signaling cascade, and then suppressed cell cycle progression [31]. The TRIT1 encodes an isopentenyltransferase that is located to the mitochondrion and modifies tRNAs by adding a dimethylallyl group onto the adenine at position 37 which is considered a tumor suppressor [32]. The product of this enzyme, isopentenyladenosine significantly inhibited the BC cell lines MDAMB-361 and MCF7. The mechanism of tumor suppressor activity is associated with inhibition of cell proliferation, blocking DNA synthesis and morphological changes [33]. Overall, these seven key genes are shown to play a direct or indirect role in BC according to previous reports and merits further investigation with respect to their application against BC.
We then developed an expression signature consisting of the seven genes by the Kaplan Meier plotter online survival analysis software [19]. This 7-genes signature could significantly stratify 1660 BC patients according to relapse free survival (HR, 0.55; 95% CI, 0.46 − 0.65; Logrank p = 5.5e−13). As seen in Figure 3, the low-risk group had significantly better relapse free survival than the high-risk group. The 7-genes signature could be established as a useful predictor of disease prognosis in BC patients.
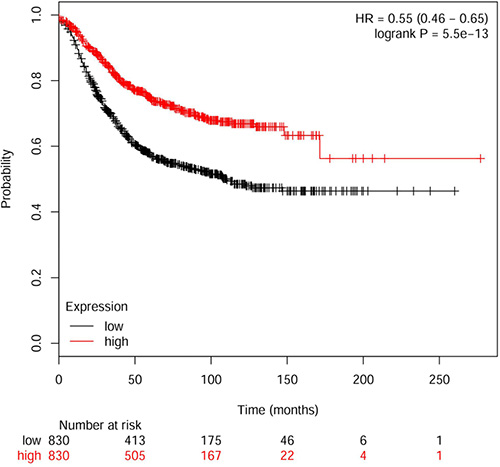
Figure 3: Kaplan–Meier plot of relapse free survival using the 7-genes signature.
Finally, we identified the smallest subnetwork connecting seven key genes derived from the common network representing both PPI and gene co-expression information (Figure 4). The other six genes in this subnetwork are all cancer related: five of them are seed genes and one is MITF gene [34]. Paired t-test analysis showed statistically significant differences in these 13 gene expression levels between BC and normal samples (p-value = 0.049) using the gene expression data of 531 BC and 62 normal tissue samples from TCGA.
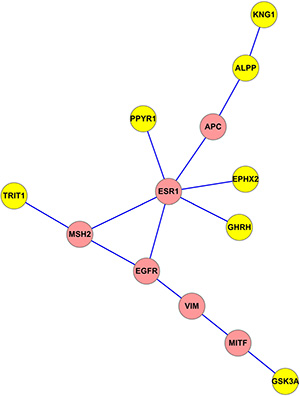
Figure 4: The smallest subnetwork connecting seven key genes derived from the common network.
In summary, the identified seven candidate genes may help to reveal the underlying molecular mechanisms of BC and provide guidance orientation on possible personalized therapeutic regimen selection. It has not escaped our notice that the approach of this study may give a new insight to understand BC as well as other cancers. Besides the PPI and gene co-expression information used in this study, epigenetic and mutation level information also play an important role in BC. In the future work, we will identify driver mutations that have direct effect on a selective growth advantage of BC cells using these multidimensional data sets.
MATERIALS AND METHODS
Protein-protein interaction network
Protein-protein interaction (PPI) networks provide a lot of valuable information to understand cellular function and biological processes. Numerous studies have shown that proteins in the same interaction, that is, they are adjacent in the constructed PPI network, always have some common features [21, 35–38]. It can be further deduced that proteins in the subnetwork connecting known BC related genes are most likely to share similar biological functions as shown in many studies [39–40]. In this study, the PPI network was constructed based on the protein interaction information retrieved from STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://string-db.org/) (9606.protein.links.v9.1) [15], a well-known online interaction repository which include direct (physical) and indirect (functional) associations.
Gene co-expression network
Recently, gene co-expression network has emerged as a new tool for microarray analysis [41–42]. The transcript levels of two co-expressed genes rise and fall together across samples. It is showed that functionally related genes are frequently co-expressed constituting conserved transcription modules [43–44]. Here, gene expression microarray and RNA-Seq data of 531 BC tumor samples were collected from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov) which aims to generate comprehensive, multi-dimensional maps of the key genomic changes in major types and subtypes of cancer [16]. Then a gene co-expression network was constructed using R software package “WGCNA” (Weighted correlation network analysis) which is a comprehensive collection of R functions for performing various kinds of weighted correlation network analysis [17]. The similarity was computed to evaluate the distance between each pair of genes using the function adjacency(). Pearson’s correlation coefficient is used as the co-expression measure. The RNA-Seq data were transformed into log2 scale after adding a constant +1 as described in Meißner [45].
Seed genes
BC related genes were collected from the Breast Cancer Gene Database (BCGD) which is a sub-database of Tumor Gene Family of Databases (TGFD) (http://www.tumor-gene.org/tgdf.html). After removing 11 gene which are absent in the common network representing both PPI and gene co-expression information, we obtained 57 genes as BC related genes (seed genes) with a high level of confidence (Table 2).
Subnetwork extraction
Here, we used a method to discover candidate genes related to BC by constructing a common network contained both PPI and gene co-expression information. So far there are numerous methods that can be used to find the subnetworks. In this study, GenRev [18], a standalone and platform independent software for exploring the functional relevance genes, was used to identify subnetwork. The input files were comprised of seed genes and the common network. The Pearson’s correlation coefficients in co-expression network were used as the weights of edges. Since the common network imported into GenRev contained only weights of edges but not weights of all nodes, we used the not-weighted Klein-Ravi algorithm, not-weighted and edge-weighted limited k-walk algorithm in GenRev [18]. As a result, GenRev was used mapped the genes to the common network and extracted three subnetworks. The subnetwork and the linker genes were visualized using Cytoscape software [22].
Klein–Ravi algorithm
The Klein–Ravi algorithm is one of the algorithms in GenRev, which was proposed to solve the node-weighted Steiner tree problem [20]. The objective of the node-weighted Steiner tree problem was to find a subnetwork with a minimum score which connects all the seeds. The score of a subnetwork was estimated by the sum of the scores of its nodes. We can find more details from the original work [20].
Limited k-walks algorithm
The limited k-walks algorithm is another algorithm in GenRev, which can run randomly in the network by using a Markov chain and build a relevant subnetwork connecting seed nodes [21]. The relevance of an edge and a node related to the seed genes is assessed by the expected times random walk passes starting from one seed to any of the others. By default, weights of all edges were equal to 1. The Pearson’s correlation coefficients in co-expression network were set as weights of edges when the edge-weighted limited k-walk algorithm was used. More details are available in the original work [21].
Kaplan Meier plotter
In this study, an online survival analysis software, Kaplan Meier plotter was used to assess prognosis value of the signature developed by the seven key candidate genes [19]. The Kaplan Meier plotter is capable of evaluating the effect of 54,675 genes on survival using 10,188 cancer samples using the log rank test to compare the survival curves. To analyze the prognostic value of 7 genes, the 3557 relapse-free survival samples were split into two groups according to various quantile expressions of the proposed biomarker. The two patient cohorts were compared by a Kaplan-Meier survival plot, and the hazard ratio with 95% confidence intervals and logrank p-value were calculated.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by National Natural Science Foundation of China (31271817, 61272333, 31301101), the Key Project of Science and Technology of Anhui (1501031099), the Anhui Provincial Natural Science Foundation (1408085QF106), the Specialized Research Fund for the Doctoral Program of Higher Education (20133401120011), and the Technology Foundation for Selected Overseas Chinese Scholars from Department of Human Resources and Social Security of Anhui Province (No. [2014]-243).
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
1. American Cancer Society. Cancer Facts & Figures 2015. Atlanta: American Cancer Society. 2015.
2. Kabel AM, Baali FH. Breast cancer: insights into risk factors, pathogenesis, diagnosis and management. J Cancer Res Treat. 2015; 3:28–33.
3. Zhang Y, Lv F, Yang Y, Qian X, Lang R, Fan Y, Liu F, Li Y, Li S, Shen B. Clinicopathological features and prognosis of metaplastic breast carcinoma: experience of a major chinese cancer center. PLoS One. 2015; 10:e0131409.
4. Santana-Davila R, Perez EA. Treatment options for patients with triple-negative breast cancer. J Hematol Oncol. 2010; 3:b15.
5. Ma H-W, Zeng A-P. The connectivity structure, giant strong component and centrality of metabolic networks. Bioinformatics. 2003; 19:1423–1430.
6. Dale JM, Popescu L, Karp PD. Machine learning methods for metabolic pathway prediction. BMC Bioinformatics. 2010; 11:15.
7. Chen L, Huang T, Shi X-H, Cai Y-D, Chou K-C. Analysis of protein pathway networks using hybrid properties. Molecules. 2010; 15:8177–8192.
8. Zhu L, You Z-H, Huang D-S, Wang B. t-LSE: a novel robust geometric approach for modeling protein-protein interaction networks. PLoS One. 2013; 8:e58368.
9. Huang D-S, Zhang L, Han K, Deng S, Yang K, Zhang H. Prediction of protein-protein interactions based on protein-protein correlation using least squares regression. Curr Protein Pept Sci. 2014; 15:553–560.
10. Deng S-P, Zhu L, Huang D-S. Predicting Hub Genes Associated with Cervical Cancer through Gene Co-expression Networks. IEEE/ACM Trans Comput Biol Bioinform. 2015; 13:27–35.
11. Aure MR, Steinfeld I, Baumbusch LO, Liestøl K, Lipson D, Nyberg S, Naume B, Sahlberg KK, Kristensen VN, Børresen-Dale A-L. Identifying in-trans process associated genes in breast cancer by integrated analysis of copy number and expression data. PLoS One. 2013; 8:e53014.
12. Glas AM, Floore A, Delahaye LJ, Witteveen AT, Pover RC, Bakx N, Lahti-Domenici JS, Bruinsma TJ, Warmoes MO, Bernards R. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 2006; 7:1.
13. Sparano JA, Paik S. Development of the 21-gene assay and its application in clinical practice and clinical trials. J Clin Oncol. 2008; 26:721–728.
14. Filipits M, Rudas M, Jakesz R, Dubsky P, Fitzal F, Singer CF, Dietze O, Greil R, Jelen A, Sevelda P. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin Cancer Res. 2011; 17:6012–6020.
15. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C. STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013; 41:D808–D815.
16. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70.
17. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:1.
18. Zheng S, Zhao Z. GenRev: exploring functional relevance of genes in molecular networks. Genomics. 2012; 99:183–188.
19. Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8:e82241.
20. Klein P, Ravi R. A nearly best-possible approximation algorithm for node-weighted Steiner trees. J Algorithms. 1995; 19:104–115.
21. Dupont P, Callut J, Dooms G, Monette J-N, Deville Y, Sainte B. Relevant subgraph extraction from random walks in a graph. Universite catholique de Louvain, UCL/INGI, Number RR. 2006; 7.
22. Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Data mining in proteomics: from standards to applications. 2011; 291–303.
23. Yuan H, Kopelovich L, Yin Y, Lu J, Glazer RI. Drug-targeted inhibition of peroxisome proliferator-activated receptor-gamma enhances the chemopreventive effect of anti-estrogen therapy. Oncotarget. 2012; 3:345–356. doi: 10.18632/oncotarget.457.
24. Dawany NB, Dampier WN, Tozeren A. Large - scale integration of microarray data reveals genes and pathways common to multiple cancer types. Int J Cancer. 2011; 128:2881–2891.
25. Garcia-Fernandez MO, Schally AV, Varga JL, Groot K, Busto R. The expression of growth hormone-releasing hormone (GHRH) and its receptor splice variants in human breast cancer lines; the evaluation of signaling mechanisms in the stimulation of cell proliferation. Breast Cancer Res Treat. 2003; 77:15–26.
26. Mancino M, Ametller E, Gascón P, Almendro V. The neuronal influence on tumor progression. Biochim Biophys Acta. 2011; 1816:105–118.
27. Iizuka N, Oka M, Yamamoto K, Tangoku A, Miyamoto K, Miyamoto T, Uchimura S, Hamamoto Y, Okita K. Identification of common or distinct genes related to antitumor activities of a medicinal herb and its major component by oligonucleotide microarray. Int J Cancer. 2003; 107:666–672.
28. Pospisil P, Iyer LK, Adelstein SJ, Kassis AI. A combined approach to data mining of textual and structured data to identify cancer-related targets. BMC Bioinformatics. 2006; 7:1.
29. von der Heyde S, Wagner S, Czerny A, Nietert M, Ludewig F, Salinas-Riester G, Arlt D, Beißbarth T. mRNA profiling reveals determinants of trastuzumab efficiency in HER2-positive breast cancer. PLoS One. 2015; 10:e0117818.
30. Kinyamu HK, Collins JB, Grissom SF, Hebbar PB, Archer TK. Genome wide transcriptional profiling in breast cancer cells reveals distinct changes in hormone receptor target genes and chromatin modifying enzymes after proteasome inhibition. Mol Carcinog. 2008; 47:845–885.
31. Lu Y, Lin Y-Z, LaPushin R, Cuevas B, Fang X, Yu SX, Davies MA, Khan H, Furui T, Mao M. The PTEN/MMAC1/TEP tumor suppressor gene decreases cell growth and induces apoptosis and anoikis in breast cancer cells. Oncogene. 1999; 18:7034–7045.
32. Spinola M, Galvan A, Pignatiello C, Conti B, Pastorino U, Nicander B, Paroni R, Dragani TA. Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene. 2005; 24:5502–5509.
33. Spinola M, Colombo F, Falvella FS and Dragani TA. N6 - isopentenyladenosine: A potential therapeutic agent for a variety of epithelial cancers. Int J Cancer. 2007; 120:2744–2748.
34. Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005; 436:117–122.
35. Wurmbach E, Chen Yb, Khitrov G, Zhang W, Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J. Genome - wide molecular profiles of HCV - induced dysplasia and hepatocellular carcinoma. Hepatology. 2007; 45:938–947.
36. Wu J, Vallenius T, Ovaska K, Westermarck J, Mäkelä TP, Hautaniemi S. Integrated network analysis platform for protein-protein interactions. Nat Methods. 2009; 6:75–77.
37. Lee I, Date SV, Adai AT, Marcotte EM. A probabilistic functional network of yeast genes. Science. 2004; 306:1555–1558.
38. Hu Z, Hung J-H, Wang Y, Chang Y-C, Huang C-L, Huyck M, DeLisi C. VisANT 3.5: multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic acids res. 2009; gkp406.
39. Kourmpetis YA, van Dijk AD, Bink MC, van Ham RC, Ter Braak CJ. Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS One. 2010; 5:e9293.
40. Ng K-L, Ciou J-S, Huang C-H. Prediction of protein functions based on function–function correlation relations. Comput Biol Med. 2010; 40:300–305.
41. Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003; 302:249–255.
42. Bergmann S, Ihmels J, Barkai N. Similarities and differences in genome-wide expression data of six organisms. PLoS Biol. 2003; 2:e9.
43. Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004; 14:1085–1094.
44. Van Noort V, Snel B, Huynen MA. The yeast coexpression network has a small - world, scale - free architecture and can be explained by a simple model. EMBO Rep. 2004; 5:280–284.
45. Meißner T, Fisch KM, Gioia L, Su AI. OncoRep: An n-of-1 reporting tool to support genome-guided treatment for breast cancer patients using RNA-sequencing. BMC Med Genomics. 2015; 8:1.